 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid phosphine-free synthesis of CdSe quantum dots: promoting the generation of Se precursors using a radical initiator†
Bo
Hou
a,
David
Benito-Alifonso
a,
Richard
Webster
b,
David
Cherns
b,
M. Carmen
Galan
a and
David J.
Fermín
*a
aSchool of Chemistry, University of Bristol, Cantocks Close, Bristol BS8 1TS, UK. Web: http://www.chm.bris.ac.uk/pt/electrochemistryE-mail: David.Fermin@bristol.ac.uk; Tel: +44 (0)117 9288981
bSchool of Physics, University of Bristol, Tyndall Avenue, Bristol BS8 1TL, UK
First published on 19th February 2014
Abstract
The replacement of phosphine containing compounds in the synthesis of II–VI quantum dots (QDs) via the ‘hot-injection’ method has received considerable attention in recent years, in particular toward scaling-up production. A key bottleneck in current approaches is the poor solubility of elemental Se in solvents such as 1-octadecene (1-ODE) or oleylamine requiring a heating temperature of 200 °C for several hours, or the introduction of additives such as alkylthiols or NaBH4, or replacement by SeO2. In the present work, we elucidate the fundamental steps in the dissolution of elemental Se in 1-ODE and oleylamine with the view of facilitating the large-scale synthesis of CdSe QDs. The main organoselenium species generated during the solubilisation of elemental Se in 1-ODE and oleylamine were identified by 1D and 2D NMR spectroscopy (1H, 13C and 77Se). Experimental evidence suggests that the rate determining step is the formation of Se radicals, via homolytic cleavage of the Se–Se bond, that attack the allylic proton in 1-ODE and oleylamine. Plausible reaction pathways in both systems are proposed. Finally, we demonstrate that the radical-mediated solubilisation of Se can be significantly accelerated by the addition of azobisisobutyronitrile (AIBN), a common radical initiator used in the polymer industry. In this way, a highly concentrated Se precursor was prepared. The “hot-injection” of the Se precursor into CdO containing 1-ODE solution leads to the formation of highly luminescent CdSe QDs with a well-defined cubic structure.
1. Introduction
The optical properties of CdSe quantum dots (QDs) have generated a tremendous amount of interest due to their potential exploitation in a variety of fields including light-emitting devices,1 imaging probes2 and absorber layers in photovoltaic devices.3 Colloidal synthesis is widely recognized as key for large-scale fabrication of highly monodispersed and stable QDs. The seminal work by Murray et al.4 introduced the methodology of injecting phosphine–chalcogenide precursors into an organometallic solution at temperatures above 300 °C.4b Another important contribution in the field was made by Peng and co-workers, replacing the highly unstable organometallic precursors by metal oxides (e.g. CdO) in the presence of stearic acid.5 With regard to the solubilization of the chalcogenide component, Lewis bases such as tributylphosphine (TBP) or trioctylphosphine (TOP) have been extensively used.6 However, these phosphines are highly flammable and corrosive,7 prompting the search for alternative approaches.Mulvaney and co-workers reported the first phosphine-free synthesis of CdSe, based on the dissolution of Se in 1-octadecene (1-ODE) after prolonged periods of heating at high temperatures.8 Subsequently, other research groups have employed similar approaches to synthesize phosphine-free nanocrystals.9 Solvents such as olive oil,10 paraffin,11 and oleylamine12 have also been implemented. In addition to long time heating protocols aimed at facilitating the dissolution of Se, additives including NaBH413 and alkylthiols14 have been introduced. The disadvantage of the latter approaches is the introduction of additional elements (e.g. Na, B, and S) which could appear as impurities in the final product. Recent studies have demonstrated that the synthesis of QDs can be carried out by employing insolubilized Se15 or SeO216 powder with good yields. In these approaches, the reported Se concentration in precursor solution is not higher than 0.2 mol dm−3, which represents a bottleneck towards scale-up. In order to make the reaction suitable to the industrial scope, a good understanding of the Se dissolution process is crucial. Raston and co-workers proposed that Se–Se and Se–C bonding compounds are the main species after the dissolution of Se into 1-ODE.17 However, the mechanism of formation and nature of the Se precursors in these solvents remain to be fully elucidated.
In this work, we investigate the process involved in the dissolution of Se in 1-ODE and oleylamine by NMR, FTIR and mass spectroscopy, with the aim of optimizing this key step which is considered the bottleneck in the phosphine-free synthesis of CdSe QDs.12b,14 Our results confirm that the process is initiated by the attack of thermally generated Se radicals to the allylic position of 1-ODE upon heating at temperatures above 200 °C, in agreement with previous studies.17 We show that this reaction manifests itself by the migration of the terminal olefin group in 1-ODE and cis/trans isomerisation in oleylamine, generating a range of organoselenium structures. Furthermore, we show for the first time that Se dissolution in 1-ODE can be significantly accelerated by addition of trace amounts of the radical initiator azobisisobutyronitrile (AIBN, I). This approach allows not only the preparation of Se precursor solutions with concentrations as high as 1.0 mol dm−3, but also the facile synthesis of highly luminescent CdSe QDs under phosphine-free conditions yielding phase-pure monodispersed cubic nanocrystals.

