 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A fluorene based covalent triazine framework with high CO2 and H2 capture and storage capacities†
Stephan
Hug
abc,
Maria B.
Mesch
d,
Hyunchul
Oh
e,
Nadine
Popp
d,
Michael
Hirscher
e,
Jürgen
Senker
d and
Bettina V.
Lotsch
*abc
aDepartment of Chemistry, Ludwig-Maximilians-Universität München, Butenandtstr. 5-13, 81377 Munich, Germany. E-mail: bettina.lotsch@cup.uni-muenchen.de; Fax: +49 89 2180 77440; Tel: +49 89 2180 77429
bMax Planck Institute for Solid State Research, Heisenbergstr. 1, 70569 Stuttgart, Germany. E-mail: b.lotsch@fkf.mpg.de; Fax: +49 711 689 1612; Tel: +49 711 689 1610
cNanosystems Initiative Munich (NIM) and Center for Nanoscience, Schellingstr. 4, 80799 Munich, Germany
dInorganic Chemistry III, Universität Bayreuth, Universitätsstr. 30, 95447 Bayreuth, Germany
eMax Planck Institute for Intelligent Systems, Heisenbergstr. 3, 70569 Stuttgart, Germany
First published on 5th February 2014
Abstract
Porous organic polymers have come into focus recently for the capture and storage of postcombusted CO2. Covalent triazine frameworks (CTFs) constitute a nitrogen-rich subclass of porous polymers, which offers enhanced tunability and functionality combined with high chemical and thermal stability. In this work a new covalent triazine framework based on fluorene building blocks is presented, along with a comprehensive elucidation of its local structure, porosity, and capacity for CO2 capture and H2 storage. The framework is synthesized under ionothermal conditions at 300–600 °C using ZnCl2 as a Lewis acidic trimerization catalyst and reaction medium. Whereas the materials synthesized at lower temperatures mostly feature ultramicropores and moderate surface areas as probed by CO2 sorption (297 m2 g−1 at 300 °C), the porosity is significantly increased at higher synthesis temperatures, giving rise to surface areas in excess of 2800 m2 g−1. With a high fraction of micropores and a surface area of 1235 m2 g−1, the CTF obtained at 350 °C shows an excellent CO2 sorption capacity at 273 K (4.28 mmol g−1), which is one of the highest observed among all porous organic polymers. Additionally, the materials have CO2/N2 selectivities of up to 37. The hydrogen adsorption capacity of 4.36 wt% at 77 K and 20 bar is comparable to that of other POPs, yet the highest among all CTFs studied to date.
Introduction
Lately, the development of covalent triazine frameworks (CTFs), a subclass of porous organic polymers (POPs), garnered significant attention owing to their high surface areas combined with a highly robust nature, that is, high thermal (>300 °C) and chemical stabilities towards concentrated acids and bases.1–5 Therefore, CTFs are considered ideal scaffolds for applications such as heterogeneous catalysis,6–9 gas storage and separation.9–16 Recently, low temperature syntheses of CTFs have set the stage for further applications such as for chemo- and size-selective membranes17 and in optoelectronics.10The development of potent gas storage systems has been fueled by the need for highly selective gas capture materials apt to selectively filter out or enrich relevant gases such as methane or carbon dioxide. The anthropogenic emission of CO2 is known to be a major source of global warming. The global emission of CO2 by power plants and public transportation has risen remarkably in the last decades.18 According to the International Energy Agency (IEA), appropriate capture and storage of postcombusted CO2 (CCS) has the potential of decreasing the emissions up to 20%.19 Typically, flue gas of a coal-fired power plant consists of approximately 15% CO2, 5% H2O, 5% O2 and 75% N2 and is emitted at 40–80 °C and 1 bar.20,21 Therefore, materials suitable for CCS require a high preference for adsorption of CO2 under these conditions. Amine scrubbing and cryogenic cooling are the only established technologies for CO2 capture up to date, which, however, have the disadvantage of increasing the energy requirements of a power plant by 25–40%.20,22 Especially amine scrubbing needs large volumes of solvents, high temperatures for regeneration and costly disposal when expired.22,23 The disposal and the formation of toxic byproducts during the regeneration step additionally raise environmental concerns about this technology. Lately, the use of solid physical adsorbents for CO2 capture, such as metal–organic frameworks (MOFs) and POPs, was part of several reviews.20,21,24–27 Main advantages of solid adsorbents are their long lifetimes and recovery at moderate temperatures. Especially POPs are promising because of their high chemical and thermal stabilities as well as synthetic versatility, giving rise to a large variety of functional and structural designs.
In this work we present the synthesis and characterization of a new fluorene-based covalent triazine framework (fl-CTF) at different temperatures. We tested the material properties regarding CO2 adsorption at 273 K, 298 K and 313 K, along with the CO2 over N2 selectivity, showing high capture capacities and good selectivities. Additionally, we measured high-pressure adsorption of H2 at 77 K and 298 K, yielding uptakes at the forefront of polymeric hydrogen storage systems.
Experimental section
Materials and methods
All starting materials and solvents were obtained from commercial sources and were used without further purification. 2,7-Dibromo-9H-fluorene (99%) was purchased from Acros Organics. Zinc cyanide (98%) and trifluoromethanesulphonic acid (99%) were obtained from ABCR. Anhydrous zinc chloride (99.995%), 2-bromo-9H-fluorene (95%) and tetrakis(triphenyl-phosphine)palladium(0) (99%) were purchased from Sigma Aldrich. Dimethylformamide (99%) and 1,5-bis(diphenyl-phosphino)pentane (97%) were obtained from Alfa Aesar.Argon, carbon dioxide and nitrogen adsorption/desorption measurements were performed at 87, 273, 298 and 313 K with an Autosorb iQ surface analyzer (Quantachrome Instruments, USA). Samples were outgassed in a vacuum (10−7 mbar) at 200–300 °C for 6 h to remove all guests. Pore-size distributions were determined using the calculation model for Ar at 87 K on carbon (slit pore, QSDFT equilibrium model) or for CO2 at 273 K on carbon (NLDFT model) of the ASiQwin software (v2.0) from Quantachrome. For BET calculations pressure ranges of the Ar isotherms were chosen with the help of the BET Assistant in the ASiQwin software. In accordance with the ISO recommendations multipoint BET tags equal or below the maximum in V(1 − P/P0) were chosen. The isosteric heats of adsorption were calculated from the CO2 adsorption isotherms using the Quantachrome software ASiQwin (v2.0) based on the Clausius–Clapeyron equation (see ESI,† section 6c).
