 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nitrogen-enriched and hierarchically porous carbon macro-spheres – ideal for large-scale CO2 capture†
Bingjun
Zhu
a,
Kaixi
Li
b,
Jingjing
Liu
c,
Hao
Liu
c,
Chenggong
Sun
c,
Colin E.
Snape
c and
Zhengxiao
Guo
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, England, UK. E-mail: z.x.guo@ucl.ac.uk
bInstitute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan, Shanxi 030001, China
cDepartment of Chemical and Environmental Engineering, University of Nottingham, University Park, Nottingham, NG7 2RD, England, UK
First published on 26th February 2014
Abstract
A facile and efficient “spheridization” method is developed to produce nitrogen-enriched hierarchically porous carbon spheres of millimeters in diameter, with intricate micro-, meso- and macro-structural features. Such spheres not only show exceptional working capacity for CO2 sorption, but also satisfy practical requirements for dynamic flow in post-combustion CO2 capture. Those were achieved using co-polymerized acrylonitrile and acrylamide as the N-enriched carbon precursor, a solvent-exchange process to create hierarchically porous macro-sphere preforms, oxidization to induce cyclization of the polymer chains, and carbonization with concurrent chemical activation by KOH. The resulting carbon spheres show a relatively high CO2 uptake of 16.7 wt% under 1 bar of CO2 and, particularly, an exceptional uptake of 9.3 wt% under a CO2 partial pressure of 0.15 bar at 25 °C. Subsequent structural and chemical analyses suggest that the outstanding properties are due to highly developed microporous structures and the relatively high pyridinic nitrogen content inherited from the co-polymer precursor, incorporated within the hierarchical porous structures.
1. Introduction
Anthropogenic CO2 emission from the burning of fossil fuels is considered to be a significant contributor to global climate change. Fossil fuels still provide over 85% of the world's energy consumption today and are likely to dominate energy supply for many decades to come.1 Therefore, developing a practical CO2 capture technology is critically important to mitigate climate change. Porous carbon materials have been under intensive study as CO2 sorbents, due to their high moisture/chemical stabilities and low costs. These can also be regenerated after CO2 adsorption either by a relatively small change of temperature or pressure with a minimum energy penalty.2,3 Such porous carbon sorbents are usually derived from natural substances or polymeric precursors and KOH is frequently used as the activation agent to enhance porosity. A sawdust-based porous carbon activated by KOH at a KOH/precursor weight ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 600 °C shows a high CO2 adsorption capacity of 21.1 wt% (4.8 mmol g−1) under 1 bar CO2 and 25 °C,4 whereas a polypyrrole-derived porous carbon activated and tested under the same conditions yields a CO2 adsorption capacity of 16.9 wt% (3.9 mmol g−1).5 Porous carbon sorbents derived from resorcinol-formaldehyde,6 yeast,7 fungi8 and urban wastes9 also show a wide range of capacities from 1 to 21 wt%. However, these studies only focused on relatively fine and irregular powder of tens of microns. For practical applications in large scale CO2 capture, such as CO2 capture in a fluidized-bed, the ideal sorbent is in the form of porous spheres of several millimeters in diameter for optimum flow dynamics and CO2 sorption.10 Spherical carbon beads also reduce material abrasion due to smooth rotational impact. Furthermore, the flow resistance can be readily maintained, because the dynamic stacking density of carbon spheres does not change under a given set of flow conditions. A recent report shows phenolic resin-based activated carbon spheres with specific surface area of 2400 m2 g−1 and a total CO2 uptake of 20.2 wt% (4.6 mmol g−1) at 23 °C and 1 bar CO2,11 but the size of the carbon spheres is only about 0.6 μm, limited by the manufacturing method. The activated carbon spheres also rely on a rather large weight ratio of KOH/polymer (4:1) to produce a highly porous structure, leading to an extremely low carbon yield and low material strength. Such micro- and nano-sized carbon spheres are too buoyant to be effectively controlled under flue-gas conditions, and may readily clot filters during CO2 removal.10 Therefore, millimeter-sized carbon spheres with effective CO2 capture capabilities are desirable for practical CO2 sorption applications, e.g., via a fluidized bed.
:1 and 600 °C shows a high CO2 adsorption capacity of 21.1 wt% (4.8 mmol g−1) under 1 bar CO2 and 25 °C,4 whereas a polypyrrole-derived porous carbon activated and tested under the same conditions yields a CO2 adsorption capacity of 16.9 wt% (3.9 mmol g−1).5 Porous carbon sorbents derived from resorcinol-formaldehyde,6 yeast,7 fungi8 and urban wastes9 also show a wide range of capacities from 1 to 21 wt%. However, these studies only focused on relatively fine and irregular powder of tens of microns. For practical applications in large scale CO2 capture, such as CO2 capture in a fluidized-bed, the ideal sorbent is in the form of porous spheres of several millimeters in diameter for optimum flow dynamics and CO2 sorption.10 Spherical carbon beads also reduce material abrasion due to smooth rotational impact. Furthermore, the flow resistance can be readily maintained, because the dynamic stacking density of carbon spheres does not change under a given set of flow conditions. A recent report shows phenolic resin-based activated carbon spheres with specific surface area of 2400 m2 g−1 and a total CO2 uptake of 20.2 wt% (4.6 mmol g−1) at 23 °C and 1 bar CO2,11 but the size of the carbon spheres is only about 0.6 μm, limited by the manufacturing method. The activated carbon spheres also rely on a rather large weight ratio of KOH/polymer (4:1) to produce a highly porous structure, leading to an extremely low carbon yield and low material strength. Such micro- and nano-sized carbon spheres are too buoyant to be effectively controlled under flue-gas conditions, and may readily clot filters during CO2 removal.10 Therefore, millimeter-sized carbon spheres with effective CO2 capture capabilities are desirable for practical CO2 sorption applications, e.g., via a fluidized bed.
Here, we report a facile and effective method of producing mm-sized radially porous carbon spheres with an excellent CO2 sorption capacity. Co-polymerized precursors are spheridized via the mechanism of Plateau–Rayleigh instability12 and solidified into a porous pre-form via a solvent-exchange process. The experimental results show that this method is effective in producing millimeter carbon spheres with excellent structural integrity and exceptional CO2 adsorption performance.