2. Experimental
2.1. Se dissolution in 1-ODE and oleylamine
The dissolution of elemental Se (3 mmol) into 1-ODE (9 mmol, 2.88 cm3) or oleylamine (9 mmol, 4.23 cm3) was investigated by taking aliquots of the reaction mixture during the following heating program: room temperature (step I), 60 °C for 30 min under vacuum (step II), 130 °C for 30 min under vacuum (step III), 200 °C for 30 min (step IV) and 20 h (step V) under Ar. Each aliquot was analysed by 1D 1H, 13C and 2D NMR, as well as FTIR spectroscopy. The concentration of the final Se solution in 1-ODE or oleylamine is 1.04 and 0.71 mol dm−3, respectively.2.2. Facile Se dissolution promoted by AIBN
Following a similar procedure as in the previous experiment, a mixture of Se and 1-ODE (1.04 mol dm−3) was prepared with the addition of 0.6 mg of AIBN at room temperature. The composition of the solution was investigated at room temperature under Ar (step I) and after heating at 200 °C for 1 h (step II), 2 h (step III) and 3 h (step IV).2.3. Phosphine-free synthesis of CdSe QDs
The QDs were prepared based on the method reported by Peng18 with a commonly employed Se precursor concentration (0.1 mol dm−3). Typically, Se (0.0395 g) and 1-ODE (5 cm3) are loaded into a 50 cm3 two-neck flask and heated at 60 °C under vacuum for 1 h, followed by addition of 0.001 g of AIBN (1% mol mol−1). The temperature is subsequently increased to 200 °C for 1 h under Ar, leading to the dissolution of Se (clear yellow solution). CdO (0.0254 g), stearic acid (0.228 g) and 1-ODE (20 cm3) were introduced into a 250 ml three-neck round bottom flask and dried in a vacuum at 100 °C for 30 min. The metal-precursor solution was subsequently heated to 320 °C under an Ar atmosphere. Once the solution became colourless, the Se precursor was quickly injected into the cadmium solution. The particles were allowed to grow for different times at 320 °C, after which the heating mantle was removed. The QDs were purified by dissolving in a methanol–hexane mixture (v/v = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and centrifuged for 10 min to remove 1-ODE, followed by addition of acetone and further centrifugation. The precipitate was dissolved in chloroform for further characterization.
:1) and centrifuged for 10 min to remove 1-ODE, followed by addition of acetone and further centrifugation. The precipitate was dissolved in chloroform for further characterization.
2.4. NMR, FTIR and electron microscopy methodologies and instrumentation
Reactions requiring anhydrous conditions were performed under an atmosphere of either anhydrous nitrogen or argon. All glassware was flame-dried prior to use and glass syringes and needles were placed in an oven (150 °C) for at least 2 h and allowed to cool in desiccators under an atmosphere of anhydrous nitrogen. Aliquots from the reaction mixture were collected at different stages of the heating program under an Ar atmosphere and samples were allowed to cool to room temperature before analysis. 1D, 2D 1H NMR and 13C NMR spectra from 2D proton correlation spectroscopy (COSY), carbon–proton heteronuclear single quantum correlation (HSQC) and carbon–proton heteronuclear multiple-bond correlation spectroscopy (HMBC) were recorded in a 400 MHz Varian INOVA 400 instrument. Chemical shifts are quoted in parts per million (ppm) and referenced to SiMe4 (1H NMR 0 ppm) and CDCl3 (13C NMR 77.16 ppm). The 13C NMR sequence parameters were set up as follows: a 45° pulse of 7.70 μs, τ2 = 1.0 s, and an acquisition time of 1.28 s. The 1D and 2D 77Se NMR spectra including 1D proton decoupled 77Se NMR and 2D proton–selenium HMBC were recorded in a 500 MHz Varian INOVA 500 instrument. The 77Se NMR sequence parameters were set up as follows: 45° pulse, τ2 = 1.0 s, and an acquisition time of 0.860 s. A saturated solution of diphenyldiselenide in CDCl3 was used as an external standard.19 FTIR spectra were obtained using a Perkin-Elmer Spectrum 100 FTIR Spectrometer. The structure of the CdSe dots was investigated by powder XRD (BRUKER D8, Cu Kα radiation λ = 1.54 Å) and JEOL 2011 high resolution TEM operating at 200 kV fitted with an EDX Oxford Instruments ISIS 300 system. Selected area electron diffraction (SAED) analysis was performed using a Philips EM430 instrument with a camera length set at 270 mm and the electron wavelength at 0.251 Å.2.5. Electrospray ionization (ESI) and negative ion matrix assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) analysis of Se-containing product
In the 1-ODE system, the Se containing products were isolated by silica gel flash column chromatography using hexane as the eluent. In the oleylamine system, the Se containing products are isolated through vacuum distillation using a Kugerlrohr apparatus. The isolated products were dried under vacuum and re-dissolved into dichloromethane for ESI+ analysis. MALDI analyses were recorded using a HP MALDI instrument (Applied Biosystems 4700 Proteomics Analyzer) using gentisic acid as the matrix.3. Results and discussion