High-pressure hydrogen adsorption/desorption measurements were performed on an automated Sievert's type apparatus (PCTPro-2000) with a so-called micro-doser (MD) from HyEnergy. The original setup was upgraded by a heating and cooling device to regulate the sample temperature. The adsorption and desorption isotherms (0–20 bar) were measured at various temperatures (77 to 298 K) in a sample cell volume of ≈1.3 mL using ultra high purity hydrogen gas (99.999%). Samples were outgassed in a vacuum (4.5 × 10−6 mbar) at 200 °C for 6 h to remove all guests. The isosteric heat of adsorption is calculated from the absolute adsorbed hydrogen according to a variant of the Clausius–Clapeyron equation (see ESI†).
Cryogenic hydrogen adsorption/desorption measurements at 19.5 K were measured with laboratory-designed volumetric adsorption equipment with a temperature controlled cryostat. The experimental set-up has been described in detail elsewhere.28,29 Samples were activated under vacuum (10−4 mbar) at 150 °C for 12 h, prior to each measurement. For the laboratory-designed cryostat, the temperature control was calibrated by measuring the liquefaction pressure for hydrogen and nitrogen in the empty sample chamber at various temperatures.
Infrared (IR) spectroscopy measurements were carried out on a Perkin Elmer Spectrum BX II (Perkin Elmer, USA) with an attenuated total reflectance unit.
Powder X-ray diffraction (XRD) was measured on a BRUKER D8 Advance (Bruker AXS, Madison, Wisconsin, USA) in the Bragg–Brentano geometry or on a HUBER G670 (HUBER Diffraktionstechnik, Rimsting, Germany) in the Guinier geometry equipped with an imaging plate detector.
Elemental analysis (EA) was carried out with an Elementar vario EL (Elementar Analysensysteme, Germany).
Magic angle spinning (MAS) solid-state nuclear magnetic resonance (ssNMR) spectra were recorded at ambient temperature on a BRUKER DSX500 Avance NMR spectrometer or a BRUKER AvanceIII HD 400 NMR spectrometer (Bruker Biospin, Rheinstetten, Germany) with an external magnetic field of 11.75 T and 9.4 T, respectively. The operating frequencies are 500.1 MHz, 125.7 MHz and 400.1 MHz, 100.6 MHz for 1H and 13C, respectively and the spectra were referenced relative to TMS (1H, 13C). The samples were contained either in 2.4, 3.2 mm or 4 mm ZrO2 rotors. The 1D 1H13C cross-polarization (CP) MAS spectra were acquired with a ramped-amplitude (RAMP) CP sequence and contact times between 2 and 10 ms. CPPI (cross-polarization combined with polarization inversion) experiments were carried out to get information about the number of hydrogen atoms directly attached to the carbon. An initial contact time of 2 ms was used and spectra with inversion times up to 400 μs were measured. The measurements were carried out using spinning frequencies of 10 kHz and 15 kHz for CP, and 5.1 kHz for CPPI measurements, respectively. During acquisition broadband proton decoupling using SPINAL64 or TPPM was carried out.
Solution-state NMR spectroscopy was performed on a JEOL DELTA NMR (JEOL, Japan) by single pulse experiments. The spectra were referenced against CDCl3 (δ(1H) 7.26 ppm, δ(13C{1H}) 77.16 ppm).
High resolution mass spectroscopy (HRMS) was carried out on a JEOL MS700 (JEOL, Japan), by direct electron ionization (DEI).
Differential thermal analysis and thermogravimetry (DTA/TG) were measured on a SETARAM TG-DTA92-2400 combined DTA-TG-thermobalance (SETARAM Instrumentation, France) in aluminum oxide crucibles. Heating was performed from room temperature to 800 °C with a heating rate of 5 °C min−1 under a helium atmosphere.
Microwave reactions were carried out in a Biotage Initiator (Biotage AB, Sweden) in 10–20 mL microwave vials from Biotage.
9H-Fluorene-2-carbonitrile30
A microwave vial was charged with DMF (20.0 mL) and a stream of argon was bubbled through the solvent for 30 min. 2-Bromo-9H-fluorene (1.03 g, 4.00 mmol), Zn(CN)2 (939 mg, 8.00 mmol), Pd(PPh3)4 (185 mg, 0.16 mmol), and bis(diphenyl-phosphino)pentane (72.7 mg, 0.16 mmol) were added and the stream of argon was continued for 2 min. The vial was sealed and the yellow mixture was heated in the microwave for 5 min at 150 °C. The obtained orange suspension was poured into sat. NaHCO3-sol. (200 mL) and extracted with CHCl3 (3 × 200 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. The crude product was purified by column chromatography (hexane–ethylacetate 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) to yield 9H-fluorene-2-carbonitrile as a light tanned solid (0.59 g, 3.09 mmol, 77%). δH (300 MHz; CDCl3) 7.86–7.79 (3H, m, Ph), 7.68–7.64 (1H, m, Ph), 7.67–7.57 (1H, m, Ph), 7.47–7.37 (2H, m, Ph), 3.93 (2H, s, CH2); δC (68 MHz; CDCl3) 146.3, 144.0, 143.7, 140.0, 131.3, 128.72, 128.68, 127.4, 125.4, 121.1, 120.5, 119.8, 109.8, 36.9.
:5) to yield 9H-fluorene-2-carbonitrile as a light tanned solid (0.59 g, 3.09 mmol, 77%). δH (300 MHz; CDCl3) 7.86–7.79 (3H, m, Ph), 7.68–7.64 (1H, m, Ph), 7.67–7.57 (1H, m, Ph), 7.47–7.37 (2H, m, Ph), 3.93 (2H, s, CH2); δC (68 MHz; CDCl3) 146.3, 144.0, 143.7, 140.0, 131.3, 128.72, 128.68, 127.4, 125.4, 121.1, 120.5, 119.8, 109.8, 36.9.