2. Results and discussion
2.1 Formation of carbon spheres
The sphericity of carbon spheres depend on several factors, which are optimized and presented in the Experimental section. The corresponding discussion is included in ESI.† The copolymer with the best spherical shape is with an acrylamide–acrylonitrile molar ratio of 1:3.3. Therefore, it is selected to produce the carbon spheres. The polymer sphere in Fig. 1a shows a smooth surface: the polar diameter R1 (vertical) is 3.4 mm, slightly longer than the equatorial diameter R2 (horizontal) of 3.3 mm, which gives a roundness ratio of R1/R2 = 1.03. Some scratches on the surface are indicative of the soft nature of this polymer. The cross-sectional image of the polymer sphere (Fig. 1b) shows a hierarchical porous structure, typically with three regions: a relatively thin outer mantle of 0.5 mm in thickness with radial channels of about 0.08 mm in diameter, a thick inner mantle of 1 mm in thickness with radial channels of 0.3 mm in diameter and a relatively hollow core of 0.3 mm in diameter. This hierarchically and radially channeled porous structure is a direct consequence of the solvent-exchange process that takes place between the polymer sphere and the water bath as explained below.
 | ||
| Fig. 1 SEM images of (a) a typical polymer sphere, (c) an oxidized polymer sphere and (e) a carbon sphere activated with a KOH/sphere weight ratio of 1:1. (b, d and f) are their corresponding cross-sectional images. (g) A high resolution TEM image of the activated carbon sphere. (h) A fragment from a carbon sphere activated with a higher KOH/sphere weight ratio of 2:1 – the original sample has already lost its structural integrity. | ||
Since dimethyl sulfoxide (DMSO) is highly miscible with water, due to the concentration gradients of DMSO and water in the sphere and the water bath, DMSO tends to diffuse from the sphere into the water bath, and vice versa. As the polymer has low solubility in water, the polymer chains tend to partition or “precipitate out” with the exchanged water. This “solvent-exchange” based mechanism is schematically illustrated in Fig. 2.
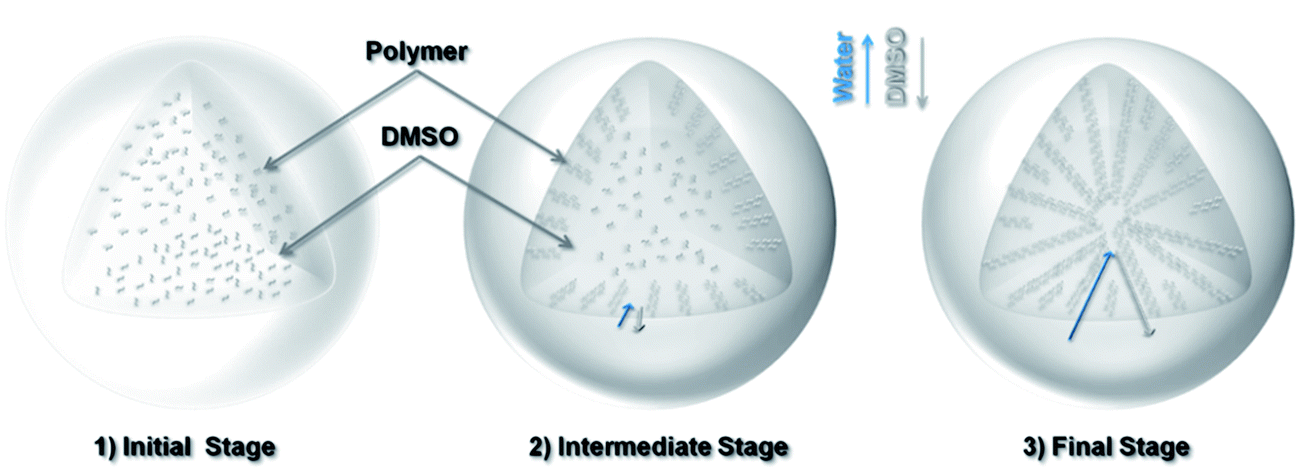 | ||
| Fig. 2 Schematic of the solvent-exchange process for the formation of a porous polymer sphere: (1) initial stage: the polymer is dissolved in a DMSO solvent drop; (2) intermediate stage: the exchange of solvents (DMSO ↔ water) and the formation of aligned channels near the surface; (3) final stage: progression of solvent exchange and the formation of larger channels and hollow core. | ||
The exchange process starts with a spherical drop of polymer solution. At the beginning, the concentration gradients are steep across the interface of the solvents or the surface of the sphere, which leads to dense sites (or “nuclei”) of polymer precipitates. The radial diffusion of the exchanging solvents results in radial flow channels, which become the porous channels at a later stage. As the exchange continues, the partitioned polymers at the surface region restrict the rate of further solvent exchange towards the center. This process gradually allows more polymers to cluster together, driven by the reduction in surface energy, leading to increasingly large porous channels towards the inner region. Towards the end of the process, there are relatively fewer polymers in the core region, which becomes hollow upon solvent removal. Fig. 1c shows the oxidized polymer sphere maintains its original spherical shape but with a reduced diameter of 2.3 mm. The cross-sectional image (Fig. 1d) indicates the radially channeled structure is retained. The interconnected porous structure allows KOH solution to diffuse across the entire structure during sample soaking. After KOH activation at 600 °C, the sample surface is significantly etched, but the shape and integrity of the sphere are preserved, thanks to the mild activation condition (KOH/sphere = 1:1). The cross-sectional image Fig. 1f shows the radially channeled structure survives from the chemical activation, acting as the supporting framework of the sphere. The high resolution TEM image (Fig. 1g) shows segmented graphitic platelets with well-aligned carbon layers. This observation differs from TEM images of carbon sorbents studied in previously mentioned literature, where carbon layers are randomly orientated. It is mentioned earlier that the solvent-exchange process helps to align the polymer chains radially. These aligned polymer chains form pyridine and pyridone rings after oxidation (ESI Fig. S2†), which directly influences the formation and alignment of carbon layers after the carbonization and activation. It can be clearly observed in the TEM image that there are nano-scale voids between edges of those graphitic platelets, which are the origins of micropores. However, too high a ratio of KOH to spheres (KOH/sphere = 2:1) leads to fragile samples and loss of sphericity (Fig. 1h).
2.2 Characterization of porous structures
The specific surface area and pore size distribution of carbon spheres were comparatively characterized by means of gas-sorption, using CO2 and N2 probes, and mercury porosimetry, respectively for ultramicro-, micro-/meso- and macro-pores (details in Section 4). The results are summarized in Table 1.| Sample | Method | |||||
|---|---|---|---|---|---|---|
| N2 sorption | CO2 sorption | Hg porosimetry | ||||
| S N2 [m2 g−1] | S Micro [m2 g−1] | V N2 [cm3 g−1] | V Micro [cm3 g−1] | V CO2 [cm3 g−1] | V Hg [cm3 g−1] | |
| CSC | 4.5 | 0.1 | 0.006 | 0 | 0.068 | 1.67 |
| CSA-600 | 520.2 | 401.4 | 0.316 | 0.149 | 0.147 | 1.64 |
| CSA-700 | 1231.4 | 1118.5 | 0.552 | 0.434 | 0.251 | 2.08 |
| CSA-800 | 2032.4 | 1704.8 | 0.992 | 0.709 | 0.262 | 3.08 |
“CSC” is the sphere carbonized at 600 °C without KOH activation. Its isotherm lies at the bottom of Fig. 3a, and shows a small specific surface area of 4.5 m2 g−1 and pore volume of 0.006 cm3 g−1. CSA-x (x = 600, 700, 800) stands for the carbon spheres activated at x °C. With KOH activation, CSA-600 starts to develop a porous structure with a specific surface area of 520.2 m2 g−1 and pore volume of 0.317 cm3 g−1.
