 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Muon studies of Li+ diffusion in LiFePO4 nanoparticles of different polymorphs†
Thomas E.
Ashton
a,
Josefa Vidal
Laveda
a,
Donald A.
MacLaren
b,
Peter J.
Baker
c,
Adrian
Porch
d,
Martin O.
Jones
c and
Serena A.
Corr
*a
aSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: serena.corr@glasgow.ac.uk; Tel: +44 (0)141 3302274
bSUPA, School of Physics and Astronomy, University of Glasgow G12 8QQ, UK
cISIS Pulsed Neutron and Muon Source, STFC Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0QX, UK
dCentre for High Frequency Engineering, School of Engineering, Cardiff University, Cardiff, CF24 3AA, UK
First published on 5th March 2014
Abstract
The lithium diffusion in nanostructured olivine LiFePO4 has been investigated for the first time using muon spectroscopy (μSR). A microwave-assisted approach has been employed for nanoparticle preparation, where the choice of solvent is shown to play an important role in determining particle morphology and crystal chemistry. Two phases have been obtained: Pnma LiFePO4 and the high pressure Cmcm phase. The Li+ diffusion behaviour is strikingly different in both phases, with DLi of 6.25 × 10−10 cm2 s−1 obtained for Pnma LiFePO4 in good agreement with measurements of bulk materials. In contrast, Li+ diffusion is impeded with the addition of the high pressure Cmcm phase, with a lower DLi of 3.96 × 10−10 cm2 s−1 noted. We have demonstrated an efficient microwave route to nanoparticle synthesis of positive electrode materials and we have also shown μSR measurements to be a powerful probe of Li+ diffusion behaviour in nanoparticles.
A Introduction
Olivine structured Pnma LiFePO4 has been the focus of much attention for the development of efficient positive insertion electrodes, as it presents an economical and non-toxic option for a rechargeable Li-ion battery cathode material.1–4 LiFePO4 exhibits a high charge density, good cyclability and is complementary to most conventional polymer electrolytes. Recently, great efforts have been made in the development of nanostructured electrodes due to potential improvements in electrochemical performance, as their small size allows for shorter diffusion pathlengths while increased surface areas improve electrode–electrolyte interactions.5,6Phase pure olivine materials can be obtained using conventional synthetic methods, such as solid-state ceramic routes, sol–gel routes and solvothermal methods.7–11 While high temperature ceramic routes will often yield bulk materials, the choice of solvent in solvothermal reactions can often play a determining role in resulting particle morphology and size. One example of a class of materials finding increasing use as solvents for the preparation of electrode materials is the use of ionic liquids, where elegant control over resulting particle size and shape has recently been demonstrated for the case of the solvothermal synthesis of LiFePO4 and LiMnPO4.12–14 In recent years, microwave-assisted solvothermal methods have appeared as a faster, efficient approach to inorganic materials.15 For example, LiMPO4 (M = Fe, Mn) has been prepared by using a benzyl alcohol approach after only 3 minutes at 180 °C.16 Microwave routes to nanostructured Li2FeSiO4 and Li2MnSiO4 using a tetraethylene glycol solvent have also been reported,17 while recently Nazar and co-workers have established a fast, microwave-assisted polyol route to the triplite LiFeSO4F phase with tetraethylene glycol.18
Here, we report the synthesis of nanoparticulate LiFePO4 using a microwave-assisted solvothermal route. We show how the crystal chemistry and resulting morphology can be controlled by the solvent and iron starting materials employed. We employ two methods combined with microwave heating: a polyol synthesis and an ionothermal route. We also report, for the first time, on the diffusive nature of Li+ through LiFePO4 nanoparticles prepared in this manner using positive muon spin relaxation (μSR). The nature of Li+ diffusion in LiFePO4 continues to attract considerable attention. A number of methods already exist for the study of Li+ diffusion, yet there is significant variation between the results obtained for DLi in LiFePO4 (ranging from ∼10−7 cm2 s−1 from Mössbauer spectroscopy19 to ∼10−14 cm2 s−1 for galvanostatic intermittent titration techniques [GITT]20). Recently, electrochemical methods employed for calculating Li+ diffusion coefficients in thin film electrodes have raised questions due to differences caused by the nature of the diffusion, the electrode surface area and the smoothness of the electrode surface.21–23 Theoretical studies have found that, for LiFePO4, Li+ diffusion is confined to a curved 1-dimension in the [010] direction, with DLi estimates of 10−8 cm2 s−1.24–26 This 1-dimensional diffusion has been shown experimentally using a combination of neutron diffraction and maximum entropy methods.27 Activation barriers for Li+ and electron mobility have also been investigated experimentally using NMR and impedance analysis.28,29
μSR has previously been employed as a sensitive probe for magnetic ordering and also in the investigation of dynamic sample effects.30 It has also been successfully applied to the study of Li+ diffusion in a number of Li-ion battery materials including lithium metal oxides and ternary lithium nitridometallates, where the Li+ diffusion perturbs the muon environment.31–34 μSR has been shown to reliably determine DLi values for LixCoO2, with values obtained close to theoretically predicted values.32,35 Recently, the use of μSR as a probe to study Li+ diffusion in olivines has also been demonstrated for bulk olivine materials, including bulk LiFePO4.36–38
Herein, we examine the Li+ diffusion in nanoparticulate Pnma LiFePO4 and the high pressure Cmcm LiFePO4 phase using μSR for the first time. We observe a thermally activated Li+ hopping regime for nanostructured Pnma LiFePO4, similar to measurements obtained for bulk samples, demonstrating the reliability of this technique for the study of Li+ diffusion. We also examine muon diffusion of the Cmcm LiFePO4 polymorph for the first time in a mixed phase sample of Pnma LiFePO4/Cmcm LiFePO4.