9H-Fluorene-2,7-dicarbonitrile31
A microwave vial was charged with DMF (20.0 mL) and a stream of argon was bubbled through the solvent for 30 min. 2,7-Dibromo-9H-fluorene (1.31 g, 4.00 mmol), Zn(CN)2 (939 mg, 8.00 mmol), Pd(PPh3)4 (277 g, 0.24 mmol), and bis(diphenyl-phosphino)pentane (109 mg, 0.24 mmol) were added and the stream of argon was continued for 2 min. The vial was sealed and the yellow mixture was heated in the microwave for 5 min at 150 °C. The obtained orange suspension was poured into H2O (300 mL) and extracted with CHCl3 (4 × 300 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. The crude product was recrystallized from CH2Cl2 to give 9H-fluorene-2,7-dicarbonitrile as light green crystals (0.66 g, 3.05 mmol, 76%). δH (270 MHz; CDCl3) 7.93 (2H, d, 3JHH = 8.1 Hz, Ph), 7.89 (2H, d, 4JHH = 0.7 Hz, Ph), 7.74 (2H, dd, 3JHH = 8.0 Hz, 4JHH = 0.7 Hz, Ph), 4.04 (2H, s, CH2); δC (68 MHz; CDCl3) 144.4, 144.3, 131.7, 129.1, 121.7, 119.1, 111.8, 36.9.Tris(9H-fluoren-2-yl)-1,3,5-triazine
In an outgassed 25 mL Schlenk tube trifluoromethanesulphonic acid (0.80 mL, 9.00 mmol) was cooled to −18 °C and under stirring 9H-fluorene-2-carbonitrile (574 mg, 3 mmol) was added in portions. The mixture was left in the icebath for 40 h giving a dark green solid. The solid was slowly added to ice, turning into an orange suspension. After neutralizing with ammonia (25% in H2O) the suspension turned light purple and the precipitate was filtered off. Washing with water and little amounts of acetone yielded tris(9H-fluoren-2-yl)-1,3,5-triazine as a light tanned solid (477 mg, 83%). MS (DEI+) m/z (relative intensity) 574 (MH+, 18%), 573 (M+, 40), 192 (C14H10N+, 100), 191 (C14H9N+, 92), 190 (C14H8N+, 79), 165 (C13H9+, 24), 164 (C13H8+, 21). HRMS (DEI) m/z: [M]+ calcd for C42H27N3 573.2205; found 573.2219. Anal. calc. for C42H27N3: C, 87.93; H, 4.74; N, 7.32. Found: C, 86.69; H, 4.86; N, 7.18%. Mp 350–356 °C (decomp.).Synthesis of covalent triazine frameworks
In a typical synthesis a Duran ampoule (1.5 × 12 cm) was charged with 9H-fluorene-2,7-dicarbonitrile (500 mg, 2.31 mmol) and ZnCl2 (1.57 g, 11.6 mmol) within a glove box. The ampoule was flame sealed under vacuum and was subjected in a tube oven to temperatures between 300 and 600 °C (for temperature programs, see Table S1, ESI†). After cooling to ambient temperature, the ampoule was opened and its content ground thoroughly. The crude product was stirred in H2O (150 mL) for 1 h, filtered, and washed with 1 M HCl (75 mL) and ethanol (75 mL). The mixture was then stirred at 90 °C in 1 M HCl (80 mL) overnight, filtered, and subsequently washed with 1 M HCl (3 × 75 mL), H2O (12 × 75 mL), THF (2 × 75 mL), and acetone (1 × 75 mL). Finally, the black powder was dried overnight at 60 °C.Results and discussion
Synthesis and characterization
The standard procedure for preparing CTFs is by ionothermal synthesis using ZnCl2 both as a “solvent” and as a Lewis acidic catalyst at temperatures above 300 °C.1–4 CTFs synthesized at low temperatures (300–400 °C) may show little crystallinity, yet at the same time may have well-defined local structures.1,5,9,32 Synthesis at higher temperatures typically leads to dramatic increases in the surface areas accompanied by the loss of structural elements, especially the triazine moieties.4,5 In this work we followed this procedure using 9H-fluorene-2,7-dicarbonitrile as a precursor. The precursor was synthesized by a Negishi crosscoupling reaction from 2,7-dibromo-9H-fluorene in the microwave with just 5 min reaction time (Scheme 1). This straightforward method to produce aromatic dinitriles is analogous to the synthesis described by us previously.5 Work-up was done by recrystallisation and gave good yields of 76%. The syntheses of the CTFs were performed at temperatures between 300 and 600 °C, yielding fluorene-CTFs at 300 °C (fl-CTF300), 350 °C (fl-CTF350), 400 °C (fl-CTF400), 500 °C (fl-CTF500) and 600 °C (fl-CTF600). The reaction times were 48 h for all samples, except for fl-CTF300 and fl-CTF350, where the reaction times were raised to 96 h after very low yields had been obtained due to unreacted monomers or oligomers. Reaction times of 168 h did not give higher yields or changes in the material properties. | ||
| Scheme 1 Synthesis route for fl-CTFs. | ||
CTF-0, CTF-1 and CTF-2 are the only three examples of CTFs which show moderate crystallinity.1,12,32 Therefore, it was little surprising that our materials were found to be largely amorphous in the XRD measurements, except for broad peaks at 14.8, 22.7 and 33.6° 2θ. The peak at 22.7° 2θ is attributed to a 00l reflection indicating a “graphitic” layer stacking with an interlayer distance of ∼3.9 Å, which is rather large and consistent with only weak interlayer coupling (Fig. S1, ESI†).
