 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correlating surface cation composition and thin film microstructure with the electrochemical performance of lanthanum strontium cobaltite (LSC) electrodes
G. M.
Rupp
a,
A.
Limbeck
a,
M.
Kubicek
a,
A.
Penn
ab,
M.
Stöger-Pollach
b,
G.
Friedbacher
a and
J.
Fleig
a
aVienna University of Technology, Institute of Chemical Technologies and Analytics, Getreidemarkt 9/164, 1060 Vienna, Austria. E-mail: ghislain.rupp@tuwien.ac.at
bVienna University of Technology, University Service Center for Transmission Electron Microscopy, Wiedner Hauptstr. 8-10, A-1040 Vienna, Austria
First published on 26th February 2014
Abstract
La0.6Sr0.4CoO3−δ thin films of varying thicknesses (20–170 nm) were prepared by pulsed laser deposition on yttria-stabilized zirconia (100) substrates, and their electrochemical electrode performance was correlated with the chemical surface composition and microstructure (e.g. porosity, surface area). The surface cation composition was analyzed by an atomic etching procedure with on-line inductively coupled plasma optical emission spectrometry detection. The surface sensitivity of the method was increased by dynamically switching the etching reagent during the on-line analysis and quantitative results for even the top atomic layer were obtained. A water-soluble Sr-rich surface species could be quantified on top of the LSC films and in combination with electrochemical analysis of the films by impedance spectroscopy an improved understanding of the surface exchange resistance could be obtained. Microstructural features such as the effective porosity of the films became accessible by a combination of these methods.
1 Introduction
Recently solid oxide fuel cells (SOFCs) have received strong interest because of their potential for highly efficient energy conversion, high power density as well as their fuel flexibility. Applications include combined power and heating systems (CPH) and auxiliary power units (SOFC-APUs), and current cells typically operate at temperatures between 800 and 1000 °C. Many research activities focus on lowering these operating temperatures, thus avoiding unfavorable side reactions of the materials.1,2 However, it remains a challenge to find suitable intermediate temperature cathode materials for efficient catalytic reduction of oxygen followed by fast ionic transport to the electrolyte. One promising cathode material for such intermediate temperature SOFCs (500–600 °C) is Sr-doped LaCoO3 (LSC), as it offers both mixed ionic–electronic conductivity and acceptable catalytic activity for the oxygen reduction.3,4 However, degradation of the electrochemical kinetics may occur and is most likely correlated with changes of the electrode surface. So far different techniques based on electron (auger electron spectroscopy) and ion (secondary ion mass spectrometry) bombardment as well as X-rays (X-ray photoelectron spectroscopy) revealed surface composition changes of perovskite-type materials, such as (La, Sr)MnO3,5–7 (La, Sr)(Co, Fe)O3 (ref. 8–13) and Sr(Ti, Fe)O3 (ref. 14 and 15), upon elevated temperatures or polarization.In particular, Sr enrichment was identified and directly linked to a severe decrease of the oxygen reduction reaction activity.8,9,12,13,16 However, the underlying mechanism for the Sr segregation is not fully understood and fundamental questions are still under debate:17,18 what is the dominant driving force for Sr segregation in thin films? Which steps of the oxygen reduction reaction are hindered? Can this effect be avoided? Further high quality measurements are inevitable to deconvolute the complex interrelationships of cathode materials' microstructure, bulk and surface chemistry and electrochemical properties. Recently, we introduced a novel method to gather quantitative results on the composition of the near-surface region of La0.6Sr0.4CoO3−δ thin films:12 an in situ solid phase extraction system based on chemical etching was coupled with an inductively coupled plasma-optical emission spectrometer (ICP-OES) giving the advantage of on-line cation quantification by a simple liquid standard calibration.