B Synthesis
Powder samples of LiFePO4 were prepared by grinding LiH2PO4 (0.263 g; 2.54 mmol) and FeC2O4·2H2O (0.456 g; 0.254 mmol) in an agate mortar for 10 min and adding to 10 ml of either ethylene glycol (Sample LFP-EG1; Alfa Aesar, 99%) or 1-ethyl-3-methyl imidazolium trifluoromethanesulfonate (EMI-TFMS) (Sample LFP-IL; Solvionic, 99.5%) in 35 ml glass reaction vessels. The mixtures were stirred for 20 minutes before irradiation with microwaves in a CEM Discover SP microwave synthesiser (2.45 GHz) for 3 hours at 250 °C. The products were washed with water (2 × 20 ml), ethanol (2 × 20 ml) and acetone (20 ml), before drying in a vacuum oven at 80 °C overnight. The pale green powders were characterised by X-ray diffraction (XRD) (PANalytical X'Pert powder diffractometer) and scanning electron microscopy (SEM) (Carl Zeiss Sigma variable pressure analytical SEM). SEM samples were prepared on adhesive stubs and coated using a plasma sputter coater with a 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Au:Pt target to avoid charging feedback. Transmission electron microscopy (TEM) was performed on a JEOL ARM instrument, operated at 200 keV. TEM samples were prepared by dispersing the sample in ethanol and dropping the suspension onto an amorphous holey carbon coated grid.
:1, Au:Pt target to avoid charging feedback. Transmission electron microscopy (TEM) was performed on a JEOL ARM instrument, operated at 200 keV. TEM samples were prepared by dispersing the sample in ethanol and dropping the suspension onto an amorphous holey carbon coated grid.
Muon spectroscopy
Spin polarised positive muons were implanted into LiFePO4 samples, where they stop at interstitial sites and decay with a mean lifetime of 2.2 μs. Whilst implanted in the sample, the muon spin direction is affected by the local magnetic field at the stopping site. When the muon decays into a positron and two neutrinos, the positron is preferentially emitted in the direction of the muon spin at the instant of decay. The muon spin polarisation can be followed as a function of time by measuring the asymmetry in the count rate of the decaying positrons, A(t), in two banks of detectors on opposite sides of the sample; (essentially, we monitor the muon's spin through its daughter positron).μSR experiments were carried out at the ISIS pulsed muon and neutron source, using the EMU instrument and data were analysed using the WIMDA program. The samples were prepared by transferring the powders of LiFePO4 (approximately 1 g) into titanium sample holders with a titanium foil window. Ti depolarises muons very weakly and so gives an easy-to-subtract background. In order to probe the lithium diffusion behaviour in two of our samples, we measured a temperature range of 100 K to 400 K at 10 K increments and at 0 G and applied longitudinal fields of 5 and 10 G. Multiple magnetic field measurements give more reliable determinations of simultaneously fitted parameters since it allows greater investigation of how the field distribution experienced by the muon is decoupled by the field applied parallel to the initial muon spin polarisation.
C Results and discussion
Microwave synthesis of nanoparticulate LiFePO4
Ethylene glycol and EMI-TFMS were chosen as solvents for the preparation of LiFePO4 nanoparticles for two reasons:(a) Choice of solvent has been previously shown to heavily influence the resulting nanoparticle shape.39
(b) Both solvents are high boiling point solvents (ethylene glycol boils at 196 °C; EMI-TFMS has a decomposition temperature of 340 °C) with dipole moments which can interact with incoming microwaves to uniformly heat reactants.
Our synthetic approach takes advantage of a solvent's ability to efficiently absorb microwave energy and convert this into heat through the dielectric heating effect.40 A material's dielectric properties can be described by its complex relative permittivity ε = ε1 − iε2, which depends on both frequency and temperature. The real part ε1 (more precisely, the quantity ε1 − 1) is a measure of the ability of the material to be polarized by an electric field, and the imaginary part ε2 is a measure of the efficiency with which the material converts electric field energy into heat. Assuming a uniform internal electric field of magnitude E within a sample of volume V, the time-averaged power dissipated P at some frequency f can be written as P = πε2ε0fE2V.
In order to assess the behaviour of the solvents we have employed in greater detail, we measured the microwave dielectric properties of ethylene glycol and the ionic liquid EMI-TFMS. Measurements were taken in the range 0.01 GHz to 10 GHz using a broadband coaxial probe connected to a microwave network analyser (N5232A PNA-L, Agilent Technologies).41 All measurements were taken at a constant temperature of 27.5 °C and values of complex permittivity were verified using a TM010 microwave cavity operating at 2.45 GHz.42 Results for the frequency dependence of the complex permittivity of both liquids are shown in Fig. 1(a) and (b). We find that ethylene glycol behaves close to that of a classical Debye liquid43 of static permittivity εs = 37.8 ± 0.4 and relaxation frequency of 1.57 ± 0.01 GHz. EMI-TFMS, on the other hand, behaves as a liquid with finite electrical conductivity, whose imaginary (i.e. lossy) permittivity ε2 exhibits the expected frequency variation below about 1 GHz of ε2 ≈ σ/2πε0f ∝ 1/f. From this, we deduce a dc electrical conductivity of σ = 0.96 ± 0.01 S m−1.