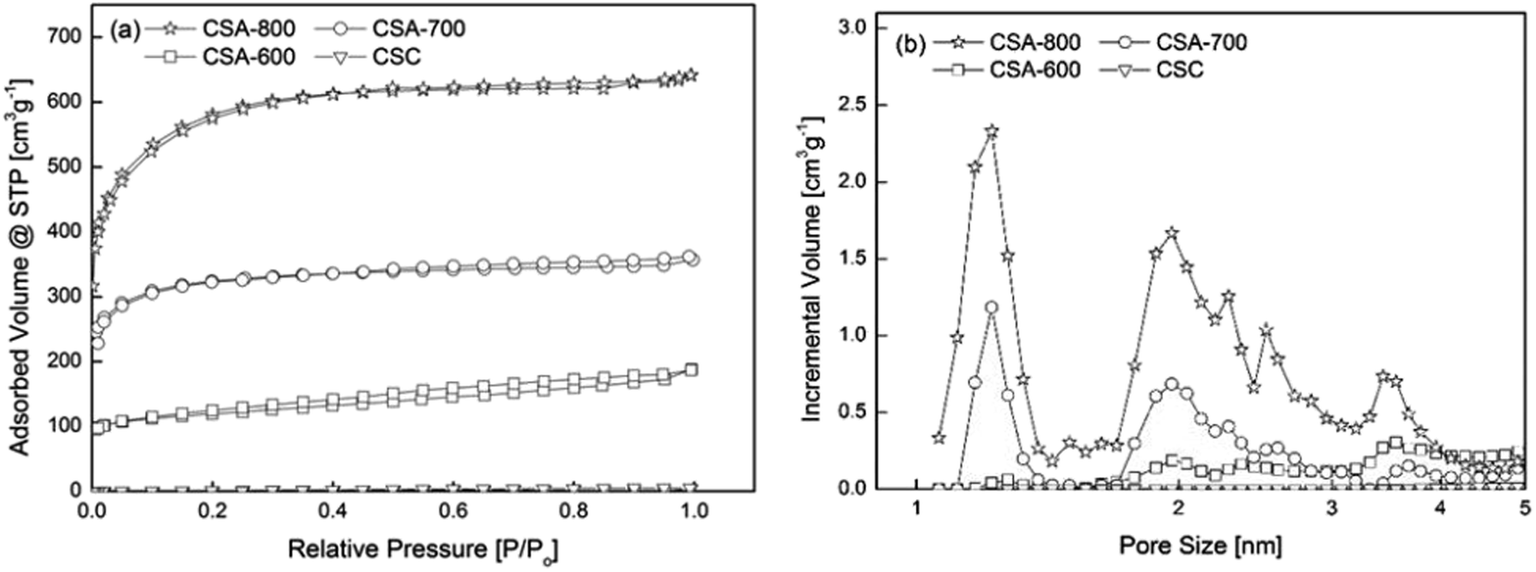 | ||
| Fig. 3 (a) Nitrogen sorption isotherms of samples prepared under different conditions and (b) their corresponding pore size distributions (log). | ||
Generally, the surface area increases with carbonization temperature, from 520 m2 g−1 at 600 °C, more than doubled at 700 °C, to over 2000 m2 g−1 at 800 °C. The emergence of the porous structure can be explained by the reaction between KOH and carbon, which is a well-studied chemical activation method of producing activated carbon. The development of porous structure at different temperatures can be attributed to different reaction mechanisms (see ESI for details†).13,14 For the sample activated at 800 °C, besides the removal of carbon atoms, earlier literature suggests the formation of elementary potassium and its vaporization make it possible for elementary potassium atoms to penetrate graphene layers, expanding the distance between the layers, further enhancing the surface area and the pore volume.15
In addition, Fig. 3b shows that the pore size distributions of CSA-600, 700 and 800 are mainly centered at 1.2 and 2 nm, indicating pore development at both micro- and meso-pore ranges. CSA-600 possesses a relatively small volume of micro- and meso-pores, while CSA-700 shows a marked increase in the micropore volume, almost three times greater than that of CSA-600. CSA-800 exhibits an even higher peak at 1.2 nm but with an apparently enhanced peak at 2 nm and several other weak peaks distributed in the mesopore range.
In order to assess the ultra-micropore (<0.7 nm) characteristics, CO2 was used as a probe for sorption analysis. The results show that the ultra-micropore volume (VCO2 in Table 1) is greater at a higher activation temperature, but the magnitude of increase is higher from CSA-600 to CSA-700 (by 71%) than from CSA-700 to CSA-800 (by 4.4%). The finding indicates that a higher activation temperature does not just result in an increased volume of ultra-micropores but also leads to more rapid growth of pore sizes. In addition, for the sample CSC, though its micropore volume is negligible from N2 sorption, CO2 sorption analysis shows that it possesses a small level of ultra-micropores, which cannot be accurately analyzed by the N2 sorption technique.
In addition to the micro- and meso-pore analysis, Table 1 also summarizes the macropore volumes (VHg) of the carbon spheres. The large macropores originate from the hierarchical macroporous channeled structure formed during the solvent-exchange process. It is further enlarged due to more aggressive activation reactions at elevated temperatures. Such macropores should facilitate diffusion of CO2 within the carbon spheres. However, an excess level of such macropores may reduce the volume capacity of CO2 sorption in the sphere. Further study is under way to optimize the distribution of the pore sizes in those hierarchical porous carbon spheres.