In this contribution, we first describe how this method can be further improved by using different solvents. Then we apply this technique to analyze cation surface segregations of La0.6Sr0.4CoO3−δ thin films of different thicknesses and correlate the results with electrochemical film properties. Conclusions on the chemical composition of the surface and bulk are drawn. Moreover, on-line in situ ICP-OES analysis proves to be a useful method for analyzing open nano-porosity and can be an efficient alternative to ellipsometric porosimetry19 or high resolution electron imaging techniques20 combined with a rather time consuming evaluation of the images for surface area calculations.21,22 In the context of this work the importance of geometrically well-defined electrodes for electrochemical investigation23 is again emphasized and consequences are pointed out when this prerequisite is not fulfilled.
2 Experimental
2.1 Thin film preparation
Targets for deposition of LSC thin films by pulsed laser deposition (PLD) were obtained from powders prepared by Pechini synthesis.24 La2O3, SrCO3 and Co powders (all Sigma Aldrich, 99.995%) were individually dissolved in nitric acid and mixed in appropriate ratios. Citric acid (TraceSELECT®, ≥99.9998%) was added for chelation. A calcination step was performed at 1000 °C, followed by isostatic pressing (∼150 MPa) of the powder to a pellet and a sintering procedure at 1200 °C for 12 hours in air, thus yielding a LSC target for PLD. The target composition was determined from a deposited thin film, which was completely dissolved in 0.12 mol l−1 hydrochloric acid and analyzed by non-dynamic ICP-OES. The target composition was determined to be La0.62±0.02Sr0.45±0.02Co0.95±0.04O3−δ.The LSC thin films were deposited onto (100) oriented, one side epipolished yttria stabilized zirconia (YSZ, 9.5 mol% Y2O3, CrysTec GmbH, Germany) single crystals with a thickness of 0.5 mm and a size of 5 × 5 mm2. Ablation of the target material was carried out by a KrF (λ = 248 nm) excimer laser (Lambda COMPexPro 201F) operated at a pulse repetition rate of 5 Hz, a pulse duration of 50 ns and a laser fluence of approximately 1.5 J cm−2 at the target. The substrate temperature was 450 °C according to a pyrometer (Heitronics KT19.99) with the emissivity of YSZ being set to 0.9 for the pyrometer-relevant wavelength range of 9.6 to 14.1 μm. The atmosphere during deposition consisted of 0.4 or 0.04 mbar oxygen, respectively (the resulting samples are abbreviated “4500.4” and “4500.04” in the following). The target–substrate distance was varied between 5.2 and 6.2 cm depending on the atmosphere – lower partial pressure led to a larger plasma plume and thus to a larger target–substrate distance. After deposition, thin films were cooled in the deposition atmosphere at a cooling rate of 12 °C min−1 to avoid crack formation. The LSC thin film thickness was varied by simply adapting the deposition time (pulse count).
2.2 Electrochemical characterization
Circular LSC microelectrodes of 200 μm diameter were fabricated by photolithography and subsequent chemical etching (0.24 mol l−1 hydrochloric acid) of the thin films.12 For the impedance measurements, an alternating voltage of 10 mV (rms) was applied between a microelectrode and an extended counter electrode made from the same LSC film. Any contribution of the counter electrode to the total electrode resistance could be neglected because of its 500-fold larger electrode area. The impedance was measured by a Novocontrol Alpha A High Performance Frequency Analyzer in the frequency range from 106 Hz to 10−1 Hz with a resolution of five points per decade. All spectra were recorded at an oven set temperature of 600 °C in air atmosphere. For a more detailed description of the measurement setup see ref. 25.2.3 Chemical characterization
The near-surface composition of the LSC films as well as their bulk composition was determined by continuous analysis of the eluate resulting from chemical etching. 5 × 5 × 0.5 mm3 YSZ single crystals with continuous LSC films (without micro-patterning) deposited on one side were installed into a custom-made polytetrafluoroethylene etching compartment26 with a volume of 289 μl. Thin film layers were then dissolved in a continuous flow of ultrapure water (produced by Barnstead™ Easypure™ II (18.2 MΩ cm−1)) or hydrochloric acid (EMSURE® hydrochloric acid 37% for analysis) solutions of different concentrations (0.006–0.024 mol l−1 HCl) and concentrations of the dissolved cations were subsequently analyzed.For all analytical measurements, a Thermo Scientific iCAP 6500 ICP spectrometer was employed, equipped with a peristaltic pump for sample introduction, a quartz torch, an echelle spectrometer, and a charge injection device detector. The RF-power was set to 1200 W, pump rotating speed to 15 rpm. A 0.8 l Ar min−1 auxiliary and a 12 l Ar min−1 cooling gas flow were applied. The liquid flow was set to 0.6 ml min−1 and the nebulizer gas flow to 0.7 l Ar min−1. A compact sample introduction system (Apex E – elemental scientific) was installed upstream the ICP-OES, containing a heated cyclonic spray chamber and a Peltier cooled desolvation system, thus enhancing the efficiency of the sample introduction system.