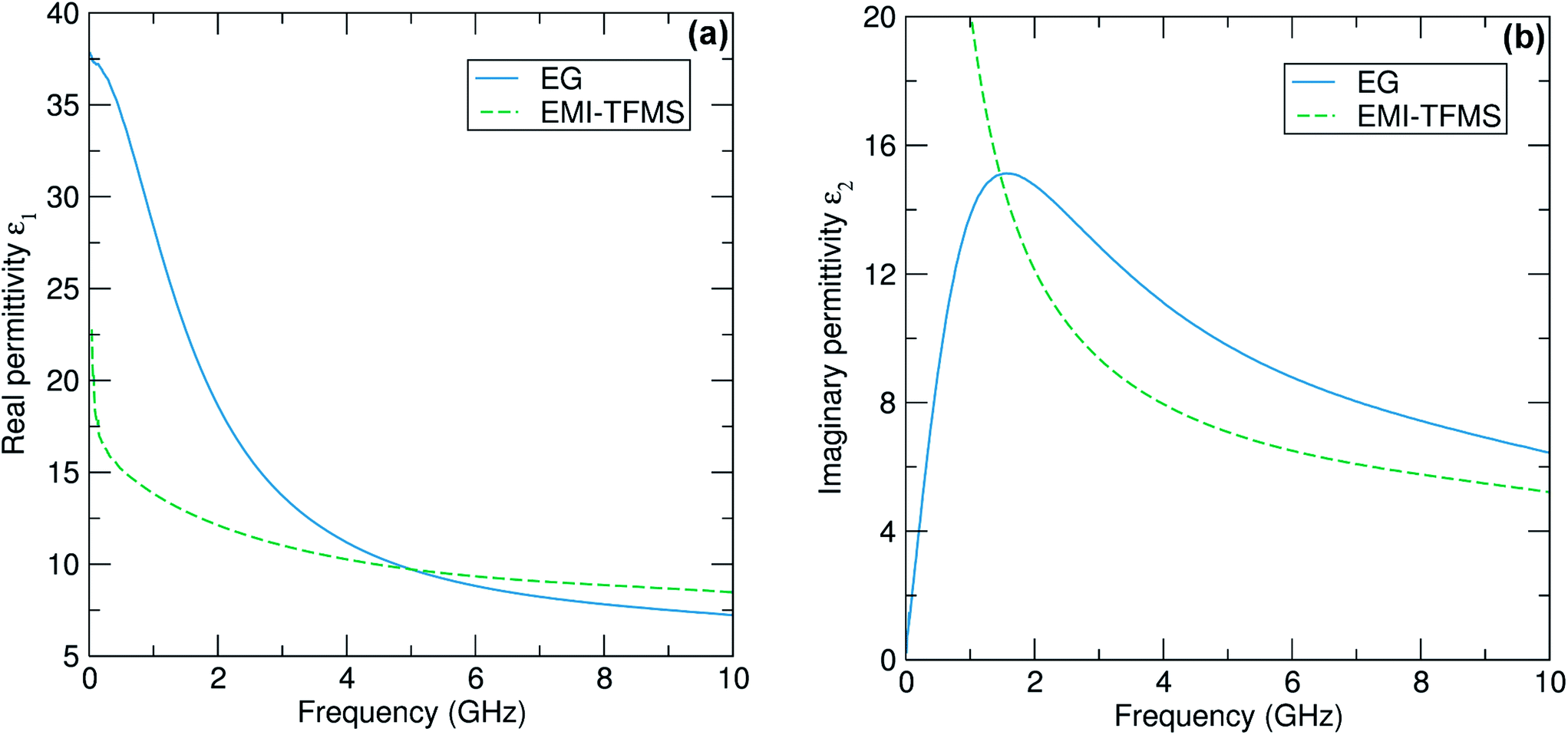 | ||
| Fig. 1 (a) Experimental data for the real part ε1 of the complex permittivity ε measured as a function of frequency for the polar liquid ethylene glycol (EG) and the ionic liquid EMI-TFMS. (b) The same plot, only this time for the imaginary part ε2. No errors bars are shown but all data are subject to a systematic error of ±1% imposed by the aperture module of the coaxial probe used to extract the permittivity data.41 | ||
Some numerical values of complex permittivity of both liquids at spot frequencies of importance for microwave heating applications (namely 915 MHz, 2.45 GHz and 5.8 GHz) are shown in Table 1. The results obtained for ethylene glycol compare well with previously reported values.44 Note that the errors quoted in our data are systematic errors of around ±1% associated with the simple quasi-static model41 used to model the aperture admittance of the coaxial probe to convert microwave reflectance data into complex permittivity values. We find that both solvents have large measured values of ε2, confirming their effectiveness as microwave absorbers. In Fig. 2 we plot the rms power dissipated P (expressed in W cm−3 of solvent) calculated for a fixed internal electric field of 10 kV m−1, as is typical in a microwave heating application, using our measured complex permittivities shown in Fig. 1. We find that that the dissipated power densities are 72 and 95 W cm−3 at 2.45 GHz for EMI-TFMS and ethylene glycol, respectively, which are sufficient to drive the high temperatures required for our reactions.
| 915 MHz | 2.45 GHz | 5.8 GHz | |
|---|---|---|---|
| Ethylene glycol | ε 1 = 29.4 ± 0.3, ε2 = 13.3 ± 0.1 | ε 1 = 16.0 ± 0.2, ε2 = 14.0 ± 0.1 | ε 1 = 9.0 ± 0.1, ε2 = 9.0 ± 0.1 |
| EMI-TFMS | ε 1 = 14.0 ± 0.1, ε2 = 21.8 ± 0.2 | ε 1 = 11.6 ± 0.1, ε2 = 10.6 ± 0.1 | ε 1 = 9.4 ± 0.1, ε2 = 6.6 ± 0.1 |
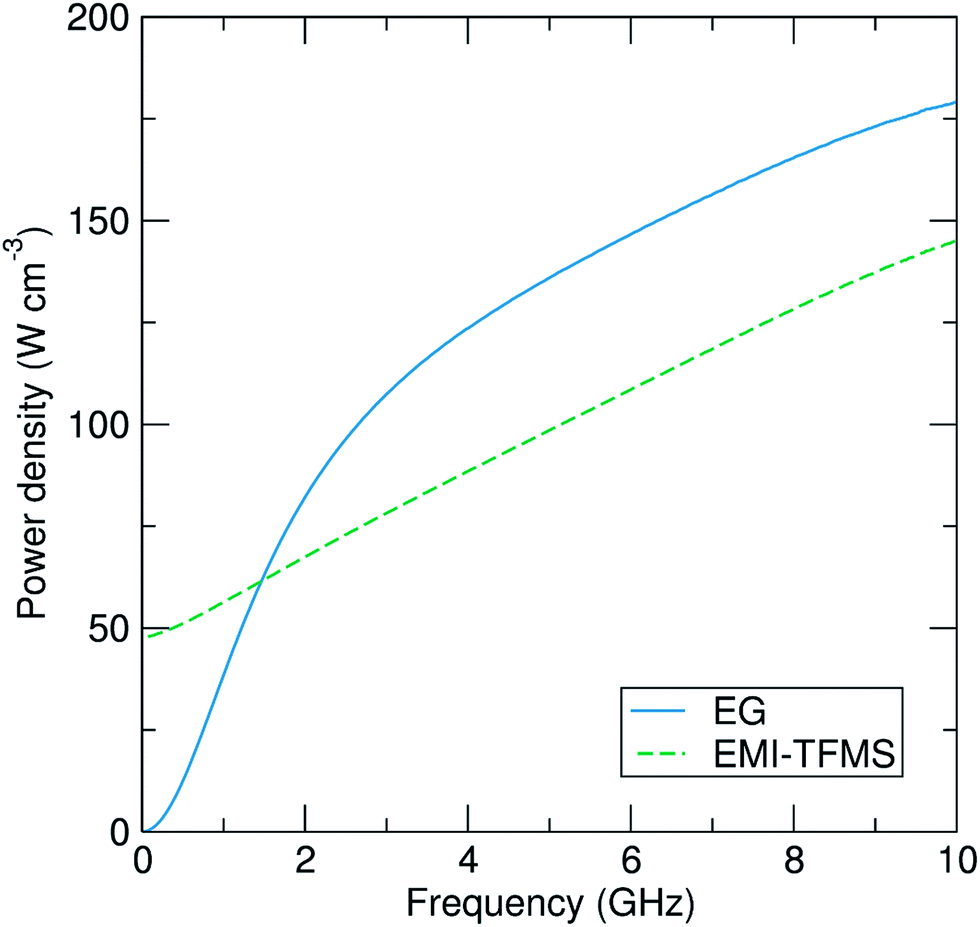 | ||
| Fig. 2 The rms dissipated power density P (expressed in W cm−3), calculated from the permittivity data of Fig. 1 for a uniform internal electric field of magnitude 10 kV m−1, plotted as a function of frequency. | ||
In a typical synthesis, stoichiometric amounts of iron precursor and LiH2PO4 were mixed with 10 ml solvent for 20 min at 30 °C before a heat treatment in the microwave chamber at 250 °C for 3 hours. The results from three experiments are presented here: (1) LFP_EG1 from iron oxalate dihydrate, LiH2PO4 and ethylene glycol solvent, (2) LFP_EG2 from iron acetylacetonate, LiH2PO4 and ethylene glycol (note, temperature is 220 °C here) and (3) LFP_IL from iron oxalate dihydrate, LiH2PO4 and ionic liquid EMI-TFMS solvent.