To probe whether trimerization in these samples was completed with the fluorene units still being intact, we used IR spectroscopy and ssNMR measurements and compared the results with the model compound tris(9H-fluoren-2-yl)-1,3,5-triazine. Fig. 1 depicts the IR and Fig. 2 and S2† the 13C ssNMR spectra of the as-synthesized materials. fl-CTF300 shows well resolved signals in the IR spectrum, whereas the signals weaken at higher synthesis temperatures and are flattened out to a large extent for fl-CTF600, indicating graphitization of the networks. This is confirmed by the 13C ssNMR measurements, which show a broadening and weakening of the signals with rising temperatures. Remarkably, the NMR spectrum of fl-CTF600 could not be measured owing to the high degree of graphitization. The IR and ssNMR spectra of fl-CTF300 and fl-CTF350 show the presence of a nitrile band (2223 cm−1, 109.5 ppm), indicating that there are still unreacted nitrile groups in the polymer. Those signals are not visible at higher synthesis temperatures. The strong IR band in the fl-CTF300 spectrum at 1511 cm−1 can be assigned to the C–N stretching mode of the triazine ring,32,33 whereas the band at 1352 cm−1 is due to in-plane triazine ring stretching vibrations.3,34 This and the corresponding NMR shift at 168.6 ppm evidence successful formation of the triazine moieties (Fig. 2, bottom left). The signals between 144 and 118 ppm and the peak at 36.9 ppm belong to the fluorene unit, which accordingly stays intact at 300 °C. The signal at 50 ppm is assigned to a sp3 CH group, which was corroborated by a CPPI measurement (Fig. S4†). This resonance suggests the formation of 9,9′-bifluorenyl units via crosslinking. A broadening of the bands between 1500 and 1100 cm−1 and the decrease of the triazine bands in the IR spectrum of fl-CTF350 indicates commencing degradation of the system. Again, ssNMR measurements confirm these results. The bands at 1607 and 814 cm−1 appear both in the precursor and in fl-CTF300 and can hence likely be assigned to the fluorene species. These bands are still retained in the spectra of fl-CTF350 and fl-CTF400, indicating intact fluorene units in those samples. The spectra of fl-CTF500 and fl-CTF600 do not show the fluorene signature anymore, which confirms the ongoing degradation of the system at elevated temperatures.
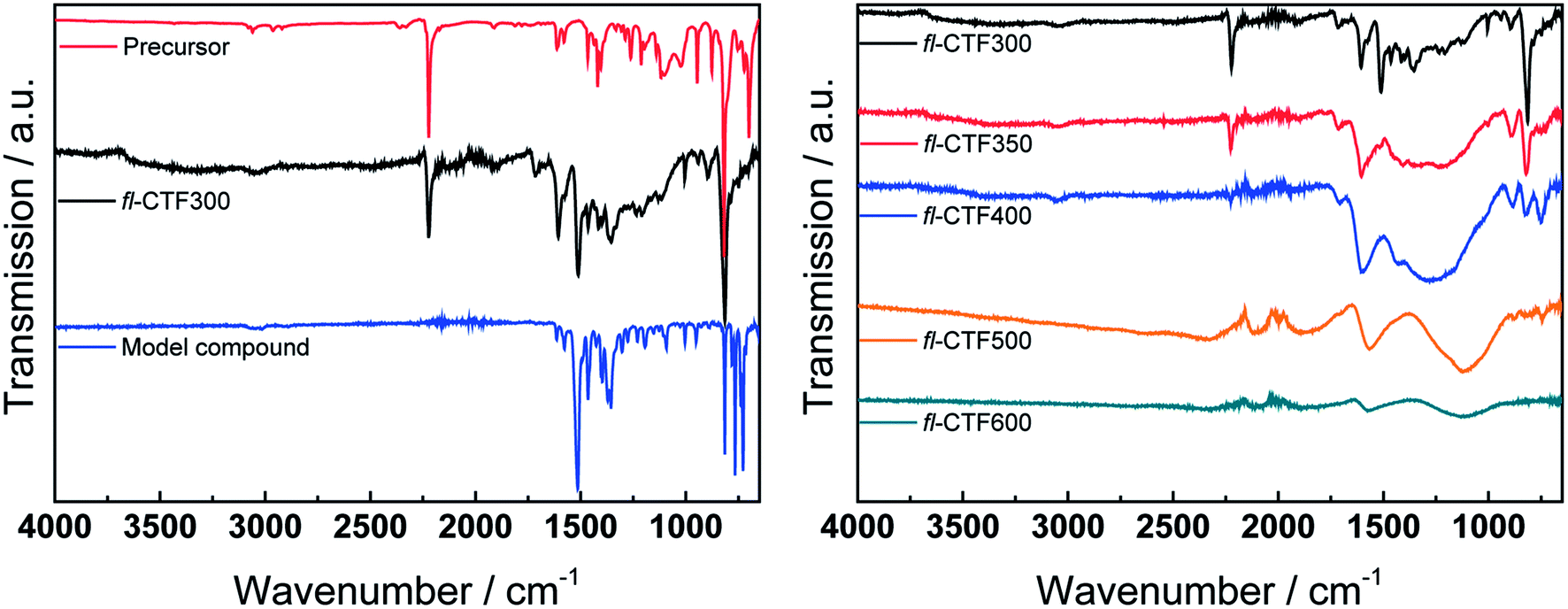 | ||
| Fig. 1 (Left) IR spectra of 9H-fluorene-2,7-dicarbonitrile, fl-CTF300 and tris(9H-fluoren-2-yl)-1,3,5-triazine. (Right) IR spectra of the samples fl-CTF300, fl-CTF350, fl-CTF400, fl-CTF500 and fl-CTF600. | ||
 | ||
| Fig. 2 13C MAS ssNMR spectra of tris(9H-fluoren-2-yl)-1,3,5-triazine (top left), fl-CTF300 (bottom left), fl-CTF350 (top right) and fl-CTF400 (bottom left). Asterisks mark rotational side bands. | ||
In summary, both IR spectroscopy and ssNMR measurements indicate degradation of the networks at temperatures higher than 300 °C, which is in line with observations made for all synthesized CTFs so far. Temperatures in excess of 400 °C are not tolerated by the triazine rings and likely induce retro-trimerization reactions with subsequent rearrangements, giving rise to irreversible formation of C–C bonds accompanied by a loss of N2.
To gain more information about the degree and course of degradation, we utilized EA measurements. EA data shown in Table S2† reveal a trend which is very similar to many synthesized CTFs: by raising the synthesis temperature, the nitrogen and hydrogen content decreases and the carbon content increases. The loss of nitrogen is very significant with a nitrogen loss of more than 40% for fl-CTF600 compared to fl-CTF300. Notably, the nitrogen content of fl-CTF300 is already lower than the calculated value, thus confirming the IR results, which reveal smaller amounts of triazine compared to the model compound.
To confirm the thermal stability of the materials we carried out DTA/TG measurements on the samples fl-CTF300 and fl-CTF600. The results (Fig. S25 and S26†) show degradation temperatures close to the synthesis temperatures and a rather low weight loss, especially for the fl-CTF600 sample.