3.1. Analysis of the Se precursor in 1-ODE
Fig. 1 displays NMR and FTIR spectra of the Se–1-ODE mixture at various heating steps. 13C NMR spectra show the emergence of new signals at δ = 124.4 and 131.7 ppm (Fig. 1a) after heating at 200 °C for 20 h under an Ar atmosphere (step V). 1H NMR data also revealed the appearance of a new multiplet signal at δ = 5.42 ppm at step V (Fig. 1b). The appearance of these signals occurs only after the complete dissolution of Se in 1-ODE. A comparison of the 2D HSQC NMR spectra of the solutions at steps I (Fig. 1c) and V (Fig. 1d) reveals the appearance of cis and trans carbon–proton correlations in the latter step, suggesting the migration of the terminal olefin moiety to form 2-octadecene (2-ODE). This analysis is supported by the 2D COSY NMR spectrum of sample V showing that the olefinic protons (δ = 5.41 and 5.43 ppm) are correlated with protons found at δ = 1.63 and 1.94 ppm (see ESI [S3]†), while the HSQC spectrum in Fig. S4† indicates that the protons at δ = 1.63 and 1.94 ppm are bonded with carbon atoms at δ = 18.0 and 32.7 ppm, which are attributed to CH2 and CH3 moieties. Furthermore, the decrease of the IR bands at ν = 991 and 908 cm−1 and the emergence of a band at ν = 964 cm−1 (Fig. 1e) are also consistent with the formation of 2-ODE. | ||
| Fig. 1 13C-NMR analysis of Se in 1-ODE at room temperature (step I), 60 °C for 30 min under vacuum (step II), 130 °C for 30 min under vacuum (step III), 200 °C for 30 min (step IV) and 20 h (step V) under Ar (a). 1H-NMR spectra of the solution precursor at steps I and V, as well as of 1-ODE after heating at 200 °C for 20 h under Ar in the absence of Se (b). The highlighted areas in (a) and (b) correspond to the signal associated with the olefin functional group. HSQC 2D NMR spectra of Se containing solutions at step I (c) and step V (d). The arrows indicate the correlation between 13C–1H from cis and trans olefin moieties. FTIR analysis of the precursor solutions at steps I to V (e). The arrows indicate a vibrational mode associated with the olefin moiety in trans 2-octadecene, while the dotted line indicates the position of the equivalent vibrational model in 1-ODE. | ||
Fig. 2 shows the 77Se NMR analysis of the Se precursor at step V after purification by chromatography. The NMR signals with chemical shifts of 416.4 (singlet), 373.1 (singlet), 373.3 (singlet), 307.4 (singlet) and 259.2 ppm (singlet) are consistent with dialkyl polyselenides species.201H, 13C, 77Se 1D and 2D NMR analyses (Fig. 3 and ESI [S4]†) confirmed the formation of a carbon–selenium bond during the solubilisation process. The species formed are a mixture of structural isomers with similar properties, complicating their isolation. A thorough study of the mixture shows, as a common feature, the presence of an olefinic group alpha to the C–Se bond.

 | ||
| Fig. 2 Proton decoupled 77Se NMR spectrum of the purified Se intermediate generated after dissolution of 1-ODE after 20 h at 200 °C under Ar. The inset shows two closed peaks with chemical shifts of 373.3 and 373.1 ppm. | ||
 | ||
| Fig. 3 1H and 77Se HMBC NMR analysis of Se containing species. As indicated in the image, 77Se NMR signals at δ = 373.3 and 373.1 ppm correlate with the 1H multiplet at δ = 1.73 and 1.54 ppm. | ||
Fig. 3 shows the 2D 1H and 77Se HMBC NMR spectra of Se containing species, confirming the Se bonding to a secondary carbon. The 2D 1H and 13C HMBC NMR spectra in Fig. S5 and S6 (see ESI [S4]†) suggest that the secondary carbon bound to Se is next to an olefinic carbon and a saturated carbon. The 2D 1H and 13C HSQC NMR spectra in Fig. S7 (see ESI [S4]†) further confirm that the olefinic moiety is vicinal to the C–Se bond and (II) is proposed to be the general core structure.
Mass values collected from ESI and MALDI MS analyses suggested the species (III) as plausible structures being formed. Detail mass spectra can be found in the ESI (S1 and S2).† A correlation of NMR and mass spectrometry analysis results is listed in Table S1.†
Based on the analysis of the NMR, FTIR, and MS data shown in Fig. 1–3 and Table S1,† the dissolution of Se in 1-ODE can be rationalized in terms of a radical process thermally initiated by the homolytic cleavage of the Se–Se bond as highlighted in Scheme 1. The key step is the attack of the allylic proton in 1-ODE by Se radicals leading to the migration of the double bond and subsequent H transfer from the Se intermediate species.17,21 This is further supported by subsequent experiments where no isomerization is observed upon extensive heating of 1-ODE in the absence of Se (shown in Fig. 1b and also ESI [S5]†). Moreover, no dissolution of Se takes place under similar conditions if 1-ODE is replaced by octadecane, which lacks the alkene functionality.