XRD patterns collected for each dried powder sample are shown in Fig. 3. For sample LFP1_EG with iron oxalate as a starting material and ethylene glycol as a solvent, a two-phase system is found with the pattern plotted in Fig. 3(a) matched to Pnma LiFePO4 and a high pressure LiFePO4 phase which crystallizes in the Cmcm space group. Heating this sample in a tube furnace under Ar at 600 °C for one hour completely transforms the high pressure phase to Pnma LiFePO4. This high pressure Cmcm phase has been realised before by García-Moreno and co-workers at high pressures (tens of kbar) and temperatures (hundreds of degrees).45 Very recently Niederberger and co-workers have observed this phase at much lower reaction temperatures and times (195 °C, 3 min) for a nonaqueous microwave synthesis, whereby a change in the benzyl alcohol–2-pyrrolidinone solvent ratio can be used to tailor the phase obtained.46 In our experiments, the solvent volume is held constant while the solvent itself is changed. Using a controlled synthesis, we can monitor the vessel pressure during synthesis. For the LFP_EG1 reaction, the observed pressure is approximately 5.86 bar once the reaction temperature of 250 °C is reached. This build-up of pressure is due to the removal of the waters of crystallisation from the iron starting material, which occurs between 170 °C and 230 °C.47 We believe it is this change in pressure which drives the formation of the high pressure phase in the ethylene glycol reaction. By employing Fe(acac)3 which has no water of crystallisation instead of Fe(C2O4)·2H2O, we can obtain pure, single phase Pnma LiFePO4 at 220 °C using an ethylene glycol solvent. The X-ray pattern of this sample, LFP_EG2, was fit by Rietveld profile analysis to the orthorhombic Pnma LiFePO4 structure and is shown in Fig. 4(a). The solubility of starting materials is also different, with Fe(acac)3 more soluble in ethylene glycol than the oxalate salt, as observed by the deep red colour of the solution prior to microwave treatment. The nature of the solvent is also of great importance in determining what phase is obtained, as demonstrated by the ionic liquid sample, LFP_IL. Using Fe(C2O4)·2H2O as a starting material and EMI-TFMS as solvent, which has a greater dissipated power density than ethylene glycol, single phase Pnma LiFePO4 is obtained after 3 hours [Fig. 4(b)]. We have also studied the effect of reaction temperature on the phase obtained. For increasing reaction temperatures using ethylene glycol as a solvent and an iron oxalate starting material, we observe a two phase product made up of α- and β-LiFePO4 even up to reaction temperatures of 300 °C (see XRD patterns in ESI, Fig. S1a†). In the case of EMI-TFMS as a solvent, we do not see the formation of the β-LiFePO4 phase and only obtain α-LiFePO4 at temperatures above 250 °C (see XRD patterns in ESI, Fig. S1b†). We are currently investigating the use of several commercial and tailored precursors to examine the effect of starting material and solvent on crystal chemistry in greater detail.
 | ||
| Fig. 3 (a) XRD data for LFP_EG1 reveals this is a two-phase system, comprising the Pnma structured LiFePO4 (α-LiFePO4) and the high pressure Cmcm phase (β-LiFePO4). These phases are depicted in (b) Cmcm high pressure LiFePO4 and (c) Pnma LiFePO4. | ||
 | ||
| Fig. 4 Rietveld analysis was performed on single phase materials (a) LFP_EG2 [Rwp 14.8%, Rexp 4.15%, a = 10.327 Å, b = 5.999 Å, c = 4.697 Å] and (b) LFP_IL [Rwp 14.3%, Rexp 8.12%, a = 10.327 Å, b = 6.003 Å and c = 4.693 Å]. | ||
SEM images taken of dried powders of LFP_EG1 and LFP_IL reveal a dependence of particle morphology on the choice of solvent. In the case of LFP_EG1, large platelets are noted, with a typical platelet diameter of 6 μm. The thickness of these platelets is of the order of 20 nm and they appear as clusters of stacked particles as shown in Fig. 5(a). A dramatic difference is noted for the LFP_IL sample, where more nanoparticulate material, which often adopts geometric forms, is found to form under the same reaction conditions. The typical particle size in this case is 200 nm and in some cases the particles appear faceted [Fig. 5(b)]. High resolution TEM images confirm the highly crystalline nature of the LFP_IL sample [Fig. 5(d)], with lattice spacings consistent with Pnma LiFePO4. Larger, sheet-like particles are again observed for the LFP_EG1 sample [Fig. 5(c)].