Porosity measurements
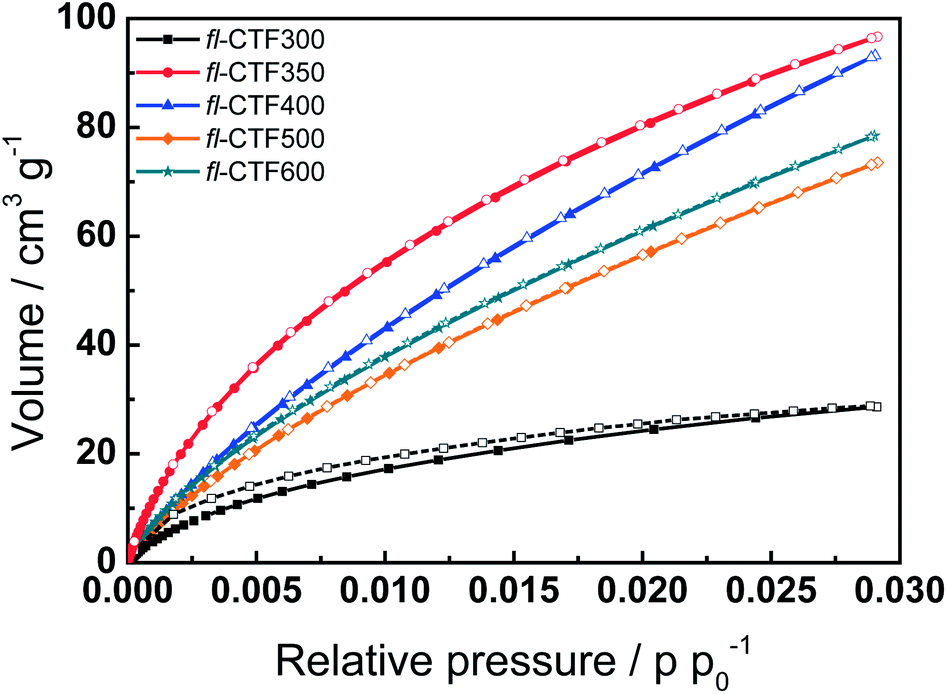
Although an increase in the synthesis temperature leads to materials with less local order and nitrogen content, it typically entails materials with higher surface areas (SAs).2,3,5 The porosities of the fl-CTFs were determined by argon and carbon dioxide physisorption measurements. Fig. 3 (left) shows the argon isotherms of the samples fl-CTF350, fl-CTF400, fl-CTF500 and fl-CTF600. It should be mentioned that fl-CTF300 (Fig. S6†) adsorbed only small amounts of argon and therefore showed overall poor porosity. The isotherm of fl-CTF350 is typical for microporous materials in that it shows rapid argon uptake at low relative pressures (p p0−1 < 0.05). Nevertheless, it cannot be described as a type I isotherm because of continuous adsorption of argon at higher pressures, indicating additional mesoporosity with a broad mesopore size distribution and a hysteresis almost spanning the entire range of p p0−1 = 0–0.9. The hysteresis resembles type H4, typical for materials with slit-shaped pores. The low pressure character of the hysteresis indicates pores within the size range of the adsorbate, causing delayed desorption.35 The isotherm of fl-CTF400 shows rapid argon adsorption at low relative pressures as well. The continuous adsorption at higher relative pressures is higher than in fl-CTF350, which should be ascribed to an increased fraction of mesopores. The isotherms of fl-CTF500 and fl-CTF600 show similar shapes and can be described as a combination of isotherms of type I and IV. The micropore filling is followed by mesopore filling showing a type H2 hysteresis around p p0−1 = 0.4. H2 hystereses describe systems with rather ill-defined pore sizes and shapes and are often observed for amorphous materials. | ||
| Fig. 3 (Left) Argon adsorption (filled symbols) and desorption (open symbols) isotherms at 87 K of fl-CTF350, fl-CTF400, fl-CTF500 and fl-CTF600. (Right) Pore size distributions of fl-CTF350, fl-CTF400, fl-CTF500 and fl-CTF600 from DFT calculations. The curves are shifted vertically in steps of 1 cm3 nm−1 g−1. | ||
The SAs of the materials were calculated from the isotherms based on the BET model and are listed in Table 1.36 The highest SA of 2862 m2 g−1 is found for fl-CTF400, which is the third highest for all CTFs after CTF-1 (400/600 °C, 3270 m2 g−1)2 and bipy-CTF (700 °C, 3219 m2 g−1).5 For fl-CTF500 and fl-CTF600 the values decrease, which is unexpected, since for all published CTFs a rise in synthesis temperature showed an increase in the SA, accompanied by a continuous decomposition of the materials (“foaming”). This phenomenon can be explained by the pore size distribution of the materials which was calculated by QSDFT methods (Fig. 3, right). For fl-CTF350 mainly micropores are observed with two main peaks at 1.05 and 0.5 nm. The latter indicates pores smaller than 0.5 nm (ultramicropores), which are not observable by argon measurements. For fl-CTF400 the peak at 1.05 nm increases and additionally a wide distribution of pores from 1.2 to 3 nm is observed, with mainly pores in the micropore region. fl-CTF500 and fl-CTF600 show comparable distributions, with an additional peak around 2.5 nm, which can be assigned to small mesopores. Overall, an increasingly mesoporous character with rising synthesis temperature is typical for CTFs, yet normally the increasing amount of mesopores is not accompanied by a significant decrease of the micropores.2,5
| Sample | BET SA [m2 g−1] | Pore volume [cm3 g−1] | CO2 uptakea [mmol g−1] | N2 uptakeb [mmol g−1] | Q st [kJ mol−1] | Selectivityd | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| V Ar,mic,DFT | V Ar,tot,DFT | V Ar,mic,DFT/VAr,tot,DFT | 273 K | 298 K | 313 K | Henry | IAST | ||||
| a At 1 bar. b At 1 bar and 298 K. c At zero coverage. d At 298 K. e Pore volume for pores smaller than 2 nm calculated from the Ar QSDFT model. f Total pore volume from the Ar QSDFT model. | |||||||||||
| fl-CTF300 | 15 | — | — | — | 1.27 | 0.71 | 0.42 | 0.04 | 43.1 | 35 | 37 |
| fl-CTF350 | 1235 | 0.57 | 0.67 | 0.85 | 4.28 | 2.29 | 1.59 | 0.18 | 32.7 | 27 | 23 |
| fl-CTF400 | 2862 | 1.13 | 1.45 | 0.78 | 4.13 | 1.97 | 1.31 | 0.17 | 30.7 | 15 | 16 |
| fl-CTF500 | 2322 | 0.77 | 1.29 | 0.60 | 3.26 | 1.65 | 1.11 | 0.21 | 31.7 | 13 | 12 |
| fl-CTF600 | 2113 | 0.70 | 1.16 | 0.60 | 3.48 | 1.80 | 1.22 | 0.22 | 32.4 | 14 | 12 |
The amount of micropores compared to mesopores can be calculated by the ratio of the microporous volume Vmic to the total pore volume Vtot. These pore volumes can be calculated from the amount of vapor adsorbed at chosen relative pressures assuming the pores are filled with a liquid adsorbate, and from DFT calculations. The results for the fl-CTFs are listed in Tables 1 and S4† and confirm the previous observations. fl-CTF400 features the largest absolute amount of micropores followed by fl-CTF500 and fl-CTF600. Remarkably, fl-CTF350 has the highest fraction of micropores with respect to the total pore volume (85%), which is clearly higher than for fl-CTF500 and fl-CTF600 (both 60%). The widening of the pores can favorably be explained by the degradation of the networks at elevated synthesis temperatures, leading to a “swelling” (“foaming”) of the materials.