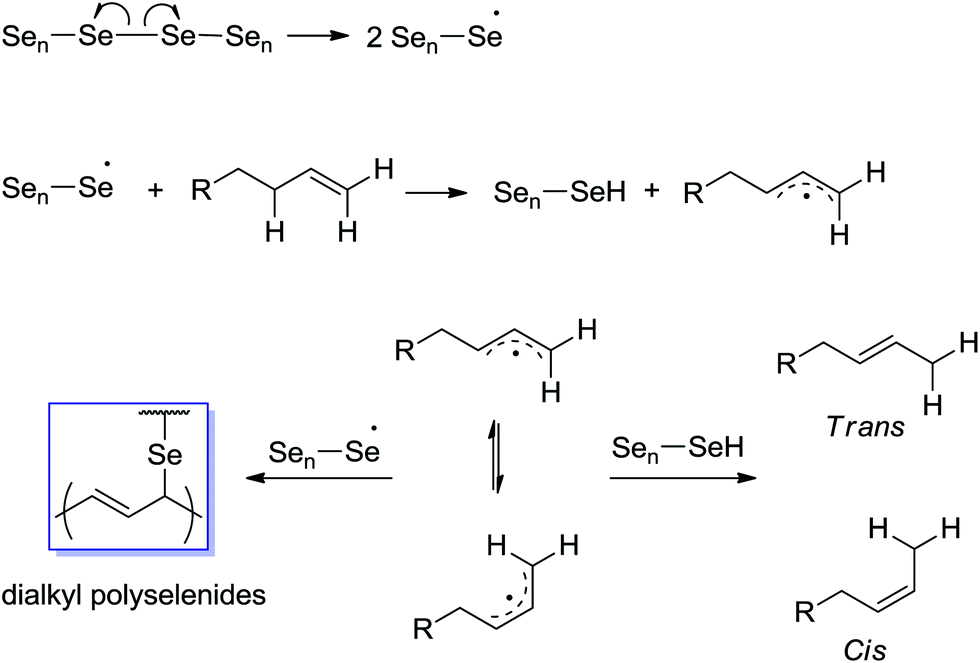 | ||
| Scheme 1 Proposed mechanism for the thermally induced Se radical attack of the allylic proton in 1-ODE, leading to the dissolution of Se and the migration of the terminal olefin. Highlighted in the blue frame is the olefinic group alpha to the C–Se bond which is a common feature present in the organoselenium species. | ||
3.2. Dissolution of Se into oleylamine
In addition to 1-ODE, oleylamine is another solvent often used for the preparation of Se precursors.12a In our recent work, organoselenium species were detected by employing mass spectroscopy and 1H and 13C NMR spectroscopy, during the Se dissolution in oleylamine.22 Although isomerization of the double bond was identified in this instance, the depletion of the olefin moiety was also observed, which did not occur in the case of 1-ODE.22Fig. 4 shows the HSQC NMR analysis of Se containing species at steps I and IV. A significant decrease in the signals at 13C δ = 130 ppm and 1H δ = 5.35 ppm upon heating at 200 °C over prolonged periods of time (step IV, highlighted in Fig. 4b) is consistent with our previous reported results.22 The disappearance of this signal occurs concomitantly with the dissolution of Se. | ||
| Fig. 4 2D HSQC NMR analysis of (a) Se and oleylamine mixture before heating; (b) Se and oleylamine after heating at 200 °C for 20 h. | ||
Fig. 5 displays FTIR spectra of Se containing species at steps I to IV. The cis and trans isomerization is detected in the case of Se dissolution. As shown in Fig. 5, cis hydrogen out of plane deformation (as highlighted, ‘band F’, step I) almost disappeared and an increase of trans (band ‘E’) is observed after the total dissolution of Se (step IV). This observation is consistent with the HSQC NMR results shown in Fig. 4 and our previous report which further confirmed the disappearance of the olefinic moiety during the Se dissolution in oleylamine.22
 | ||
| Fig. 5 FTIR analysis of the reaction of oleylamine with Se (I) oleylamine mixed with Se before heating; (II) oleylamine and Se mixture after heating at 60 °C under vacuum for 30 min; (III) oleylamine and Se after heating at 130 °C under vacuum for 30 min. (IV) Oleylamine and Se after heating at 200 °C under Ar for 30 min. (V) Oleylamine and Se after heating at 200 °C under Ar for 20 h. | ||
Fig. 6 shows the 77Se NMR spectrum of the Se precursor generated at step V. Two major signals at δ = 336.2 and 525.6 ppm show the characteristic peaks of polyalkyl polyselenides compounds.20 This result is similar to our previous findings on Se dissolution into 1-ODE. Moreover, the 77Se NMR spectrum shows the presence of a multiplet (highlighted in the inset image, δ = 336.2 ppm) indicating the formation of the polyalkyl polyselenides structure.