The following background corrected emission lines were chosen for evaluation: Sr (346.446 nm), La (379.478 nm), Co (228.616 nm; 238.892 nm). A maximum integration time of 1 second for the transient signal was chosen.
The schematic procedure for the standard calibration and the actual thin film analysis is shown in Fig. 1. A six-port valve allowed proper switching between the load and measurement position in both cases. A constant flow of two internal standards (0.8 ml min−1 0.12 mol l−1 HCl, spiked with 1 ppm Cu (Cu-Standard Titrisol®), and 0.6 ml min−1 0.5 ppm Mn (Mn-Standard, Titrisol®) spiked 0.24 mol l−1 HCl) was applied to overcome non-spectral interferences. For standard calibration (l.h.s., load position (green)), a standard solution (mixture of La (ICP-Multi-Element Standard, Aristar®), Co and Sr (both Plasma Emission Standard, Prolabor®) was injected into a sample loop with 333 μl volume, whereas the Mn and Cu spiked solutions were redirected to the Apex E. Once the tube was filled completely with the standard solution, the six-port valve was switched (l.h.s. measurement position (red)). The measurement started and the tube content was pushed into the sample introduction system of the ICP-OES.
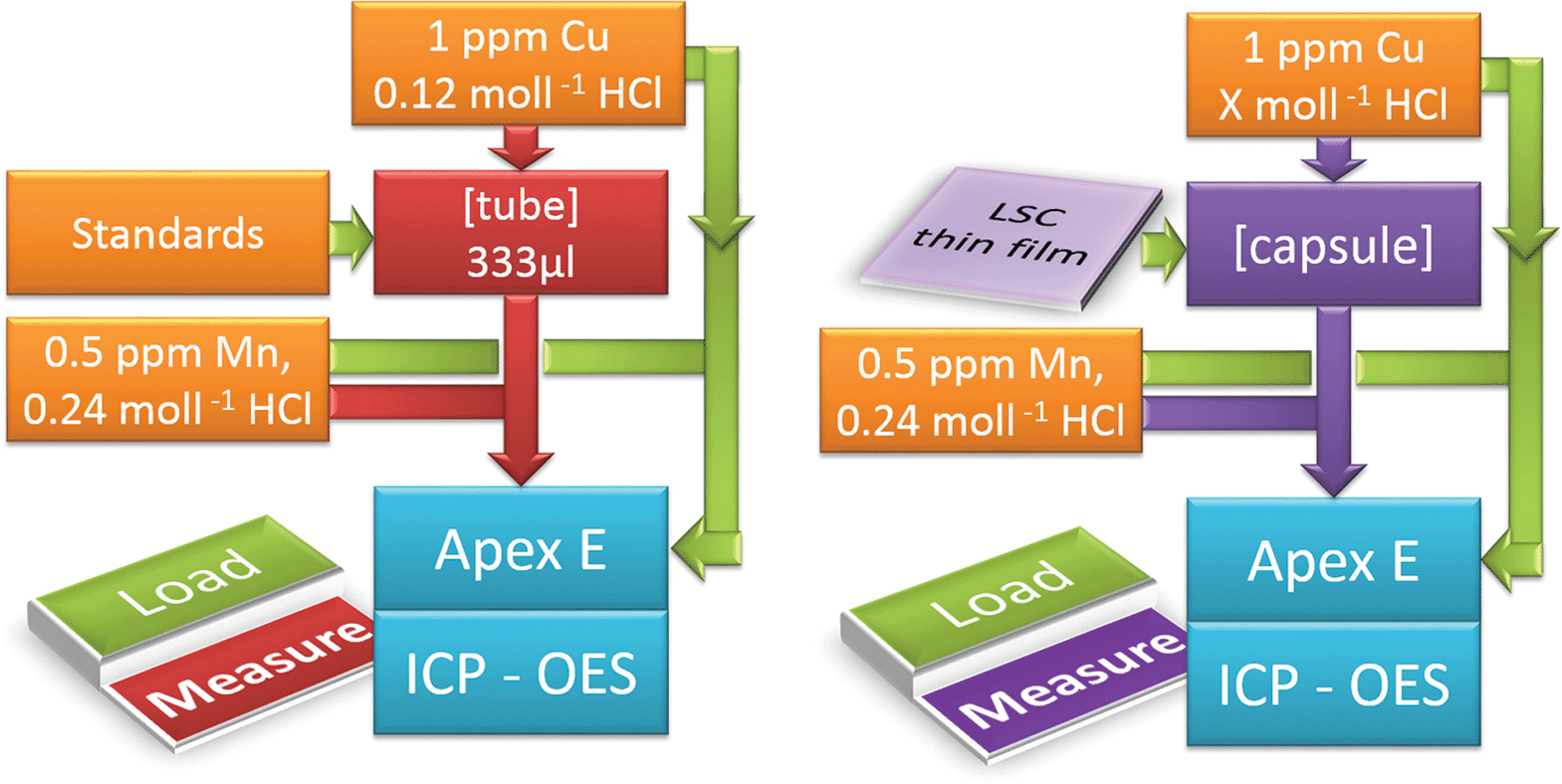 | ||
| Fig. 1 ICP-OES on-line analysis depicting the load and measurement position for standard calibration (l.h.s.) and sample analysis (r.h.s.). | ||
2.4 Geometrical and microstructural investigation
Atomic Force Microscopy (AFM) was performed on LSC thin films to analyze the sample topography and to calculate the surface roughness. A Nanoscope® V multimode setup was utilized in tapping mode, equipped with silicon tips. In general, a scan rate of 2 Hz and a resolution of 512 × 512 pixels over a scan area of 1 × 1 μm2 were chosen. The collected data were evaluated and plotted by Bruker's NanoScope Analysis 1.3 software. A Lyncée Tec Digital Holographic Microscope (DHM) R1101 provided information about the microelectrode thickness and their lateral dimensions. Secondary electron microscopy was performed on a FEI Quanta 200 field emission gun scanning electron microscope (FESEM) equipped with an Everhart–Thornley detector. The microstructure of the thin films was analyzed by Transmission Electron Microscopy (TEM) using an FEI Tecnai F 20 equipped with a field emission gun. Bright field imaging and high angle annular dark field detection (HAADF) were performed at acceleration voltages of 200 kV.3 Results and discussion