2.3 Chemical compositions of carbon spheres
The results of chemical composition analysis by XPS are summarized in Table 2. The relative FTIR and N-1s XPS spectra are presented in Fig. 4. Fig. 4a compares the FTIR spectra of the polymer, oxidized polymer (CSO) and activated carbon spheres. All FTIR spectra show one wider peak between 3700 and 3000 cm−1, which corresponds with the N–H and O–H stretching vibration bands. The polymer sphere shows three sharper peaks from 2240 to 1450 cm−1. The peak at 2244 cm−1 is attributed to the stretching vibrations of nitrile groups (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) from the acrylonitrile units in the polymer sphere, while the peak at 1670 cm−1 is attributed to the stretching vibrations of primary amide groups from the acrylamide (–CO–NH2) units.16 There is a small shoulder right beside the 1670 cm−1 peak at 1640 cm−1, which originates from the stretching vibrations of carbonyls (C
N) from the acrylonitrile units in the polymer sphere, while the peak at 1670 cm−1 is attributed to the stretching vibrations of primary amide groups from the acrylamide (–CO–NH2) units.16 There is a small shoulder right beside the 1670 cm−1 peak at 1640 cm−1, which originates from the stretching vibrations of carbonyls (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) from the primary amide groups. The peak around 1452 cm−1 is assigned to the in-plane bending vibrations of methylene bridges (–CH2–) from both acrylonitriles and acrylamides. After the oxidation reaction, the peak around 2244 cm−1 completely disappears. In addition, the intensity of the amide peak at 1670 cm−1 is reduced and shifted to 1614 cm−1 and the shoulder at 1640 cm−1 disappears as well. It is consistent with the mechanism of the oxidation process whereby the nitrile and amide groups are removed through the formation of pyridinic rings in the cyclization reaction. The peak at 1614 cm−1 is the evidence of the emerging pyridinic rings. The intensity of the methylene peak at 1452 cm−1 is increased and shifted to 1440 cm−1, which can be attributed to the formation of carbon double bonds in the pyridinic rings.16 After the activation reaction, the intensities of both peaks are greatly reduced due to further loss of nitrogen content and regular pyridinic structure due to chemical attack by KOH. Besides, there exist a range of weak peaks below 1300 cm−1 in the spectra. The spectrum of the polymer shows a relatively broad and weak band from 1200 to 1000 cm−1, which can be assigned to C–N stretching vibrations. There are also two peaks at 858 cm−1 and 695 cm−1, which can be assigned to the out-of-plane bending of N–H and deformation of C–H. When it comes to the oxidized polymer CSO, multiple weak peaks appear in the range of 1300 to 1000 cm−1, due to the ester and ether groups introduced by the oxidation process. For CSA-600, all the previously mentioned peaks below 1300 cm−1 disappear due to carbonization and activation reactions at the elevated temperature.16
O) from the primary amide groups. The peak around 1452 cm−1 is assigned to the in-plane bending vibrations of methylene bridges (–CH2–) from both acrylonitriles and acrylamides. After the oxidation reaction, the peak around 2244 cm−1 completely disappears. In addition, the intensity of the amide peak at 1670 cm−1 is reduced and shifted to 1614 cm−1 and the shoulder at 1640 cm−1 disappears as well. It is consistent with the mechanism of the oxidation process whereby the nitrile and amide groups are removed through the formation of pyridinic rings in the cyclization reaction. The peak at 1614 cm−1 is the evidence of the emerging pyridinic rings. The intensity of the methylene peak at 1452 cm−1 is increased and shifted to 1440 cm−1, which can be attributed to the formation of carbon double bonds in the pyridinic rings.16 After the activation reaction, the intensities of both peaks are greatly reduced due to further loss of nitrogen content and regular pyridinic structure due to chemical attack by KOH. Besides, there exist a range of weak peaks below 1300 cm−1 in the spectra. The spectrum of the polymer shows a relatively broad and weak band from 1200 to 1000 cm−1, which can be assigned to C–N stretching vibrations. There are also two peaks at 858 cm−1 and 695 cm−1, which can be assigned to the out-of-plane bending of N–H and deformation of C–H. When it comes to the oxidized polymer CSO, multiple weak peaks appear in the range of 1300 to 1000 cm−1, due to the ester and ether groups introduced by the oxidation process. For CSA-600, all the previously mentioned peaks below 1300 cm−1 disappear due to carbonization and activation reactions at the elevated temperature.16
| Sample | Chemical composition [at.%] | ||||||
|---|---|---|---|---|---|---|---|
| C | O | N | K | Pyridinic | Pyrrolic | Pyridinic N-oxide | |
| CSO | 67.32 | 13.99 | 18.69 | 0 | 43.0 | Pyridonic: 46.99 | 10.01 |
| CSC | 82.68 | 4.24 | 13.08 | 0 | 42.53 | 42.75 | 14.72 |
| CSA-600 | 78.60 | 12.30 | 6.81 | 2.29 | 30.31 | 57.59 | 12.10 |
| CSA-700 | 80.79 | 11.49 | 5.94 | 1.78 | 24.27 | 62.31 | 13.42 |
| CSA-800 | 84.22 | 9.90 | 4.76 | 1.12 | 17.56 | 67.62 | 14.82 |
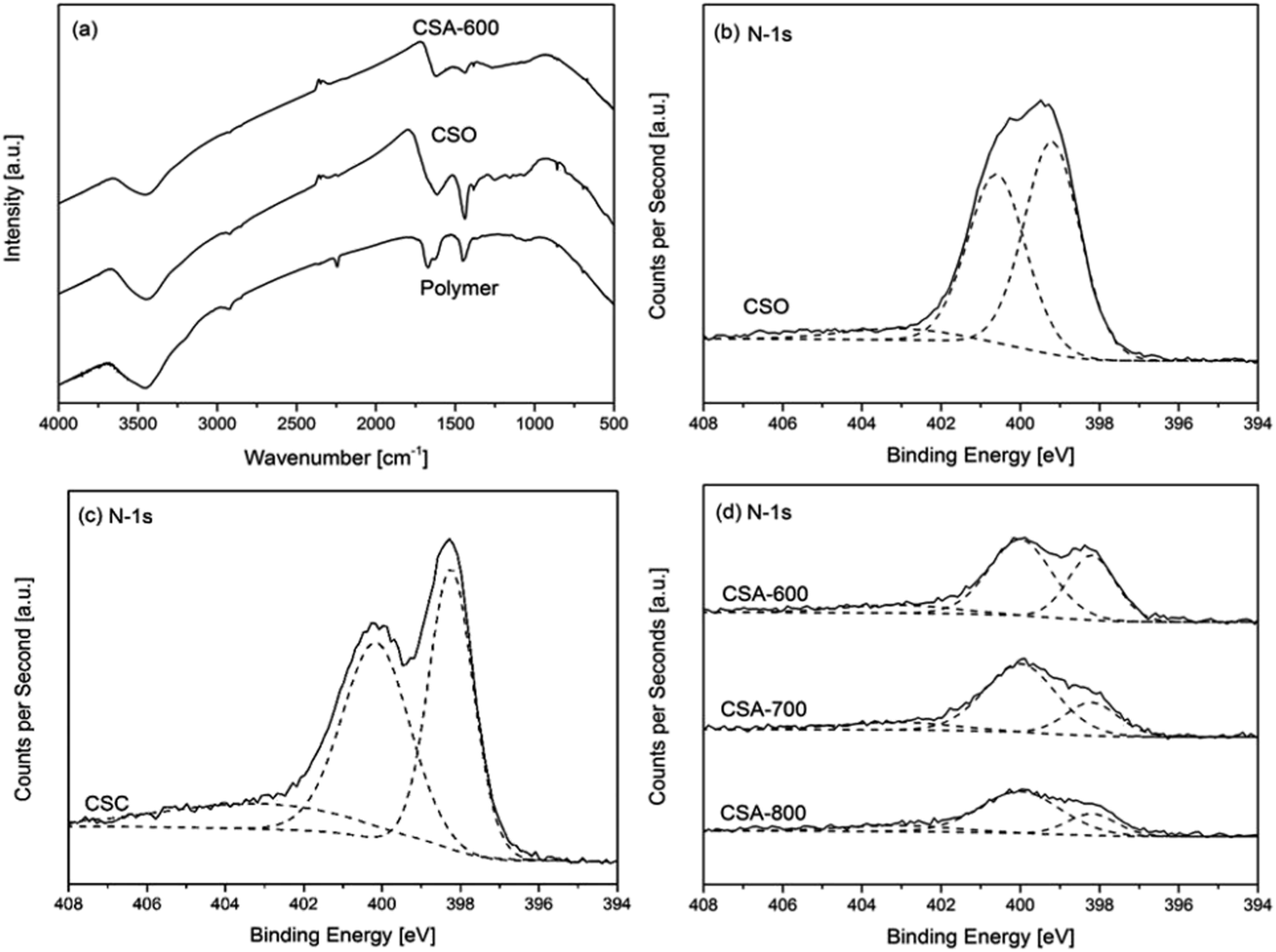 | ||