 | ||
| Fig. 5 SEM and TEM images of (a and c) LFP_EG1 and (b and d) LFP_IL, respectively. Large, stacked platelets and rods, with a typical thickness of 20 nm are noted for the EG1 sample, while more crystalline particles are seen for the IL sample. Long-range crystallinity throughout particles of both samples was confirmed by HRTEM images, with (inset) Fourier transforms demonstrating long-range crystallographic ordering (c and d). | ||
μSR studies of Li+ diffusion in nanoparticulate LiFePO4
In terms of structure, the Pnma LiFePO4 phase is characterised by open channels running in the b-direction through which Li+ ions can diffuse during electrochemical cycling, as shown in Fig. 3(c). The structure of the Cmcm phase, shown in Fig. 3(b), is made up of rows of edge-sharing octahedral along the c axis, with PO4 and LiO4 tetrahedra running in the a direction.45 As demonstrated previously, the major structural difference between these polymorphs is in the Li–Li distances, with the high pressure phase increasing to a point at which the lithium hopping mechanism is no longer viable.46 The electrochemical properties of this phase have been investigated and it has been shown to be electrochemically inactive, with theoretical predictions in agreement with experiment.45,46In order to probe the Li+ diffusion in the pure Pnma and Cmcm-containing nanosized LiFePO4 samples prepared here, we recorded μSR data at zero field (ZF) and applied longitudinal fields (LF) of 5 G and 10 G. The typical raw data obtained for the LFP_IL sample, recorded at 300 K, are shown in Fig. 6. The initial positron asymmetry, regardless of applied field, is approximately 17%. These measurements, which are taken above the antiferromagnetic ordering at TN (LFP_IL, 51 K; LFP_EG, 49 K), contain a fast initial relaxation likely due to interactions with the paramagnetic iron moments and a slow relaxation from interactions with nuclear magnetic fields from 7Li, 6Li and 31P. By applying a longitudinal field parallel to the direction of the beam, any interactions between the muon and the local nuclear magnetic field distribution that it probes can be eliminated. Fig. 6 demonstrates this decoupling, where it can be seen that the application of progressively larger LF (from 5 G to 10 G) reduces this slower relaxation rate. Similar observations have been reported for bulk LiFePO4.36–38
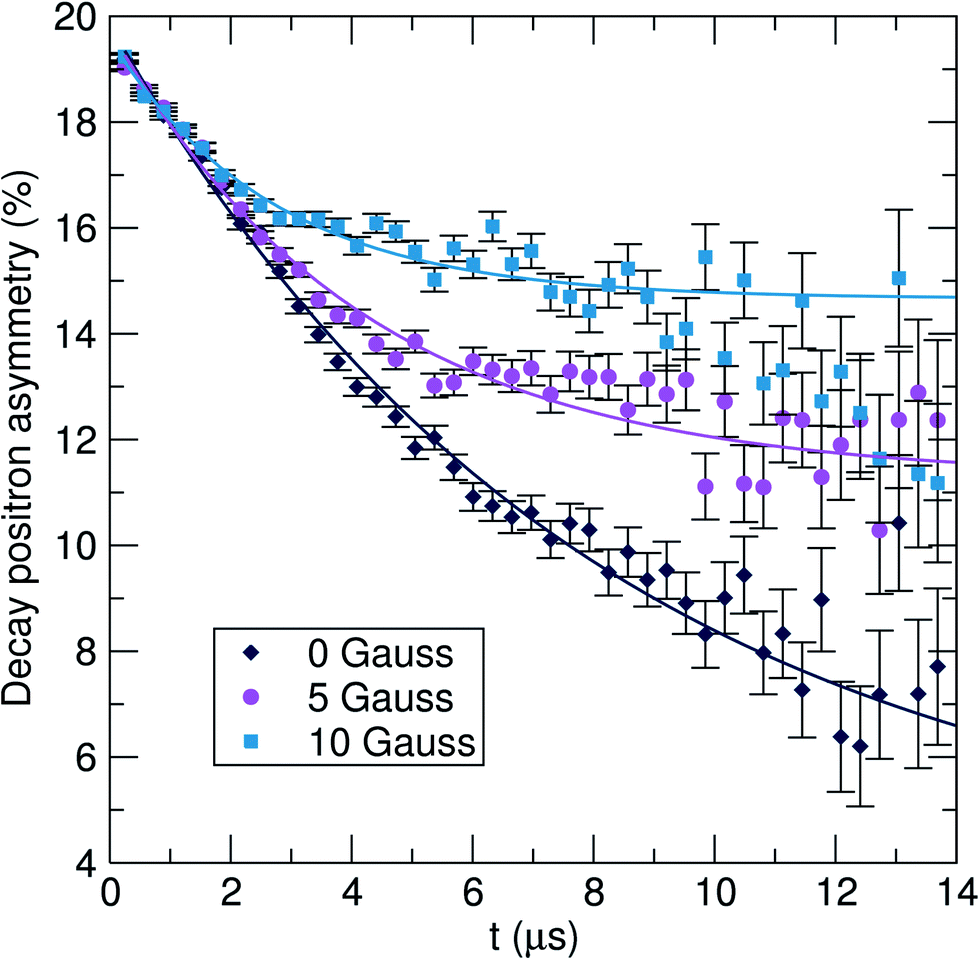 | ||
| Fig. 6 Raw μSR data for LFP_IL collected at 300 K at zero field (ZF) [diamonds] and applied longitudinal fields of 5 G [circles] and 10 G [squares]. | ||
In order to probe the Li+ diffusion dynamics in our samples, data were collected over a temperature range of 100 K to 400 K at ZF and LF of 5 G and 10 G. All data were fit using three parameters: a combination of an exponentially relaxing signal to account for the initial fast relaxation from the iron magnetic moments, a baseline asymmetry and an exponentially relaxing dynamic Kubo–Toyabe function,30 which has been modified to account for fluctuations due to muon or lithium diffusion and can be employed for an assumed Gaussian distribution of local fields.48 From these fits, we can extract parameters