3.1 In situ ICP-OES on-line analysis: identification of the water soluble surface phase
An in situ ICP-OES on-line analysis technique using 0.06 mol l−1 hydrochloric acid was already shown to be applicable for investigation of the cation composition of near-surface regions in LSC thin films.12 Here we first demonstrate how to improve the method by exchanging the eluent and/or the eluent concentration and therefore manipulating the solubility of different solid phases. In Fig. 2 an ICP-OES etching profile of a freshly deposited 4500.4 LSC thin film of approx. 160 nm thickness is displayed. During the etching and analysis procedure the eluent was switched several times without interrupting the continuous eluent flux. Fig. 2 (top) shows the cation concentration extracted by the different eluents plotted vs. the sum of all the cations detected from the beginning of the measurement (total cation amount). The dominating eluent etching the LSC thin film is also highlighted. An overlap of the different eluent regimes is unavoidable, even for an instantaneous change of the eluent flux from H2O to 0.012 mol l−1 hydrochloric acid (and back). It takes a certain time to completely flush out the previous etching reagent and solute due to wall friction of the tubes and turbulent fluxes in the etching compartment. This is sketched in the top part of Fig. 2 but the exact eluent composition during that transition is not known. | ||
| Fig. 2 ICP-OES on-line analysis of a 4500.4 thin film of approx. 160 nm thickness, displaying the concentration of detected cations (top) and the calculated cation molar ratio (bottom) versus the total amount of dissolved cations (corresponding to a depth profile). Applied eluent chronology: H2O (240 s) → 12 mmol l−1 HCl (280 s) → H2O (200 s) → 12 mmol l−1 HCl (480 s). The sketch on top displays the sequence of continuous eluent flow, the arrows indicate the moment of eluent change. | ||
A clearer insight into the etching process becomes accessible when relating the calculated molar cation ratio to the total cation amount, Fig. 2 (bottom). The first part of the etching profile with ultrapure H2O is mainly characterized by a high Sr content which will be addressed in detail below, followed by noise due to a strongly decreasing amount of cations in the eluate, partly even below the detection limit. This suggests that only a certain near-surface region of the LSC films is water-soluble. Further this Sr peak endures depending on the etching condition up to 200 seconds.