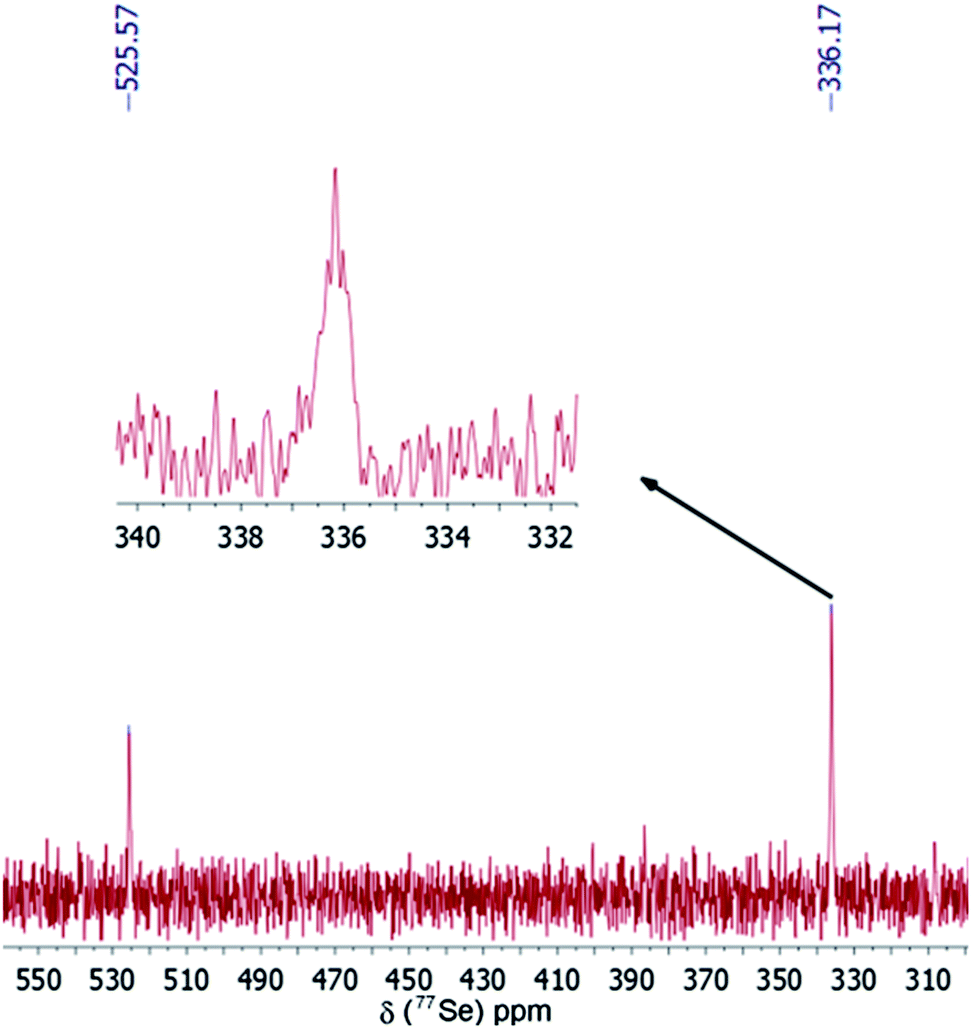 | ||
| Fig. 6 Proton decoupled 77Se NMR spectrum of the purified Se intermediate generated after dissolution in oleylamine after 20 h at 200 °C under Ar. The inset shows the enlarge image of the multiplet peak at δ = 336.2 ppm. | ||
Fig. 7 displays the 2D 1H and 77Se HMBC analysis of the Se precursor in oleylamine. The main 77Se signal at δ = 336.2 ppm correlates with protons at chemical shifts of δ = 2.81, 1.55 and 1.32 ppm. It should be noted that these results are comparable to those in Fig. 3, except for the new signal at 2.81 ppm. Moreover, the 1H and 13C HMBC NMR spectra in Fig. S9a (see ESI [S6]†) suggest that the secondary carbon bound to Se is next to carbon atoms with chemical shifts at δ = 26.79 and 47.24 ppm. The HSQC spectrum in Fig. S9b† shows these carbon atoms are CH and CH2 moieties. The analysis also shows that CH is attached to nitrogen (1H at δ = 2.76 and 13C δ = 26.78 ppm). Therefore, besides structure II and polyalkyl polyselenide structures proposed previously,22 structure IV is proposed to be one of the other core structures formed after consumption of the olefin moiety.
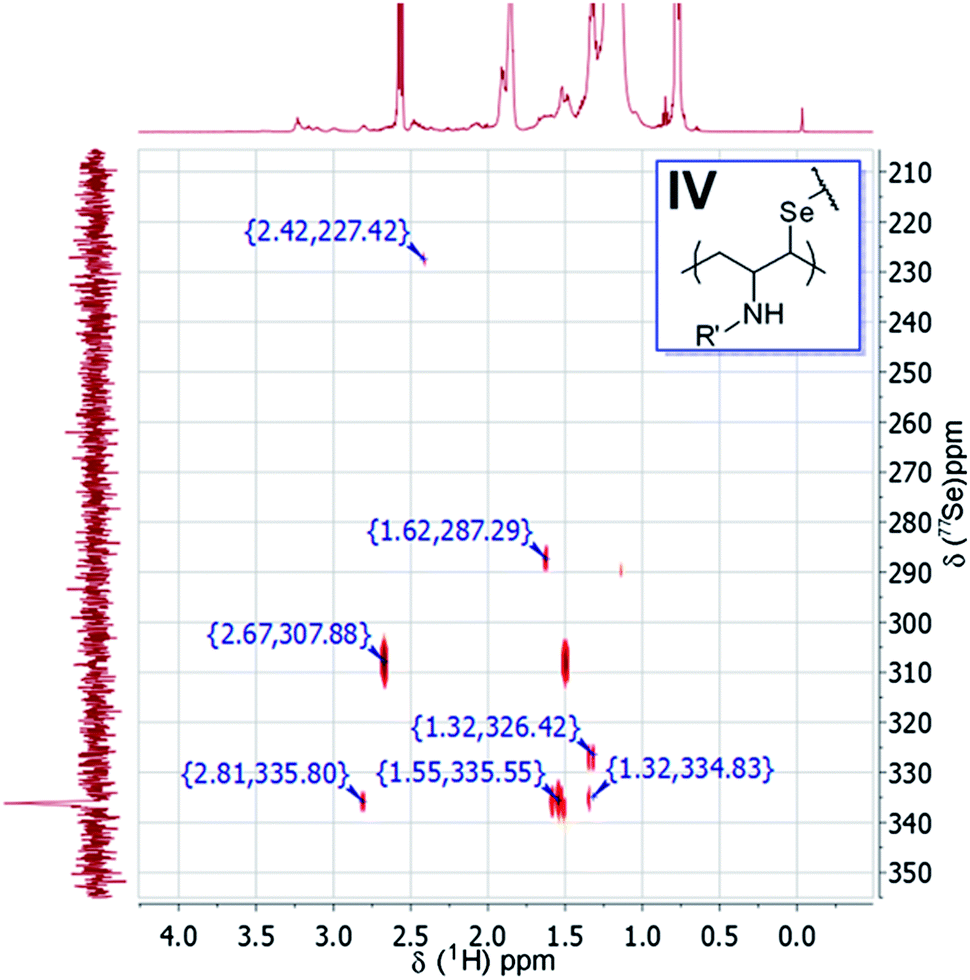 | ||
| Fig. 7 1H and 77Se HMBC NMR spectra of the Se precursor in oleylamine. As indicated in the image, 77Se (multiplet, δ = 336.17 ppm) correlates with protons 1H (multiplet) at δ = 2.81, 1.55 and 1.32 ppm. | ||
The disappearance of the olefin group during Se dissolution in oleylamine can be explained by nucleophilic attack of oleylamine. Besides the double bond, oleylamine also possesses a nucleophilic amine group capable of reacting with Se23 forming N–Se intermediates able to form N-centred radical species. Therefore, the organoselenium structures generated in the oleylamine approach are expected to be more complicated. Based on the NMR and FTIR analyses and previous results, a plausible reaction mechanism for the depletion of the olefin moiety during Se dissolution in oleylamine is proposed in Scheme 2. A radical process is thermally initiated by the homolytic cleavage of the Se–Se bond as highlighted in Scheme 2. The key step is the attack on the cis allylic proton in oleylamine by Se radicals, leading to the isomerization of the double bond and subsequent H transfer from the Se intermediate species.21 Concomitantly, nucleophilic addition from the amine occurs with subsequent addition to the alkyl chain as shown in Scheme 2.