which provide us with insight into the Li+ diffusion mechanism in our materials. In Fig. 7 and 8, we show the values of ν, the field fluctuation rate, and Δ, the local field distribution at the muon stopping site, for data collected over the full temperature range. Data extracted for the single phase LiFePO4 sample LFP_IL are shown in Fig. 7. The values obtained for Δ are very similar to those observed for bulk LiFePO4 samples reported previously, i.e. a low temperature plateau followed by a smooth decrease to higher temperatures [Fig. 7(b)]. In the case of the fluctuation rate, ν, we again observe similar behaviour as seen for bulk LiFePO4. From 160 K, we see a steady increase until 230 K after which there is a sharp drop. The observed decrease in ν above 240 K likely results from the Li+ diffusion being too fast for μ+SR.38 To evaluate the diffusion coefficient for Li+, we consider only jumps of Li+ to interstitial sites and we take the primary hopping axis to be in the b-direction. The distance travelled for each hop will be therefore b/2, giving an estimation of the Li+ diffusion coefficient, DLi, from b2ν/4. For the LFP_IL sample, we can extrapolate fits of DLiversus 1/T to obtain a Li+ diffusion coefficient at 300 K of 6.25 × 10−10 cm2 s−1. This is in close agreement to bulk sample measurements.36,37
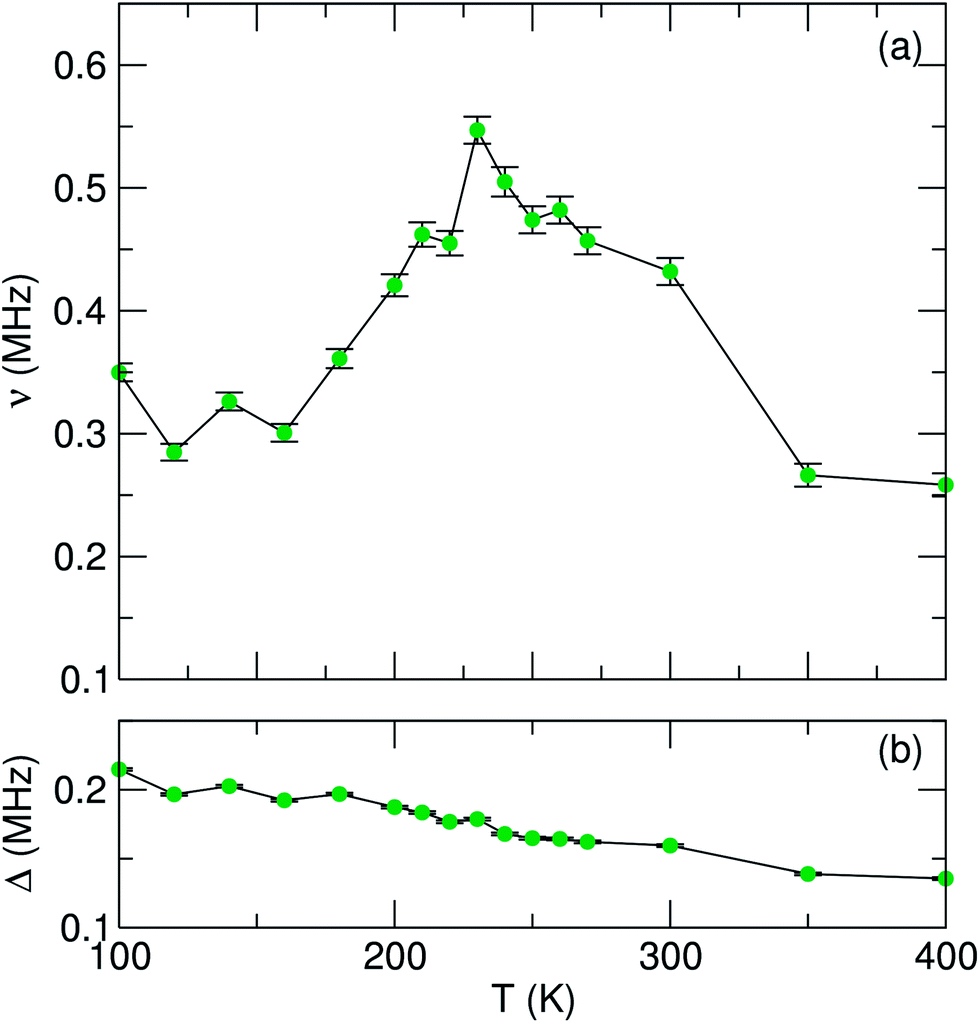 | ||
| Fig. 7 Temperature dependence of (a) fluctuation rate (ν) and (b) field distribution width (Δ) parameters derived from fitting μSR data to a dynamic Kubo–Toyabe function for the LFP_IL sample, measured from 100 K to 400 K. | ||
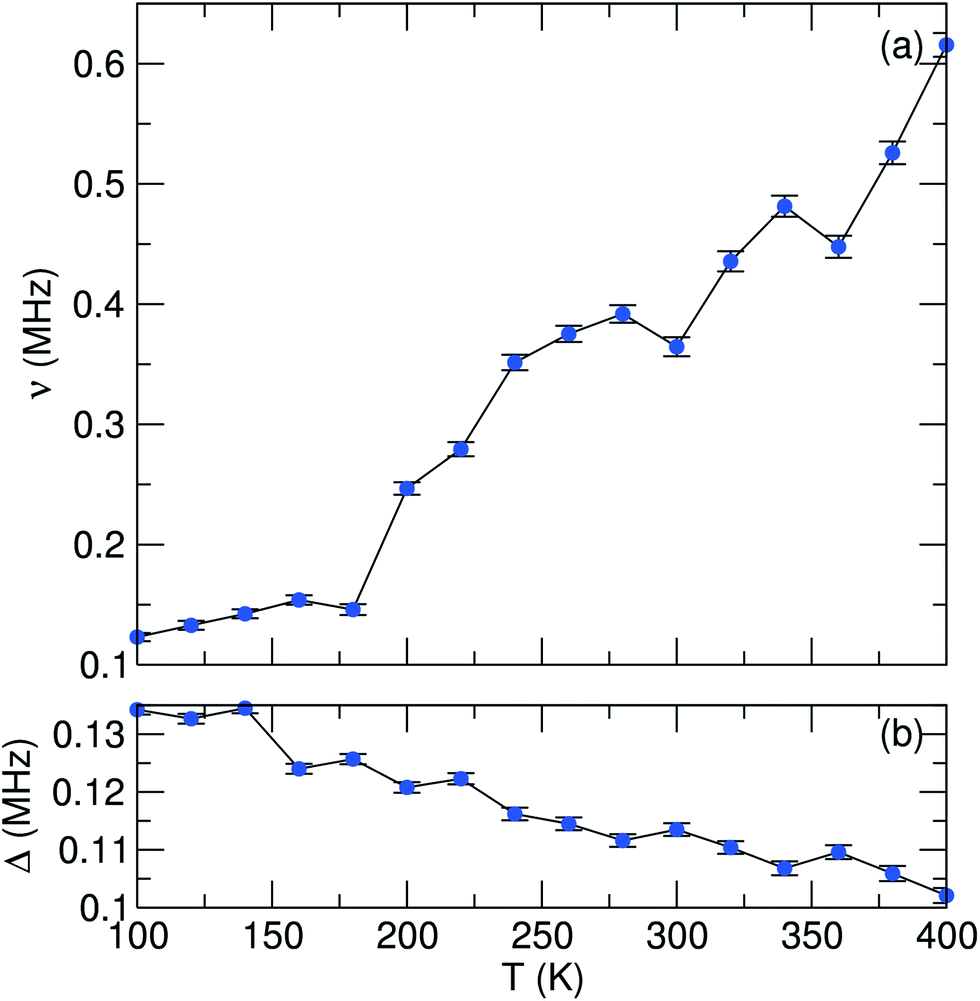 | ||
| Fig. 8 Temperature dependence of (a) fluctuation rate (ν) and (b) field distribution width (Δ) parameters derived from fitting μSR data to a dynamic Kubo–Toyabe function for the LFP_EG1 sample, measured from 100 K to 400 K. | ||
To determine the activation energy, we plot an Arrhenius fit to ν over the thermally activated region to give an estimated Ea of 58 meV for the LFP_IL sample. This value is close to the bulk reported value from Baker et al. who employed a Keren fitting function to data and obtained an Ea value of 60 meV.36 For bulk LiFePO4 prepared by ceramic methods and using similar Kubo–Toyabe fitting methods, Sugiyama et al. have found Ea values close to 100 meV.37,38 The similarities in values obtained demonstrate the robustness of this method for determining Li+ diffusion behaviour.
In the case of the LFP_EG1 sample (Fig. 8), we initially observe similar behaviour to the LFP_IL case, albeit with smaller Δ and ν values. An increase in ν is noted with increasing temperature, but now a decrease after 230 K is not seen. Instead, a steady increase is observed over the remaining temperature range. Given that this is a two phase system comprising Pnma and Cmcm LiFePO4, it is reasonable to assume that the initial increase up to 230 K is due to Li+ diffusion, similar to the case of the LFP_IL sample and previous observations for bulk samples. Previous reports on the high pressure Cmcm phase have shown that this phase is inactive electrochemically, with DFT simulations establishing the poor Li+ mobility, with no