The pore size distributions of the fl-CTFs indicate ultramicropores, which cannot be detected by argon measurements, but by carbon dioxide physisorption measurements, which allow us to probe pore sizes down to ≈0.35 nm. The isotherms shown in Fig. 4 have comparable shapes, which is consistent with the literature. In contrast to its non-porous character established by Ar physisorption, fl-CTF300 adsorbs moderate amounts of CO2, which can be rationalized by the presence of pores that are accessible to CO2 but not to Ar. A comparable phenomenon is observed for fl-CTF350, which adsorbs more CO2 than all other fl-CTFs, although having around half as much accessible SA as determined by argon physisorption. The accessible CO2 SAs were calculated with DFT simulations and are shown in Table S6.† The values are much lower than those for argon measurements, since the calculations only take pores smaller than 1.45 nm into account. Nevertheless, fl-CTF300 shows a SA of 297 m2 g−1 which is nearly half as much as that of fl-CTF500 and fl-CTF600. The highest SA is observed for fl-CTF400, followed by fl-CTF350. All samples show a broad pore size distribution ranging from 0.35–1.00 nm (Fig. S16†). A hydrogen physisorption measurement at 20 K validated the SA of fl-CTF400 giving a BET SA of 2829 m2 g−1 (Fig. S12 and S13†).
 | ||
| Fig. 4 Carbon dioxide adsorption (filled symbols) and desorption (open symbols) isotherms at 273 K of fl-CTF300, fl-CTF350, fl-CTF400, fl-CTF500 and fl-CTF600. | ||
Gas storage and selectivity studies
The high capacities of the fl-CTFs for CO2 sorption at 273 K are promising for the usage as CCS materials. Therefore, temperature-dependent sorption studies at 298 and 313 K were carried out and the heats of adsorption were calculated (Fig. 5).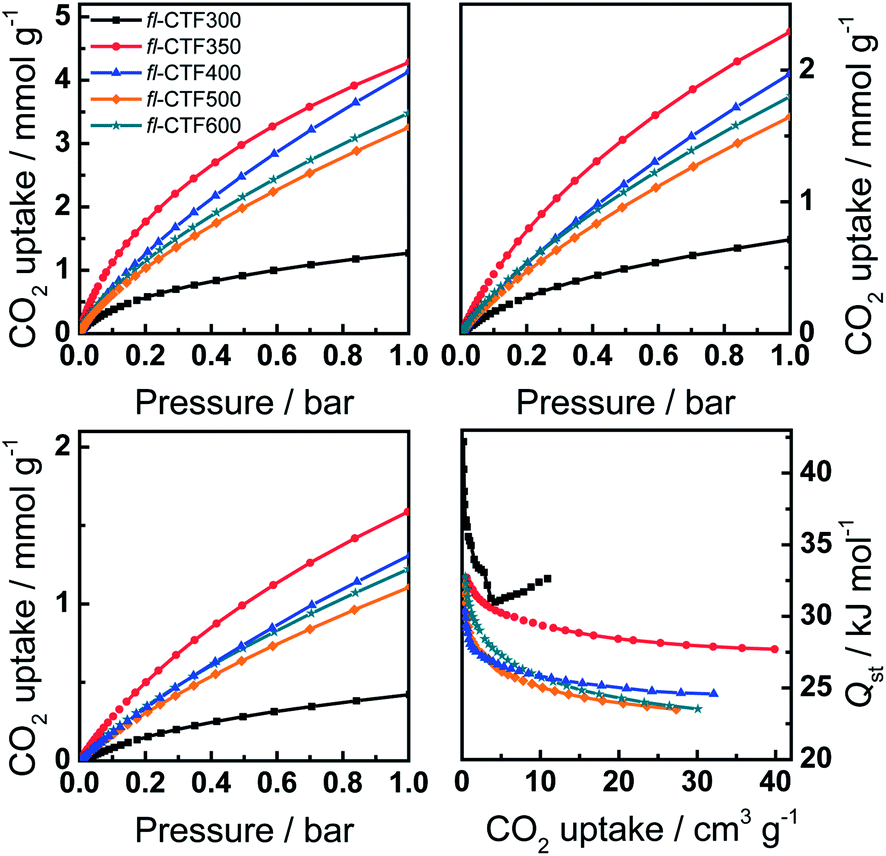 | ||
| Fig. 5 Carbon dioxide adsorption isotherms of fl-CTF300, fl-CTF350, fl-CTF400, fl-CTF500 and fl-CTF600 at 273 K (top left), 298 K (top right), 313 K (bottom left) and corresponding heats of adsorption (bottom right). | ||
The uptakes at 1 bar and the heats of adsorption at zero coverage are summarized in Table 1. With 4.28 mmol g−1 at 273 K fl-CTF350 shows the highest uptake of all materials. This value is lower than that of recently reported FCTF-1 (4.67–5.53 mmol g−1),16 but higher than the reported values for all other CTFs such as CTF-0 (4.22 mmol g−1),9 CTF-1 (2.47–3.82 mmol g−1),16 CTF-P2–P6 (1.88–3.39 mmol g−1),10 CTF-P1M–P6M (0.94–4.20 mmol g−1),10 MCTF300–500 (2.25–3.16 mmol g−1),12 PCTF-1–7 (1.84–3.23 mmol g−1)11,15 and TPI-1–7 (0.68–2.45 mmol g−1).34 In addition, they are higher than the uptake capacities of numerous POPs such as covalent organic frameworks (COFs; 1.21–3.86 mmol g−1),37,38 microporous polyimides (MPIs; 2.25–3.81 mmol g−1),39 and hyper-crosslinked organic polymers (HCPs; 1.91–3.92 mmol g−1)40,41 (for an expanded list see Table S7†). Higher values were reported so far for benzimidazole-linked polymers (BILPs; 2.91–5.34 mmol g−1),42–44 and porous polymer frameworks (PPFs; 2.09–6.12 mmol g−1).45 For the amounts adsorbed at 298 K similar results were found.