| Fig. 4 (a) Infrared spectra of the polymer, oxidized polymer (CSO) and activated carbon sphere. XPS N-1s spectra of (b) the oxidized polymer spheres, (c) carbonized sphere (CSC) and (d) activated carbon spheres CSA prepared under different conditions. | ||
Fig. 4b–d reveal how nitrogen content varies with different sample preparation conditions. The spectrum of the oxidized polymer can be deconvoluted into three major peaks at 399.3, 400.7 and 403.1 eV (Fig. 4b), which correspond to the existence of pyridinic, pyridonic and pyridinic N-oxide, respectively.17 The emergence of these two peaks is consistent with the mechanism of the oxidation reaction.18Table 2 shows the variation of chemical composition with different preparation conditions. The carbonized sample CSC shows a high nitrogen content of 13.08 at.%. Chemical activation by KOH reduces the nitrogen content of CSA-600 to 6.81 at.%, although only a mild activation condition of KOH/sphere = 1 is applied. Fig. 4c and d reveal that all carbon spheres exhibit three major peaks at 398.2, 400.0 and 403.1 eV, respectively. These peaks suggest pyridinic nitrogen, pyrrolic nitrogen and pyridinic nitrogen oxides are the three major forms of nitrogen in the carbon spheres.5,17 The former two exist at the edge of the carbon structure, which increases the affinity of CO2 towards the carbon structure due to the acidic nature of CO2 and basic nature of the nitrogen content. Fig. 4d shows all activated carbon spheres suffer severe reductions in peak intensities, compared with those of the carbonized sphere in Fig. 4c. With the increasing activation temperature, the nitrogen content of CSA-600, 700 and 800 drops from 6.81 to 4.76 at.%. In addition, Fig. 4d shows that the intensities of the 398.2 eV peaks are severely reduced compared with those of the 400.0 eV peaks. Table 2 also summarizes of the composition of different forms of nitrogen in carbon spheres. It is clear that the pyridinic nitrogen content is reduced with increasing temperatures from 600 to 800 °C. This implies that pyridinic nitrogen is less stable than pyrrolic nitrogen at high temperatures with the same activation conditions. The loss of pyridinic nitrogen can be attributed to more aggressive chemical activation reactions at a higher temperature as discussed in the previous paragraph. Besides the nitrogen content, another interesting observation is the existence of residual potassium. An earlier study suggests that it can help to enhance CO2 binding with the sorbent.19 The corresponding discussion is included in the ESI.†
2.4 CO2 adsorption on carbon spheres
Fig. 5 depicts CO2 adsorption isotherms at 0 and 25 °C from 0.1 to 1 bar of CO2. The corresponding CO2 uptakes at 1 bar CO2 are summarized in Table 3. All samples have higher CO2 uptakes at 0 °C, which can be explained by the reduced kinetic energy of CO2 gas molecules at a lower temperature. At 25 °C, CSA-700 presents the best CO2 uptake of 16.7 wt% (3.8 mmol g−1). It has a better CO2 uptake than CSA-600 of 9.4 wt% (2.1 mmol g−1), due to much enhanced porous structure (from 520.2 to 1231.4 m2 g−1), even its nitrogen content is reduced (from 6.81 to 5.94 at.%). However, even with higher surface area and larger pore volume, CSA-800 shows a lower CO2 uptake of 14.0 wt% (3.2 mmol g−1) due to further reduction in the nitrogen content, especially the loss of pyridinic nitrogen (from 24.27 to 17.56 at.% in Table 2). Earlier studies have shown pyridinic nitrogen possesses higher basicity than pyrrolic nitrogen.20 This is because pyridinic nitrogen has a lone pair of electrons with one electron donated to the conjugated π-system, while pyrrolic nitrogen donates two p-electrons that delocalize into the conjugated π-system. Therefore, pyridinic nitrogen has a higher affinity towards acidic CO2 molecules than pyrrolic nitrogen.21 Considering the experimental results from both structural and chemical analyses, it can be concluded that both porous structures and nitrogen content influence the CO2 uptake of carbon spheres. The maximum CO2 capacity is achieved by balancing both porous structure and pyridinic nitrogen content.| Sample | CO2 uptake/wt% | |
|---|---|---|
| 0 °C | 25 °C | |
| CSC | 7.2 | 4.5 |
| CSA-600 | 14.6 | 9.4 |
| CSA-700 | 25.3 | 16.7 |
| CSA-800 | 29.0 | 14.0 |
The isotherms of CSA-600 and CSA-800 intersect with each other at both 0 and 25 °C (Fig. 5a and b), which shows CSA-600 has higher CO2 adsorption capacities than those of CSA-800 at relatively low pressures. The results indicate that the level of nitrogen in the samples plays a more important role than that of porosities in the low pressure regime, because CSA-600 contains a higher amount of nitrogen than CSA-800, though the former possesses smaller specific surface area and pore volume than the latter. Moreover, the intersection point changes from 0.18 to 0.35 bar when temperature is raised from 0 to 25 °C. Similarly, the 0 °C isotherm of CSA-700 intersects with that of CSA-800 slightly above 0.5 bar but its 25 °C isotherm is entirely above CSA-800. This further implies that the nitrogen content helps to enhance CO2 adsorption at a low pressure and an elevated temperature.