 | ||
| Scheme 2 Proposed mechanism of the thermal induced Se radical attack on oleylamine, leading to the dissolution of Se and the disappearance of the olefin group. | ||
3.3. Se dissolution into 1-ODE promoted by AIBN
On the basis that Se solubilisation in 1-ODE occurs via radical species, we investigated whether trace amounts of AIBN could accelerate this process. For instance, recent studies by Yu et al. have shown that radical cycloaddition reactions involving elemental chalcogens can be accelerated in the presence of AIBN.24Fig. 8 shows clear evidence of migration of the olefin group, and the associated Se dissolution, just after 1 h heating at 200 °C in the presence of AIBN. Indeed, the 13C NMR signals at δ = 124.4 and 131.7 ppm (Fig. 8a) as well as the 1H NMR multiplet at δ = 5.42 ppm (Fig. 8b) can be observed after 1 h as opposed to 20 h in the absence of AIBN (Fig. 1). The migration of the double bond is further confirmed by HSQC NMR (Fig. 8c and d) as well as FTIR spectroscopy (Fig. 8e), suggesting that AIBN effectively promotes the dissolution of Se. AIBN decomposes at temperatures above 65 °C, eliminating molecular nitrogen and generating 2-cyanoprop-2-yl radicals acting as initiators.25 It should be mentioned that, in the absence of Se, the migration of the olefin moiety is not observed when AIBN is present. Consequently, it could be concluded that the radical initiator assists the generation of Se radicals which subsequently attack γ-H in 1-ODE as suggested in Scheme 1. | ||
| Fig. 8 13C-NMR (a) and 1H-NMR (b) analyses of the Se–1-ODE mixture at room temperature (step I) and after loading AIBN and heating at 200 °C for 1 (step II), 2 (step III) and 3 h (step IV) under Ar. HSQC 2D NMR spectra of the mixture at steps I (c) and IV (d). FTIR spectra of the solutions at various stages (e). The main features of the various spectral responses are identical to those highlighted in Fig. 1, although AIBN containing solution promotes Se solubilisation within 1 h. | ||
Reported times for Se dissolution in 1-ODE at various concentrations and temperatures are summarised in Table S2 (ESI†). The time required for solubilisation of 0.1 mol dm−3 significantly decreases from 48 h at 150 °C to between 1 and 2 h at 200 °C. Our measurements in the presence of 1 mg of AIBN show slightly shorter times for this Se concentration. The remarkable point is observed at concentrations in the range of 1.0 mol dm−3, where the dissolution time is decreased from 20 to 3 h in the presence of AIBN. This temperature is still below the melting point of Se (221 °C).26 Furthermore, the main by-products of AIBN decomposition are traces of short chain volatile alkanes which do not compromise the purity of the final material.
3.4. CdSe QD synthesis employing Se precursors generated in the presence of AIBN
CdSe QDs showing a high degree of monodispersity and strong absorption-luminescence overlap were obtained employing Se precursors in 1-ODE in the presence of AIBN as shown in Fig. 9. The TEM micrograph in Fig. 9a shows highly monodispersed 3.0 ± 0.2 nm dots obtained after 15 min growth time. The inset in Fig. 9a displays absorption and luminescence spectra featuring a strong spectral overlap and a progressive decrease of the optical band gap with increasing growth time. | ||
| Fig. 9 Characteristic TEM micrograph (scale bar 20 nm) of 3.0 ± 0.2 nm CdSe dots after 15 min growth (a). The inset shows the absorption and luminescence spectra recorded after 3 (blue), 8 (grey) and 15 min (red) growth times. Experimental and simulated selected area electron diffraction patterns (b), powder XRD (c) and high resolution TEM image (d, scale bar 2 nm) of CdSe dots. The XRD pattern of cubic CdSe (PDF 19-0191) is also shown in (c). The inset in (d) shows a 3.0 nm dot, highlighting the 70 ± 2° cross-grating originating from adjoining (111) planes in fcc crystals with a lattice spacing of 3.5 ± 0.2 Å (parallel lines). | ||
Selected area electron diffraction pattern rings in Fig. 9b are characteristic of the zinc blende (ZB) unit cell, with the 111, 220 and 311 rings clearly identifiable although broadened by the nanoscopic dimension of the crystals. The experimental data correlate closely with simulations of the diffraction patterns for 3 nm CdSe nanocrystals.27 X-ray powder diffraction (Fig. 9c) is fully compatible with phase pure ZB CdSe dots. Fig. 9d shows a typical high resolution TEM image obtained from as-prepared 3 nm CdSe QDs, featuring a cross-grating pattern generated by adjoining (111) planes with the 70 ± 2° angle. The measured lattice spacing of the (111) plane of 3.5 ± 0.2 Å is close to the value expected for cubic CdSe (the inset image of Fig. 9d).