As mentioned above, CO2 uptakes depend on the surface area, the pore sizes and the interaction energy between a sorbent and a sorbate. The latter can be expressed by the heats of adsorption Qst which we calculated for our materials (Fig. 5, bottom right). The values at the limit of zero coverage are shown in Table 1 and are the highest for fl-CTF300 and almost the same for the other fl-CTFs, ranging in the upper field compared to other POPs (see Table S7†).
For the use of fl-CTFs as potential flue gas sorbents the selectivities of CO2 over N2 need to be examined. Therefore, the Henry equation and the ideal adsorbed solution theory (IAST) were used (ESI, chapter 8†). The calculated values are listed in Table 1. As expected, fl-CTF300 and fl-CTF350 show the highest selectivities, due to the low adsorption potential towards argon compared to CO2. The selectivities range in the upper level of POPs and, remarkably, fl-CTF300 shows a higher selectivity than FCTF-1.16
Motivated by the high values of adsorbed CO2 at 273 K and the large number of micropores, we tested fl-CTF400 as a hydrogen storage material by carrying out hydrogen physisorption measurements at 77 K and 298 K up to 20 bar. The isotherm at 77 K is displayed in Fig. 6 and shows adsorption of 4.36 wt% (45.2 mmol g−1), which is higher than that observed for 2D COFs (1.46–3.88 wt%, sat. pressure)38 and polymers of intrinsic microporosity (PIMs, 1.45–2.71 wt%, 10 bar),46–50 but lower than that of the highly porous 3D COFs (6.98–7.16 wt%, sat. pressure),38 PAFs (4.2–7.0 wt%)51,52 and PPNs (8.34 wt%, 55 bar),53 which however were measured at higher pressures. To date, only three CTFs were examined for their H2 capacities and the measurements were done only up to 1 bar. CTF-1 adsorbs 1.55 wt%,1 PCTF-1 1.86 wt%11 and PCTF-2 0.9 wt%,11 which is substantially lower than the uptake capacity of our material (1.95 wt%).
 | ||
| Fig. 6 Excess hydrogen adsorption (filled symbols) and desorption (open symbols) isotherm of fl-CTF400 at 77 K. | ||
For hydrogen storage applications, knowledge of the heat of adsorption, along with the storage capacity, is of importance to better understand the microscopic host–guest interactions. Fig. S10† shows the temperature variations of the absolute hydrogen adsorption curves (77–117 K), which provides the strength of the binding potential for hydrogen in fl-CTF400. In Fig. S11,† the isosteric heat of adsorption is shown as a function of the surface coverage normalized to saturation coverage (20 bar, 77 K). Analysis of the hydrogen adsorption enthalpy gives a maximum value of 6.25 kJ mol−1 at near zero surface coverage, decreasing to 3.65 kJ mol−1 with increasing H2 loading. The overall average heat of adsorption equals 4.9 kJ mol−1. It is worth noting that the average heat of adsorption of 4.9 kJ mol−1 is relatively high after considering the rather large pore diameter (1.05–3 nm) and on comparison with most MOFs of similar pore sizes.54 This relatively high average heat of adsorption can be either due to stronger adsorption sites (possibly N sites of fl-CTF400) or due to the ultramicropores inside fl-CTF400 as detected by CO2 NLDFT simulations (0.35–1.45 nm). Table S5† summarizes the textural characteristics and hydrogen storage capacity of fl-CTF400.
Conclusions
The presented fl-CTFs were analyzed with respect to their local structure, porosity, and capacity for CO2 capture. An intact fluorene network connected by triazines could be established for fl-CTF300. The materials showed high SAs up to 2862 m2 g−1 and high capacities for the adsorption of CO2, which can be rationalized by their high fraction of ultramicropores. We find that fl-CTF350 with the highest fraction of micropores and moderate surface area shows the best CO2 uptake (4.28 mmol g−1 at 273 K) and thus ranks in the upper level among all POPs. Additionally, hydrogen adsorption of fl-CTF400 shows comparable values to other POPs. Finally, the gas selectivities of CO2 over N2 of the fl-CTFs were tested and reveal high values for fl-CTF300 and fl-CTF350, thus rendering these materials promising candidates for gas capture and storage.Acknowledgements
The authors acknowledge financial support by the Deutsche Forschungsgemeinschaft, DFG (SPP 1362, SE 1417/5-1 and LO 1801/2-1), the Nanosystems Initiative Munich (NIM), the Center for Nanoscience (CeNS) and the Fonds der Chemischen Industrie (FCI). H. Oh was supported for this research through a stipend from the International Max Planck Research School for Advanced Materials (IMPRS-AM). We thank Marius Reymann and Niklas Cordes for synthetic assistance, Stephan Werner for XRD measurements, Christian Minke for ssNMR measurements and Katrin Rudolf and Christine Pösl for DTA/TG measurements. We acknowledge Prof. Thomas Bein and Prof. Wolfgang Schnick for access to the respective measurement facilities.Notes and references
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- P. Kuhn, A. l. Forget, D. Su, A. Thomas and M. Antonietti, J. Am. Chem. Soc., 2008, 130, 13333–13337 CrossRef CAS PubMed.
- P. Kuhn, A. Thomas and M. Antonietti, Macromolecules, 2009, 42, 319–326 CrossRef CAS.
- P. Kuhn, A. Forget, J. Hartmann, A. Thomas and M. Antonietti, Adv. Mater., 2009, 21, 897–901 CrossRef CAS.
- S. Hug, M. E. Tauchert, S. Li, U. E. Pachmayr and B. V. Lotsch, J. Mater. Chem., 2012, 22, 13956–13964 RSC.