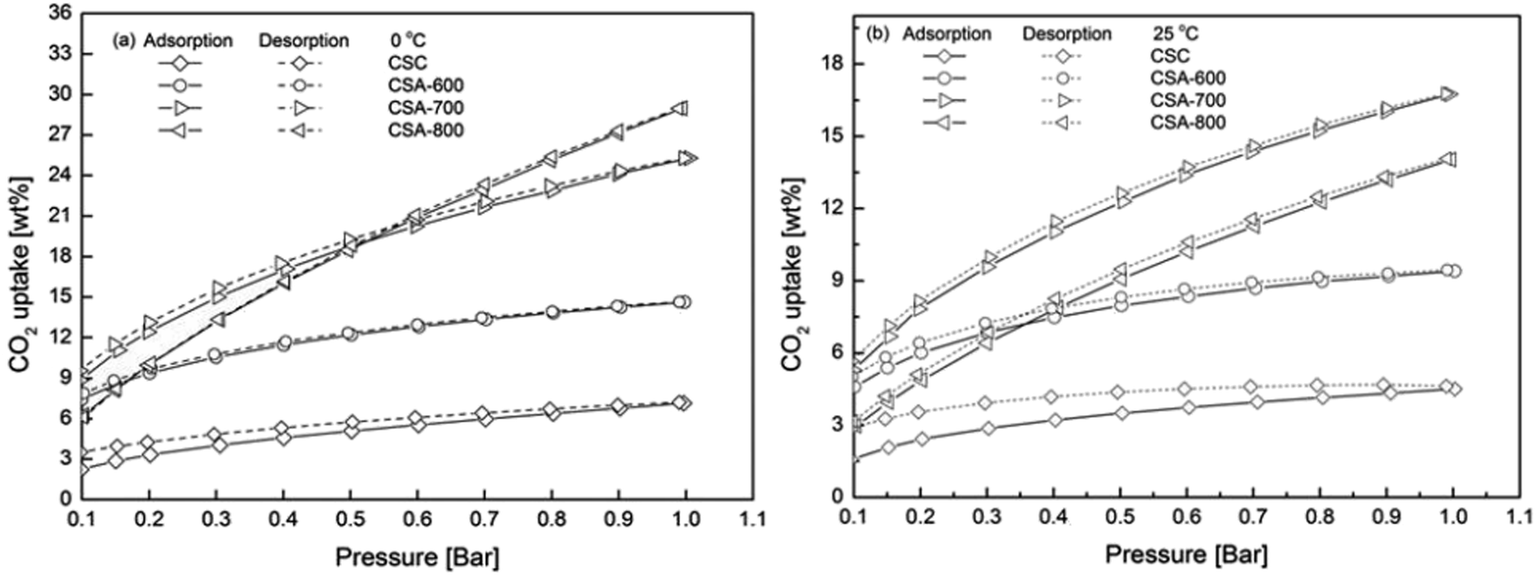 | ||
| Fig. 5 CO2 adsorption isotherms of carbon spheres prepared under different conditions at (a) 0 and (b) 25 °C from 0.1 to 1 bar. | ||
In addition, even without chemical activation by KOH, the CSC sample can adsorb 4.5 wt% (0.9 mmol g−1) CO2 at 25 °C under 1 bar CO2. This is about half of the CO2 uptake of the CSA-600, though the specific surface area of the former is two-orders of magnitude smaller. We attribute this seemingly disproportional increase to other factors that influence CO2 uptake, particularly here the nitrogen content. The preservation of a high nitrogen content (13.08 at.%) in the CSC, or the loss of the same in CSA-600, means that there is a much higher density of effective binding sites (pyridinic N-sites per surface area) in the former than the latter case. This is particularly the case for CO2 uptake at relatively low CO2 partial pressures, whereas at a high CO2 pressure, e.g. 20–30 bar, high surface area plays a more important role.22
It can be also observed in Fig. 5 that CSC cannot be fully degassed by decreasing the pressure. Furthermore, at 0 °C, when the pressure is lowered to 0.1 bar, the remaining weight of CO2 (3.5 wt%) is 59% more than the weight (2.2 wt%) at the same pressure during adsorption. This value is increased to 81% at 25 °C. This result further supports the idea that nitrogen can enhance CO2 uptake at a low pressure and an elevated temperature. By comparison, the adsorption and desorption isotherms of CSA-700 shows little hysteresis, which indicates the CO2 adsorption process can be effectively reversed by pressure swing. It can be concluded from the above observation that nitrogen plays a key role in enhancing CO2 uptake. Too high a nitrogen content may lead to excessive CO2 binding and increases the energy for CO2 desorption. Hence, it is important to optimize the nitrogen content.
Fig. 6a shows the CO2 uptake of CSA-700 in a gas mixture of 15 vol% CO2 and 85 vol% N2. The test result shows that CSA-700 adsorb about 1.3 wt% N2 when the temperature returns to 25 °C (see Fig. 6a). However, it can still adsorb 9.3 wt% CO2 even when the partial pressure of CO2 is greatly reduced. Hence, the sample demonstrates a very high selectivity of CO2 over N2. Again, this can be attributed to the relatively high nitrogen content. This is an outstanding performance, compared with materials reported in the literature and tested under similar conditions.23Fig. 6b shows the sample can maintain 91% adsorption capacity after five cycles of adsorption and desorption by temperature swing. The test condition is a simulation of the flue gas from a post-combustion power plant. Therefore, the above results further prove that such carbon spheres can exhibit exceptional performance in post-combustion CO2 capture applications.
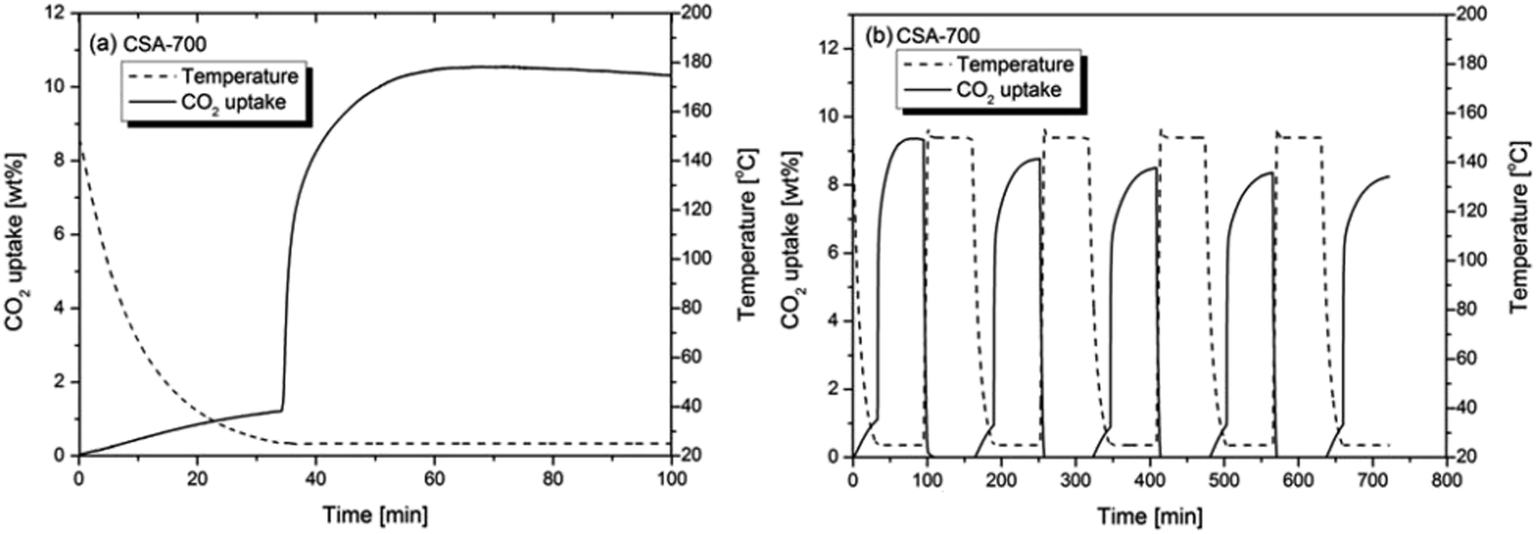 | ||
| Fig. 6 (a) The CO2 uptake of CSA-700 in a 15 vol% CO2 and 85 vol% N2 gas flow at 25 °C in a single cycle of adsorption and (b) five cycles of adsorption and desorption. | ||
3. Conclusions
An effective route is developed for the synthesis of hierarchically porous carbon spheres of millimeter scale. These are derived from an poly(acrylonitrile-co-acrylamide) precursor, leading to enriched nitrogen content and intercalated potassium cations in the carbon structures. The carbon spheres show a porous structure with a hollow core and hierarchical porous mantle. The activation temperature influences both the porous structure and nitrogen content of the carbon spheres. The carbon sphere with the highest specific surface area (2032.4 m2 g−1) and pore volume (0.992 cm3 g−1) is produced with a weight ratio of KOH/sphere = 1:1 and activated at 800 °C. This sample show a high CO2 capacity of 29 wt% (6.6 mmol g−1) at 0 °C under 1 bar CO2. However, for sorption at 25 °C, the highest CO2 uptake of 16.7 wt% (3.8 mmol g−1) is achieved with the carbon sphere activated at 700 °C, due to an optimum balance of N content and microporosity. The latter also shows an outstanding CO2 capture performance of 9.3 wt% (2.1 mmol g−1) and high selectivity of CO2 to N2, even at a low CO2 partial pressure in a mixed (CO2 and N2) environment. The results indicate that both microporosity and nitrogen content play an important role in enhancing the CO2 uptake of the carbon spheres. The shape, size, capacity and selectivity of the hierarchically porous carbon spheres are all ideal properties for practical applications in large-scale post combustion CO2 capture.