Switching to 0.012 mol l−1 hydrochloric acid causes further dissolution of the LSC film and a constant cation composition very close to the LSC target stoichiometry (highlighted by dotted lines) of La0.58±0.02Sr0.44±0.04Co0.97±0.03O3−δ is observed. Another change to ultrapure H2O leads to noise without any significant signal confirming that the water soluble film part is only present at the surface. The second etching step using diluted hydrochloric acid again reveals the LSC bulk stoichiometry.
Several conclusions can be drawn from these results: (1) a water-soluble surface phase exists on freshly prepared thin films mainly containing Sr but also Co (the latter detected only with a low signal to noise ratio); (2) LSC is barely soluble in ultrapure water (below the ICP-OES detection limit for La (most sensitive element) in this setup – 20 μg l−1); (3) preferential dissolution of certain cations of the main LSC phase by hydrochloric acid can be neglected due to the good agreement between on-line thin film stoichiometry and target stoichiometry. Assuming both a laterally uniform etching process across the whole sample and a dense thin film, one may directly transfer the total cation amount into a thin film depth. For the given example, however, the prerequisite of high density is not fulfilled, see below.
In order to verify that the Sr amount found on the surface is independent of the etching rate and that the amounts determined for ultrapure H2O are reasonable, another measurement series was conducted: four freshly deposited 4500.4 thin films were etched right from the start using 0.006 mol l−1 HCl, 0.012 mol l−1 HCl, 0.024 mol l−1 HCl or ultrapure H2O, which was exchanged by 0.012 mol l−1 HCl after 240 seconds. In Fig. 3 the amount of Sr found by ICP-OES is plotted against the total cation amount. Linear extrapolation from the range of 15 to 30 nmol to zero cation amount revealed a surplus amount of Sr on the surface. The inset in Fig. 3 illustrates that very similar strontium enrichment at the surface, between 1.37 and 1.50 nmol, results for different eluents. Integration of the Sr amount removed from the surface by ultrapure water leads to 1.50 nmol. Hence, all values coincide excellently and pure H2O can indeed be used to quantify a Sr rich surface phase without dissolving significant amounts of the LSC bulk phase. Based on these results all subsequent ICP-OES measurements were performed using ultrapure water as the etching reagent in the beginning, before switching to diluted hydrochloric acid, thus enabling us to separate information on the Sr-rich surface phase from data on LSC bulk material.