hopping observed for the ions which rattle in voids.46 From Rietveld refinement of our XRD pattern, the phase fraction of the sample is Pnma: Cmcm 80:20. Our experiments are in agreement with previous observations for the Cmcm phase, with a lower DLi value of 3.96 × 10−10 cm2 s−1 obtained for the LFP_EG1 sample (Eact = 46 meV). We can therefore rationalise our ν observations as follows for the LFP_EG1 sample. We continue to observe an increase in ν due to diffusion in the Pnma phase, which is present in excess. However, the presence of the Cmcm phase acts to limit the supply of Li+ ions which can diffuse. This impedes the lithium diffusion and results in a lower DLi value.
D Conclusions
We have shown that a microwave-assisted synthetic approach for the preparation of LiFePO4 can allow for different particle morphologies, including crystalline nanoparticles and platelets, and different phases (α- and β-LiFePO4) to be obtained in gram-scale quantities and short reaction times. The microwave dielectric measurements of ethylene glycol and EMI-TFMS reveal these as excellent microwave absorbers to generate the temperatures required for our reactions to proceed. μSR has also proved a powerful tool to examine the Li+ diffusion in these nanomaterials, with nanocrystalline Pnma LiFePO4 exhibiting similar diffusion coefficients to bulk LiFePO4. μSR has also revealed that the presence of the Cmcm phase impedes Li+ mobility and leads to a decrease in Li+ diffusion. In future, our investigations include varying the experimental conditions to allow for further tuning of the crystal chemistry and morphology, together with additional μSR experiments on mixed metal phosphates.Acknowledgements
This work was supported by funding from the EPSRC (EP/K029290/1) and Royal Society (RG100301) and we thank the STFC for beamtime allocation. We also acknowledge and thank Prof. J. M. Tarascon for introducing us to the microwave synthesis of LiFePO4 in ionic liquid media.Notes and references
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CrossRef CAS PubMed.
- A. K. Padhi, K. S. Nanjundaswamy, C. Masquelier, S. Okada and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1609 CrossRef CAS PubMed.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552 CrossRef CAS PubMed.
- M. S. Whittingham, Dalton Trans., 2008, 5424 RSC.
- A. Manthriam, A. Vadivel Murugan, A. Sarkar and T. Muraliganth, Energy Environ. Sci., 2008, 1, 621 Search PubMed.
- S. S. Zhang, J. L. Allen, K. Xu and T. R. Jow, J. Power Sources, 2005, 147, 234 CrossRef CAS PubMed.
- R. Dominko, M. Bele, M. Gaberscek, M. Remskar, D. Hanzel, S. Pejovnik and J. Jamnik, J. Electrochem. Soc., 2005, 152, A607 CrossRef CAS PubMed.
- Z. Lu, H. Chen, R. Robert, B. Y. X. Zhu, J. Deng, L. Wu, C. Y. Chung and C. P. Grey, Chem. Mater., 2011, 23, 2848 CrossRef CAS.
- B. Ellis, W. H. Kan, W. R. M. Makahnouk and L. F. Nazar, J. Mater. Chem., 2007, 17, 3248 RSC.
- J. Zhu, J. Fiore, D. Li, N. M. Kinsinger, Q. Wang, E. DiMasi, J. Guo and D. Kisailus, Cryst. Growth Des., 2013, 13, 4659 CAS.
- J.-M. Tarascon, N. Recham, M. Armand, J.-N. Chotard, P. Barpanda, W. Walker and L. Dupont, Chem. Mater., 2010, 22, 724 CrossRef CAS.