4 Experimental section
4.1. Synthesis of polymer spheres
Poly(acrylonitrile-co-acrylamide) was prepared by the copolymerization reaction of acrylonitrile (AN, 98.0%, Northern China Special Chemicals Development Center, China) and acrylamide (AM, 98.0%, Northern China Special Chemicals Development Center, China) in a 100 ml round bottom flask with dimethyl sulfoxide (25 ml, DMSO, 99.5%, Tianjin Guangfujinxi Chemical Research Institute, China). α,α′-Azoisobutyronitrile (AIBN, Shanghai no. 4 Reagent & H. V. Chemical Co., China) was used as the initiator for the radical polymerization reaction and the initiator concentration was 1 wt% of the total monomers weight, i.e., 0.091 g AIBN. A range of experiments were carried out to determine the fluidity of the polymer solution for free-forming of polymer spheres under gravity. The experimental conditions were optimized for the formation of millimeter-sized polymer spheres. The solution was magnetically stirred in a water bath at a reaction temperature of 65 °C for 3 h. Several initial monomer molar ratios were used to prepare copolymers (acrylamide–acrylonitrile = 0:1, 1:10.9, 1:3.3, 1:1.5). The ratio of 1:3.3 produced a relatively spherical shape compared with the other ratios. Therefore, the corresponding polymer spheres were selected to produce carbon spheres. Theoretically, this ratio produces a copolymer with the molar ratio of monomer units acrylamide–acrylonitrile = 0.73:1.
An in-house facility was built to produce the polymer spheres, as illustrated in Fig. 7. This set-up was adopted from an earlier work by Li et al.24 The polymer solution was dripping through a 2 mm diameter nozzle. A glass beaker filled with distilled water (1000 ml) was placed below. The distance between the nozzle and the water surface was selected to be 15 cm. Different amounts of DMSO were added to the reaction solution to adjust the viscosity for easy dripping and formation of spherical drops, and the optimum addition was identified to be 25 ml. Then, the solution was poured in the syringe. The polymer solution drops were formed below the nozzles due to Plateau–Rayleigh instability under gravity and fell into the water bath. The surface of the polymer drop solidified immediately upon contact with water and sank to the bottom of the beaker. All polymer spheres were left in the water bath for 24 h. The distilled water was refreshed every 24 h until the smell of DMSO was removed. Then, the polymer spheres were dried in a vacuum desiccator at room temperature.
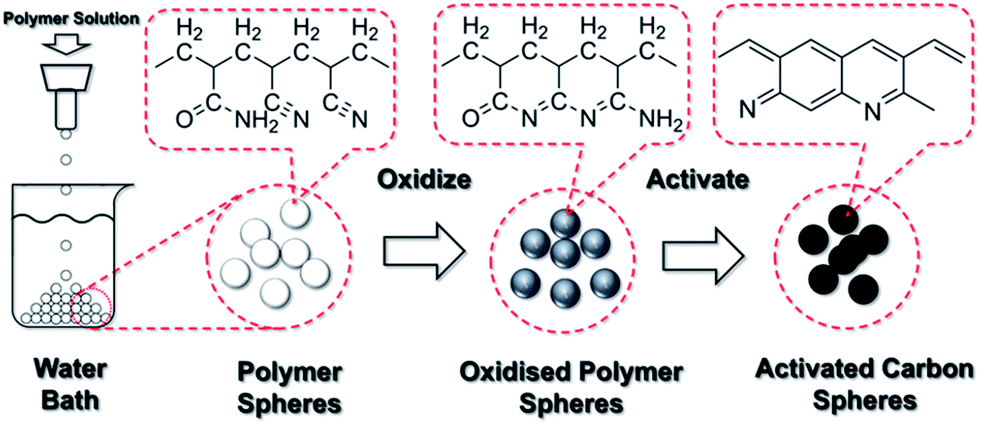 | ||
| Fig. 7 Schematic of the experimental procedures to produce activated carbon spheres. | ||
4.2. Oxidation
The dried spheres were loaded in a vertical furnace with an air compressor constantly pumping excess air through the furnace from the bottom to the top. The temperature was increased from ambient to 100 °C at a ramping rate of 2 °C min−1, then at 0.1 °C min−1 from 100 to 200 °C and finally dwelt at 200 °C for an hour. This stage was to remove residual water and DMSO from the spheres. The slow ramping rate was to avoid violent vaporization of residual gases, which would damage the sphere. Then the temperature was again raised to 300 °C at 0.2 °C min−1 and dwelt at 300 °C for the 8 hour oxidation reaction. At this stage, the slow ramping rate was to allow sufficient time for the oxidation reaction to take place and to prevent violent vaporization of un-oxidized polymer from damaging the sphere. Finally, the furnace was naturally cooled to room temperature.4.3. Carbonization and chemical activation
The oxidized polymer spheres (0.2 g) were soaked in the KOH solution (10 ml) in a 25 ml glass beaker. The concentrations of KOH gave effective KOH/sphere weight ratios of 1:1 and 2:1. The mixtures were left in a vacuum desiccator overnight and then dried in a vacuum oven at 80 °C. The dried samples were carbonized and chemically activated in a horizontal tube furnace at 600, 700 and 800 °C under constant nitrogen gas flow. The ramping rate was 3 °C min−1 and the dwelling time at the designated temperature was 1 h. After the carbonization and chemical activation, the spheres were repeatedly washed with distilled water until a pH strip showed a neutral value. Then, the wet samples were dried in vacuum at 120 °C over night.