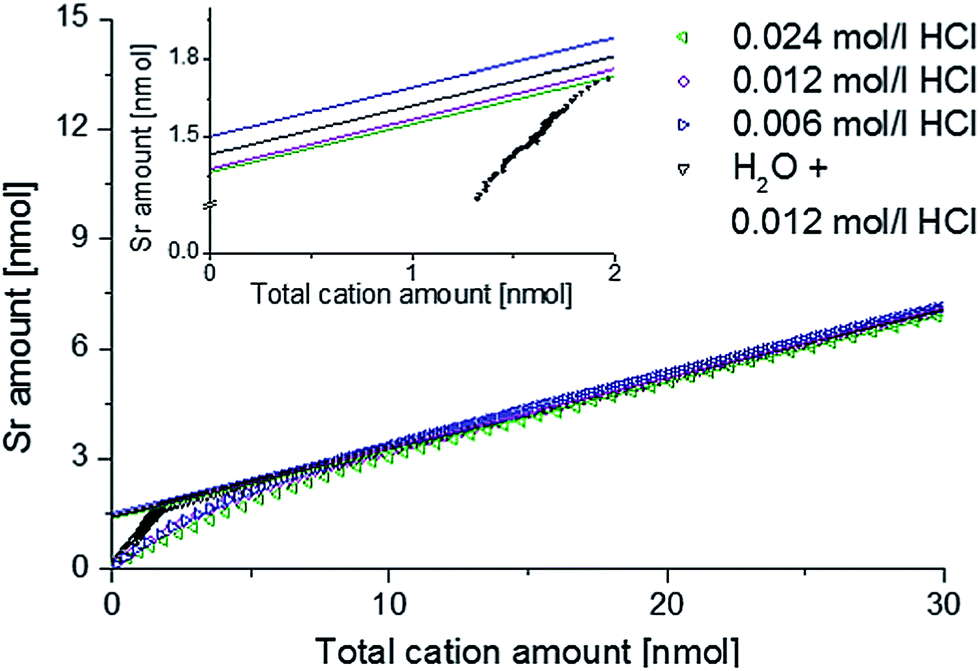 | ||
| Fig. 3 ICP-OES on-line analysis of 4500.4 thin films, showing Sr depth profiles depending on the eluent. | ||
3.2 Dependence of the Sr-rich surface phase on LSC thickness and preparation
In recent publications it was shown that LSC films prepared at low temperatures can exhibit very low polarization resistances12,27 and that the deposition temperature also affects the amounts of Sr-rich surface phases.12,13 Furthermore it was demonstrated that the microstructure and porosity of LaxSr1−xCoyFe1−yO3−δ (LSCF) films depend on the preparation conditions such as deposition temperature and oxygen partial pressure.22 In order to further analyze the correlation between Sr segregation, preparation conditions, film porosity and electrochemical properties, two series of 4500.4 and 4500.04 thin films of different thicknesses were prepared by pulsed laser deposition. By applying 1688, 3375, 6750 and 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 laser pulses to a LSC64 target film, thicknesses of 19–127 nm (4500.04) and 19–167 nm (4500.4) resulted. For each deposition pressure and film thickness, four thin films were prepared simultaneously, leading to a total of 32 samples. Two thin films of each of the eight sets were investigated by ICP-OES on-line analysis during chemical etching, one film was used for electrochemical and DHM measurements and one for FESEM and AFM imaging. Owing to problems during photolithography some additional thin films had to be prepared for the impedance measurements and their thickness was once again determined by DHM.
500 laser pulses to a LSC64 target film, thicknesses of 19–127 nm (4500.04) and 19–167 nm (4500.4) resulted. For each deposition pressure and film thickness, four thin films were prepared simultaneously, leading to a total of 32 samples. Two thin films of each of the eight sets were investigated by ICP-OES on-line analysis during chemical etching, one film was used for electrochemical and DHM measurements and one for FESEM and AFM imaging. Owing to problems during photolithography some additional thin films had to be prepared for the impedance measurements and their thickness was once again determined by DHM.