These results show that the presence of the trace amount of AIBN does not affect the nucleation of CdSe, demonstrating its suitability for the preparation of Se precursors for QD synthesis. The ability of preparing chalcogenide precursors with high concentration and short times is crucial for deployment of scalable routes for the production of thin-film photovoltaic devices. The introduction of radical initiators in phosphine-free solvents, along with the temperature and concentration, provides an additional optimisation parameter that can be exploited in this direction.
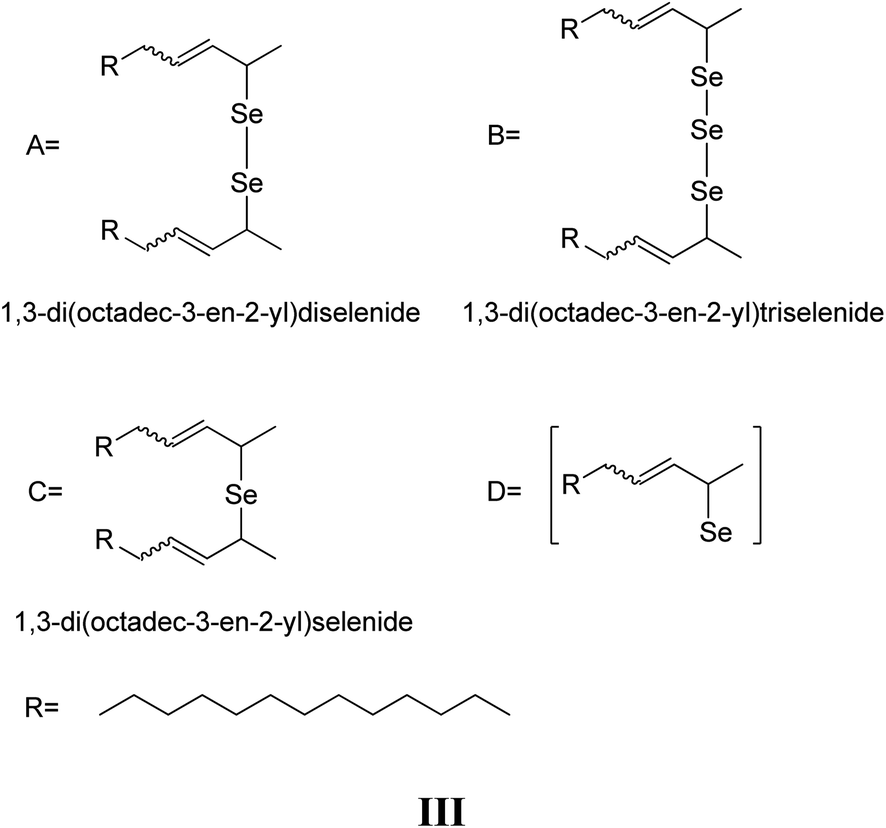
4. Conclusions
Our studies have unveiled the processes taking place during the dissolution of Se in 1-ODE and oleylamine, an important bottleneck in the scale-up of phosphine-free synthesis of CdSe QDs. The process is initiated by the generation of Se radicals which attack the allylic proton in both solvents. This reaction manifests itself by the migration of the olefin group in 1-ODE and isomerization in oleylamine.In the case of 1-ODE, the most plausible intermediates in this process are identified as 1,3-di(octadec-3-en-2-yl)monoselenide, 1,3-di(octadec-3-en-2-yl)diselenide and 1,3-di(octadec-3-en-2-yl)triselenide. On the other hand, the intrinsic nucleophilic properties of oleylamine facilitate the reaction with the double bond during Se solubilisation, inducing the formation of polyalkyl aminoselenides besides the polyalkyl polyselenide species.
For the first time, we show that addition of trace amounts of AIBN significantly accelerates the solubilisation of Se in 1-ODE in the molar concentration range. Studies carried out at various concentrations and temperatures indicate that Se solubilisation requires temperatures above 150 °C, even in the presence of AIBN. Although AIBN promotes faster Se dissolution in the 0.1 mol dm−3 concentration range, the strongest effects are observed at significantly higher Se concentrations.
The utilisation of AIBN as a promoter in the generation of Se precursors allows the facile phosphine-free synthesis of high-quality zinc-blende CdSe QDs without the need for further additives. The crystal structure of the dots contrasts with the strong wurtzite character of the materials obtained in the presence of phosphine groups.28 We strongly believe that optimization of the chalcogenide precursor's composition by adjusting the concentration of radical initiators, such as AIBN, can have a significant impact on scaling up the synthesis of these materials and their deployment in solution-processable photovoltaic technologies.
Acknowledgements
The authors are grateful to Mr Jonathan Jones, Mr Paul Lawrence and Dr Craig Butts for their support to this research. This research is supported by the Engineering and Physical Sciences Research Council (EP/G031088/1, SUPERGEN Consortium on Excitonic Solar Cells (DJF and BH) and EP/J002542/1 (MCG and DBA)).References
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulovic, Nat. Photonics, 2013, 7, 13–23 CrossRef CAS.
- (a) F. A. Esteve-Turrillas and A. Abad-Fuentes, Biosens. Bioelectron., 2013, 41, 12–29 CrossRef CAS PubMed; (b) F. Wang, X. Liu and I. Willner, Adv. Mater., 2013, 25, 349–377 CrossRef CAS PubMed; (c) X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- (a) L. Etgar, Materials, 2013, 6, 445–459 CrossRef CAS; (b) P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737–18753 CrossRef CAS.
- (a) T. Trindade, P. O'Brien and N. L. Pickett, Chem. Mater., 2001, 13, 3843–3858 CrossRef CAS; (b) C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- (a) Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2000, 123, 183–184 CrossRef; (b) L. Qu, Z. A. Peng and X. Peng, Nano Lett., 2001, 1, 333–337 CrossRef CAS.