- N. Recham, L. Dupont, M. Courty, K. Djellab, D. Larcher, M. Armand and J.-M. Tarascon, Chem. Mater., 2009, 21, 1096 CrossRef CAS.
- P. Barpanda, K. Djellab, N. Recham, M. Armand and J.-M. Tarascon, J. Mater. Chem., 2011, 21, 10143 RSC.
- I. Bilecka and M. Niederberger, Nanoscale, 2010, 2, 1358–1374 RSC.
- I. Bilecka, A. Hintennach, I. Djerdj, P. Novák and M. Niederberger, J. Mater. Chem., 2009, 19, 5125 RSC.
- T. Muraliganth, K. R. Stroukoff and A. Manthiram, Chem. Mater., 2010, 22, 5754 CrossRef CAS.
- R. Tripathi, G. Popov, X. Sun, D. H. Ryan and L. F. Nazar, J. Mater. Chem. A, 2013, 1, 2990 CAS.
- B. Ellis, L. K. Perry, D. H. Ryan and L. F. Nazar, J. Am. Chem. Soc., 2006, 128, 11416 CrossRef CAS PubMed.
- P. P. Prosini, M. Lisi, D. Zane and M. Pasquali, Solid State Ionics, 2002, 148, 45 CrossRef CAS.
- H. Xia, L. Lu and M. O. Lai, Electrochim. Acta, 2009, 54, 5986 CrossRef CAS PubMed.
- A. Eftekhari, Electrochim. Acta, 2010, 55, 3434 CrossRef CAS PubMed.
- L. Lu, Electrochim. Acta, 2010, 55, 3435 CrossRef CAS PubMed.
- D. Morgan, A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, A30 CrossRef CAS PubMed.
- M. S. Islam, D. J. Driscoll, C. A. J. Fisher and P. R. Slater, Chem. Mater., 2005, 17, 5085 CrossRef CAS.
- C. A. J. Fisher, V. H. Hart Prieto and M. S. Islam, Chem. Mater., 2008, 20, 5907 CrossRef CAS.
- S.-I. Nishimura, G. Kobayashi, K. Ohoyama, R. Kanno, M. Yashima and A. Yamada, Nat. Mater., 2008, 7, 707 CrossRef CAS PubMed.
- J. Cabana, J. Shirakawa, G. Chen, T. J. Richardson and C. P. Grey, Chem. Mater., 2010, 22, 1249 CrossRef CAS.
- C. Delacourt, L. Laffont, R. Bouchet, C. Wurm, J.-B. Leriche, M. Morcrette, J.-M. Tarascon and C. Masquelier, J. Electrochem. Soc., 2005, 152, A913 CrossRef CAS PubMed.
- S. J. Blundell, Contemp. Phys., 1999, 40, 175 CrossRef CAS.
- J. Sugiyama, K. Mukai, Y. Ikedo, H. Nozaki, M. Månsson and I. Watanabe, Phys. Rev. Lett., 2009, 103, 147601 CrossRef.
- J. Sugiyama, Y. Ikedo, K. Mukai, H. Nozaki, M. Månsson, O. Ofer, M. Harada, K. Kamazawa, Y. Miyake, J. H. Brewer, E. J. Ansaldo, K. H. Chow, I. Watanabe and T. Ohzuku, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 224412 CrossRef.
- A. S. Powell, J. S. Lord, D. H. Gregory and J. J. Titman, J. Phys. Chem. C, 2009, 113, 20758 CAS.
- A. S. Powell, Z. Stoeva, J. S. Lord, R. I. Smith, D. H. Gregory and J. J. Titman, Phys. Chem. Chem. Phys., 2013, 15, 816 RSC.
- A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2000, 3, 301 CrossRef CAS PubMed.
- P. J. Baker, I. Franke, F. L. Pratt, T. Lancaster, D. Prabhakaran, W. Hayes and S. J. Blundell, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 174403 CrossRef.
- J. Sugiyama, H. Nozaki, M. Harada, K. Kamazawa, O. Ofer, M. Månsson, J. H. Brewer, E. J. Ansaldo, K. H. Chow, Y. Ikedo, Y. Miyake, K. Oshishi, I. Watanabe, G. Kobayashi and R. Kanno, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 054430 CrossRef.
- J. Sugiyama, H. Nozaki, M. Harada, K. Kamazawa, Y. Ikedo, Y. Miyake, O. Ofer, M. Månsson, E. J. Ansaldo, K. H. Chow, G. Kobayashi and R. Kanno, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 054111 CrossRef.
- F. J. Douglas, D. A. MacLaren and M. Murrie, RSC Adv., 2012, 2, 8027 RSC.
- D. M. P. Mingos and D. R. Baghurst, Chem. Soc. Rev., 1991, 20, 1 RSC.
- A. Sulaimalebbe, A. Porch, F. J. Vidal-Iglesias and G. Attard, Microwave Symposium Digest, 2008 IEEE-MTT International, Atlanta, GA, 15–20 June 2008, pp. 1585–1588 Search PubMed.
- D. Slocombe, A. Porch, E. Bustarret and O. Williams, Appl. Phys. Lett., 2013, 102, 244102 CrossRef PubMed.
- H. Fröhlich, Theory of Dielectrics, Clarendon, Oxford, 1958 Search PubMed.
- E. Hanke, K. von Roden and U. Kaatze, J. Chem. Phys., 2006, 125, 084507 CrossRef PubMed.
- O. García-Moreno, M. Alvarez-Vega, F. García-Alvarado, J. García-Jaca, J. M. Gallardo-Amores, M. L. Sanjuán and U. Amador, Chem. Mater., 2001, 13, 1570 CrossRef.
- G. Zeng, R. Caputo, D. Carriazo, L. Luo and M. Niederberger, Chem. Mater., 2013, 25, 3399 CrossRef CAS.
- M. Hermanek, R. Zboril, M. Mashlan, L. Machala and O. Schneeweiss, J. Mater. Chem., 2006, 16, 1273 RSC.
- R. S. Hayano, Y. J. Uemura, J. Imazato, N. Nishida, T. Yamazaki and R. Kubo, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20, 850 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns of LiFePO4 products obtained for increasing temperatures for (a) ethylene glycol and (b) ionic liquid. See DOI: 10.1039/c4ta00543k |
| This journal is © The Royal Society of Chemistry 2014 |