It was not possible to give unambiguous statements on the thin film porosity based on FESEM images, as grain boundaries between the columnar grains are barely distinguishable from cracks and pinholes, see Fig. 4. Nevertheless, the visual impression suggested a denser packing of the grains for the 4500.04 thin films compared to the 4500.4 thin films. The diameter of the LSC columns growing on the substrate surface was estimated from AFM images (Fig. 5) and results, together with the roughness, are summarized in Table 1. The column diameter increases with increasing film thickness particularly for 4500.4 thin films, where additionally some protruding grains were found.
 | ||
| Fig. 4 FESEM top-views of the thickest 4500.4 and 4500.04 thin films. | ||
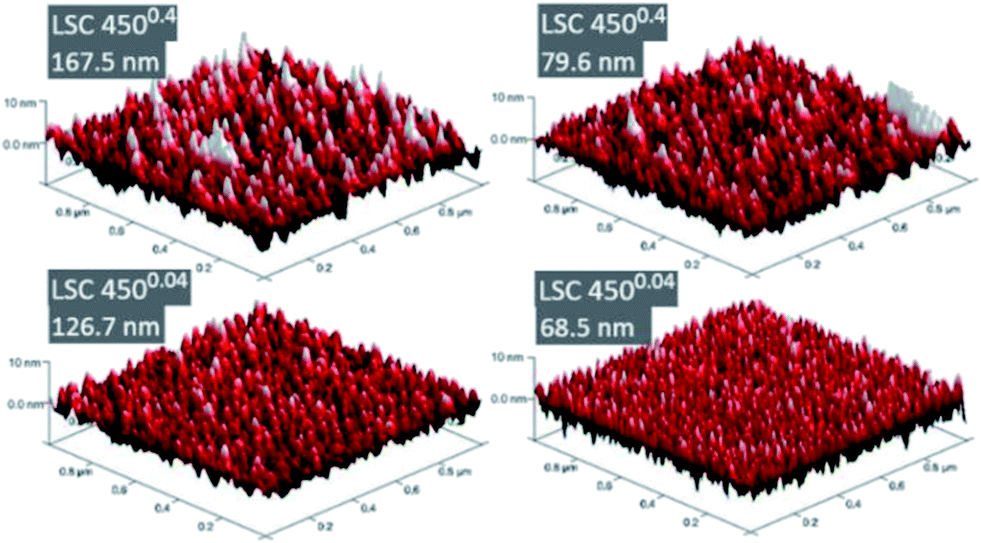 | ||
| Fig. 5 3D-AFM images of the thickest 4500.4 and 4500.04 thin films. | ||
| Sample | Mean grain size [nm] | Roughness [nm] | Surface/proj. area | |
|---|---|---|---|---|
| 4500.4 | 167.5 nm | 22.9 | 2.36 | 104.96% |
| 79.6 nm | 19.1 | 2.27 | 104.28% | |
| 41.0 nm | 14.4 | 2.12 | 117.1% | |
| 19.3 nm | 14.5 | 1.33 | 104.96% | |
| 4500.04 | 126.7 nm | 23.9 | 1.65 | 103.50% |
| 68.5 nm | 16.8 | 1.94 | 109.27% | |
| 36.4 nm | 17.7 | 1.03 | 103.24% | |
| 19.4 nm | 19.8 | 0.85 | 102.27% |
Fig. 6 displays etching profiles of all films obtained by ICP-OES and relates the cation molar ratio to the first 20 nmol of total cation amount etched off during the procedure. The time dependence of the Sr amount etched by ultrapure water, and later by HCl, is shown for all films in Fig. 7. It can be seen that the Sr was removed by ultrapure water levels after a few ten seconds and the value after 320 seconds (dashed line in Fig. 7) was taken as the total amount of strontium extracted by H2O, i.e. before HCl was added. Results are summarized in Table 2. It is very obvious that for films deposited at 0.4 mbar the Sr amount strongly increases with thickness; between 19.3 nm and 79.6 nm the increase is almost proportional to the film thickness. For films deposited at lower oxygen partial pressures, the variation of Sr amount with film thickness is much smaller, and except for the largest thickness even almost negligible. Differences between the projected and surface area (Table 1) caused by roughness can definitively not explain these differences which therefore deserve further interpretation.