4.4. Characterizations
Images of sample morphology and porous structure were obtained from a field-emission scanning electron microscope (SEM, JSM 6301F, JEOL, Japan). Samples were fixed with a piece of conductive carbon tab on a sample holder and then gold coated for imaging. The spheres were also cut in half by a scalpel for imaging of their cross-sectional morphology. High resolution images were obtained from a transmission electron microscope (TEM, JEOL 2100, Japan). Carbon spheres were ground and suspended in methanol. A drop of the suspension was placed on a carbon-film coated Cu grid (300 mesh) and then dried in air before being loaded in the TEM. A Fourier transform infrared spectroscopy (FTIR, NICOLET iS10, Thermo Scientific, USA) and an X-ray photoelectron spectrometer (XPS, K-ALPHA, Thermo Scientific, USA) were used to acquire the information of surface functional groups and chemical compositions. The porous structures of carbon spheres were analyzed by means of an automated gas sorption analyzer (Autosorb-iQ C, Quantachrome, USA). The N2 and CO2 sorption isotherms were obtained by N2 sorption at −196 °C (liquid nitrogen) and CO2 sorption at 0 °C (ice-water mixture), respectively. The specific pore volume (VN2) was determined by the amount of nitrogen adsorbed at the relative pressure 0.99. The specific surface area (SN2, pore size up to 40 nm), the specific micropore surface area (SMicro), micropore volume (VMicro) and pore size distribution were calculated using the Non-Local Density Functional Theory (slit/cylindrical pores) equilibrium model (NLDFT). The ultra-micropore volume (VCO2) was calculated by the 273 K CO2 on Carbon NLDFT model. In addition, the macropore volume (VHg, pore size up to 421 μm) was measured by means of a mercury porosimeter (AutoPore IV, Micromeritics, USA) between 0.5 and 3600 psia.The same gas sorption analyzer was also used to plot CO2 (Carbon dioxide research grade 100%, BOC, UK) adsorption and desorption isotherms from 0.1 to 1 bar at 0 and 25 °C. The samples were firstly degassed at the degassing station at 150 °C in vacuum for 2 h. Then, they were moved to the analysis station for CO2 uptake tests. Besides the uptake tests in pure CO2 environment, to simulate the practical CO2 capture condition in post combustion power plants, CSA-700 was also tested for its CO2 uptake in a gas environment with 15 vol% CO2 and 85 vol% N2 at 25 °C. The test was carried out in a thermogravimetric analyzer (TGA, Q500, TA instruments, USA). The sample was degassed at 150 °C for an hour in a constant pure N2 flow. Then, it was cooled to 25 °C before the gas flow was switched from N2 to the mixture of CO2 and N2 to measure its CO2 uptake. The sample was also tested for its CO2 uptakes over multiple cycles of adsorption and desorption by the temperature swing method. The temperatures were changed between 25 °C and 150 °C to adsorb and desorb CO2.
Acknowledgements
The authors would like to acknowledge the financial support from the UK EPSRC (EP/G063176/1, EP/G061785/1, and EP/I010955/1), the National Nature Science Foundation of China (no. 51172251 and 51061130536), Shanxi Province Science and Technology Innovation Program (no. 2012102007) and Shanxi Province International Collaboration Program (no. 2013081016). Research support was gratefully received from the Institute of Coal Chemistry, Chinese Academy of Sciences, and the Department of Chemistry, University College London, and the Department of Chemical and Environmental Engineering, University of Nottingham.Notes and references
- H. Herzog, E. Drake and E. Adams, CO2 Capture, Reuse and Storage Technologies for Mitigating Global Climate Change – A White Paper, Energy Laboratory, Massachusetts Institute of Technology, Cambridge, USA 1997, p. 5 Search PubMed.
- T. C. Drage, C. E. Snape, L. A. Stevens, J. Wood, J. Wang, A. I. Cooper, R. Dawson, Z. X. Guo, C. Satterley and R. Irons, J. Mater. Chem., 2012, 22, 2815–2823 RSC.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765–1771 CAS.
- M. Sevilla, P. Valle-Vigon and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- G. P. Hao, W. C. Li, D. Qian and A. H. Lu, Adv. Mater., 2010, 22, 853–857 CrossRef CAS PubMed.
- W. Z. Shen, Y. He, S. C. Zhang, J. F. Li and W. B. Fan, ChemSusChem, 2012, 5, 1274–1279 CrossRef CAS PubMed.
- J. C. Wang, A. Heerwig, M. R. Lohe, M. Oschatz, L. Borchardt and S. Kaskel, J. Mater. Chem., 2012, 22, 13911–13913 RSC.
- M. Olivares-Marín and M. Mercedes Maroto-Valer, Greenhouse Gases: Sci. Technol., 2012, 2, 20–35 CrossRef.
- G. Olofsson, Z. C. Ye, I. Bjerle and A. Andersson, Ind. Eng. Chem. Res., 2002, 41, 2888–2894 CrossRef CAS.
- N. P. Wickramaratne and M. Jaroniec, J. Mater. Chem. A, 2013, 1, 112–116 CAS.
- R. Mead-Hunter, A. J. C. King and B. J. Mullins, Langmuir, 2012, 28, 6731–6735 CrossRef CAS PubMed.
- J. Romanos, M. Beckner, T. Rash, L. Firlej, B. Kuchta, P. Yu, G. Suppes, C. Wexler and P. Pfeifer, Nanotechnology, 2012, 23, 015401 CrossRef CAS PubMed.
- J. C. Wang and S. Kaskel, J. Mater. Chem., 2012, 22, 23710–23725 RSC.
- A. N. A. El-Hendawy, Appl. Surf. Sci., 2009, 255, 3723–3730 CrossRef CAS PubMed.
- G. Socrates, Infrared Characteristic Group Frequencies, John Wiley & Sons, New York, USA, 1980 Search PubMed.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641–1653 CrossRef CAS.
- H. Fu, Carbon Fiber and Graphite Fiber, Chemical Industry Press, Beijing, China, 2010, pp. 145–146 Search PubMed.
- Y. F. Zhao, X. Liu, K. X. Yao, L. Zhao and Y. Han, Chem. Mater., 2012, 24, 4725–4734 CrossRef CAS.
- F. P. Richter, P. D. Caesar, S. L. Meisel and R. D. Offenhauer, Ind. Eng. Chem., 1952, 44, 2601–2605 CrossRef CAS.
- G. Liu, X. G. Li, J. W. Lee and B. N. Popov, Catal. Sci. Technol., 2011, 1, 207–217 CAS.
- G. Srinivas, V. Krungleviciute, Z. X. Guo and T. Yildirim, Energy Environ. Sci., 2014, 7, 335–342 CAS.
- N. Sun, C. Sun, H. Liu, J. Liu, L. Stevens, T. Drage, C. E. Snape, K. Li, W. Wei and Y. Sun, Fuel, 2013, 113, 854–862 CrossRef CAS PubMed.
- Y. Q. Li, K. X. Li and G. H. Sun, Mater. Lett., 2011, 65, 1022–1024 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta00438h |
| This journal is © The Royal Society of Chemistry 2014 |