- C. E. Chan-Thaw, A. Villa, P. Katekomol, D. Su, A. Thomas and L. Prati, Nano Lett., 2010, 10, 537–541 CrossRef CAS PubMed.
- R. Palkovits, M. Antonietti, P. Kuhn, A. Thomas and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6909–6912 CrossRef CAS PubMed.
- C. E. Chan-Thaw, A. Villa, L. Prati and A. Thomas, Chem. – Eur. J., 2011, 17, 1052–1057 CrossRef CAS PubMed.
- P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357–2361 CrossRef CAS PubMed.
- A. Bhunia, V. Vasylyeva and C. Janiak, Chem. Commun., 2013, 49, 3961–3963 RSC.
- X. Liu, H. Li, Y. Zhang, B. Xu, S. A, H. Xia and Y. Mu, Polym. Chem., 2013, 4, 2445–2448 RSC.
- W. Wang, H. Ren, F. Sun, K. Cai, H. Ma, J. Du, H. Zhao and G. Zhu, Dalton Trans., 2012, 41, 3933–3936 RSC.
- H. Ren, T. Ben, E. Wang, X. Jing, M. Xue, B. Liu, Y. Cui, S. Qiu and G. Zhu, Chem. Commun., 2010, 46, 291–293 RSC.
- A. Bhunia, I. Boldog, A. Moller and C. Janiak, J. Mater. Chem. A, 2013, 1, 14990–14999 CAS.
- Y. Zhao, K. X. Yao, B. Teng, T. Zhang and Y. Han, Energy Environ. Sci., 2013, 6, 3684–3692 CAS.
- X. Zhu, C. Tian, S. M. Mahurin, S.-H. Chai, C. Wang, S. Brown, G. M. Veith, H. Luo, H. Liu and S. Dai, J. Am. Chem. Soc., 2012, 134, 10478–10484 CrossRef CAS PubMed.
- M. R. Raupach, G. Marland, P. Ciais, C. Le Quéré, J. G. Canadell, G. Klepper and C. B. Field, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10288–10293 CrossRef CAS PubMed.
- Energy technology perspectives 2008, International Energy Agency, Paris, 2008 Search PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef CAS PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Polym. Int., 2013, 62, 345–352 CrossRef CAS.
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- Z. Xiang and D. Cao, J. Mater. Chem. A, 2013, 1, 2691–2718 CAS.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724–781 CrossRef PubMed.
- B. Streppel and M. Hirscher, Phys. Chem. Chem. Phys., 2011, 13, 3220–3222 RSC.
- B. Streppel, PhD thesis, Universität Stuttgart, 2011.
- M. Y. Wong and L. M. Leung, Tetrahedron, 2010, 66, 3973–3977 CrossRef CAS PubMed.
- D. Vonlanthen, A. Rudnev, A. Mishchenko, A. Käslin, J. Rotzler, M. Neuburger, T. Wandlowski and M. Mayor, Chem. – Eur. J., 2011, 17, 7236–7250 CrossRef CAS PubMed.
- M. J. Bojdys, J. Jeromenok, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 2202–2205 CrossRef CAS PubMed.
- V. G. Manecke and D. Wöhrle, Makromol. Chem., 1968, 120, 176–191 CrossRef.
- M. R. Liebl and J. Senker, Chem. Mater., 2013, 25, 970–980 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- R. Dawson, E. Stockel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239–4245 CAS.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- G. Li and Z. Wang, Macromolecules, 2013, 46, 3058–3066 CrossRef CAS.
- Y. Luo, S. Zhang, Y. Ma, W. Wang and B. Tan, Polym. Chem., 2013, 4, 1126–1131 RSC.
- C. F. Martin, E. Stockel, R. Clowes, D. J. Adams, A. I. Cooper, J. J. Pis, F. Rubiera and C. Pevida, J. Mater. Chem., 2011, 21, 5475–5483 RSC.
- M. G. Rabbani and H. M. El-Kaderi, Chem. Mater., 2011, 23, 1650–1653 CrossRef CAS.
- M. G. Rabbani and H. M. El-Kaderi, Chem. Mater., 2012, 24, 1511–1517 CrossRef CAS.
- M. G. Rabbani, A. K. Sekizkardes, O. M. El-Kadri, B. R. Kaafarani and H. M. El-Kaderi, J. Mater. Chem., 2012, 22, 25409–25417 RSC.
- Y. Zhu, H. Long and W. Zhang, Chem. Mater., 2013, 25, 1630–1635 CrossRef CAS.
- N. B. McKeown, P. M. Budd and D. Book, Macromol. Rapid Commun., 2007, 28, 995–1002 CrossRef CAS.
- N. B. McKeown, B. Gahnem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem., Int. Ed., 2006, 45, 1804–1807 CrossRef CAS PubMed.
- B. S. Gahnem, K. J. Msayib, N. B. McKeown, K. D. M. Harris, Z. Pan, P. M. Budd, A. Butler, J. Selbie, D. Book and A. Walton, Chem. Commun., 2007, 67–69 RSC.
- J.-Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670–2672 RSC.
- P. M. Budd, A. Butler, J. Selbie, K. Mahmood, N. B. McKeown, B. Gahnem, K. Msayib, D. Book and A. Walton, Phys. Chem. Chem. Phys., 2007, 9, 1802–1808 RSC.
- T. Ben, C. Pei, D. Zhang, J. Xu, F Deng, X. Jing and S. Qiu, Energy Environ. Sci., 2011, 4, 3991–3999 CAS.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 7480–7484 CrossRef CAS PubMed.
- M. Schlichtenmayer and M. Hirscher, J. Mater. Chem., 2012, 22, 10134–10143 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Temperature programs, elemental analysis, XRD patterns, 13C ssNMR of fl-CTF500, 15N ssNMR of tris(9H-fluoren-2-yl)-1,3,5-triazine, CPPI analysis, Ar isotherm of fl-CTF300, BET plots, Ar-QSDFT fittings, CO2-NLDFT distributions and fittings, CO2 isotherms at 298 and 313 K, H2 isotherms at 298 and 20 K, H2Qst plot, N2 isotherms at 298 K, Henry and IAST calculations, 1H and 13C NMR spectra of 9H-fluorene-2-carbonitrile and 9H-fluorene-2,7-dicarbonitrile, and DTA/TG measurements. See DOI: 10.1039/c3ta15417c |
| This journal is © The Royal Society of Chemistry 2014 |