- (a) C. de Mello Donegá, P. Liljeroth and D. Vanmaekelbergh, Small, 2005, 1, 1152–1162 CrossRef PubMed; (b) K. L. Sowers, B. Swartz and T. D. Krauss, Chem. Mater., 2013, 25, 1351–1362 CrossRef CAS.
- B. Stewart, A. Harriman and L. J. Higham, Organometallics, 2011, 30, 5338–5343 CrossRef CAS.
- J. Jasieniak, C. Bullen, J. van Embden and P. Mulvaney, J. Phys. Chem. B, 2005, 109, 20665–20668 CrossRef CAS PubMed.
- (a) H. Shen, H. Wang, Z. Tang, J. Z. Niu, S. Lou, Z. Du and L. S. Li, CrystEngComm, 2009, 11, 1733–1738 RSC; (b) M. Sun and X. Yang, J. Phys. Chem. C, 2009, 113, 8701–8709 CrossRef CAS; (c) L. Liu, Z. Zhuang, T. Xie, Y.-G. Wang, J. Li, Q. Peng and Y. Li, J. Am. Chem. Soc., 2009, 131, 16423–16429 CrossRef CAS PubMed.
- S. Sapra, A. L. Rogach and J. Feldmann, J. Mater. Chem., 2006, 16, 3391–3395 RSC.
- (a) Z. Deng, H. Yan and Y. Liu, J. Am. Chem. Soc., 2009, 131, 17744–17745 CrossRef CAS PubMed; (b) B. Xing, W. Li, X. Wang, H. Dou, L. Wang, K. Sun, X. He, J. Han, H. Xiao, J. Miao and Y. Li, J. Mater. Chem., 2010, 20, 5664–5674 RSC.
- (a) S. Mourdikoudis and L. M. Liz-Marzan, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS; (b) Y. Wei, J. Yang, A. W. H. Lin and J. Y. Ying, Chem. Mater., 2010, 22, 5672–5677 CrossRef CAS.
- S. C. Riha, B. A. Parkinson and A. L. Prieto, J. Am. Chem. Soc., 2011, 133, 15272–15275 CrossRef CAS PubMed.
- Y. Liu, D. Yao, L. Shen, H. Zhang, X. Zhang and B. Yang, J. Am. Chem. Soc., 2012, 134, 7207–7210 CrossRef CAS PubMed.
- (a) S. Flamee, M. Cirillo, S. Abe, K. De Nolf, R. Gomes, T. Aubert and Z. Hens, Chem. Mater., 2013, 25, 2476–2483 CrossRef CAS; (b) S. Flamee, R. Dierick, M. Cirillo, D. Van Genechten, T. Aubert and Z. Hens, Dalton Trans., 2013, 42, 12654–12661 RSC.
- M. Ibanez, R. Zamani, A. LaLonde, D. Cadavid, W. Li, A. Shavel, J. Arbiol, J. R. Morante, S. Gorsse, G. J. Snyder and A. Cabot, J. Am. Chem. Soc., 2012, 134, 4060–4063 CrossRef CAS PubMed.
- (a) C. Bullen, J. van Embden, J. Jasieniak, J. E. Cosgriff, R. J. Mulder, E. Rizzardo, M. Gu and C. L. Raston, Chem. Mater., 2010, 22, 4135–4143 CrossRef CAS; (b) R. García-Rodríguez, M. P. Hendricks, B. M. Cossairt, H. Liu and J. S. Owen, Chem. Mater., 2013, 25, 1233–1249 CrossRef.
- W. W. Yu and X. Peng, Angew. Chem., Int. Ed., 2002, 41, 2368–2371 CrossRef CAS.
- J. D. Odom, W. H. Dawson and P. D. Ellis, J. Am. Chem. Soc., 1979, 101, 5815–5822 CrossRef CAS.
- H. Eggert, O. Nielsen and L. Henriksen, J. Am. Chem. Soc., 1986, 108, 1725–1730 CrossRef CAS.
- R. M. Hoyte and D. B. Denney, J. Org. Chem., 1974, 39, 2607–2612 CrossRef CAS.
- B. Hou, D. Benito-Alifonso, N. Kattan, D. Cherns, M. C. Galan and D. J. Fermín, Chem. – Eur. J., 2013, 19, 15847–15851 CrossRef CAS PubMed.
- J. Lu, Y. Xie, F. Xu and L. Zhu, J. Mater. Chem., 2002, 12, 2755–2761 RSC.
- L. Yu, Y. Wu, T. Chen, Y. Pan and Q. Xu, Org. Lett., 2012, 15, 144–147 CrossRef PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, WILEY, 6th edn, 2007 Search PubMed.
- Y. A. Yang, H. Wu, K. R. Williams and Y. C. Cao, Angew. Chem., Int. Ed., 2005, 44, 6712–6715 CrossRef CAS PubMed.
- P. A. Stadelmann, Ultramicroscopy, 1987, 21, 131–145 CrossRef CAS.
- (a) W. W. Yu, Y. A. Wang and X. Peng, Chem. Mater., 2003, 15, 4300–4308 CrossRef CAS; (b) B. Hou, D. Parker, G. P. Kissling, J. A. Jones, D. Cherns and D. J. Fermín, J. Phys. Chem. C, 2013, 117, 6814–6820 CrossRef CAS; (c) Y. C. Li, H. Z. Zhong, R. Li, Y. Zhou, C. H. Yang and Y. F. Li, Adv. Funct. Mater., 2006, 16, 1705–1716 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta00285g |
| This journal is © The Royal Society of Chemistry 2014 |