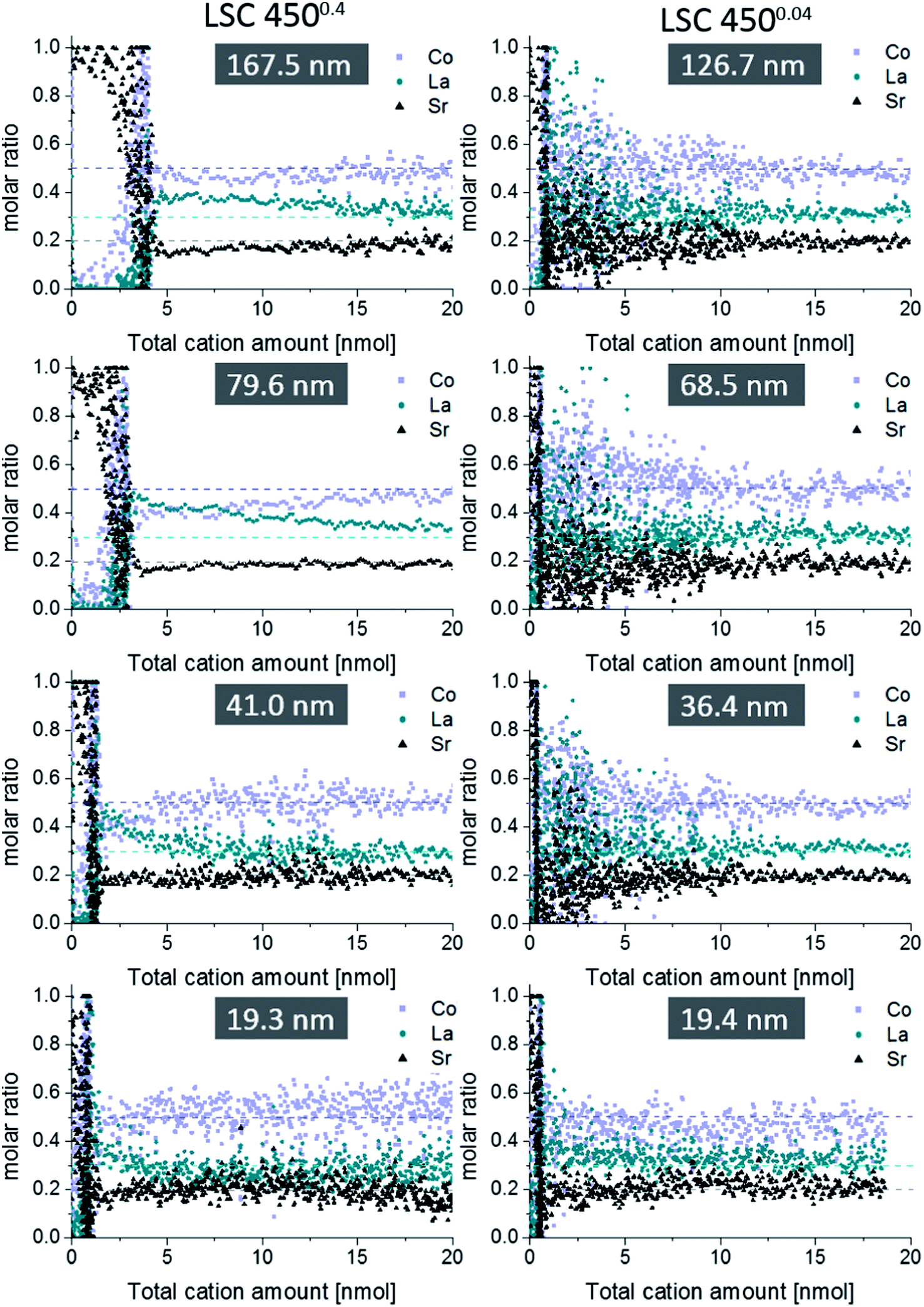 | ||
| Fig. 6 Comparison between ICP-OES on-line depth profiles of differently thin 4500.4 (l.h.s.) and 4500.04 films (r.h.s.). Only the first 20 nmol of the total cation amount are displayed, since deviations from the blank bulk stoichiometry (dashed lines) were limited to this depth. Applied eluent chronology: H2O (200 s) → 6 mmol l−1 HCl (250 s) → 12 mmol l−1 HCl (250 s). A compromise had to be reached in terms of eluent concentration in order to obtain sufficient data points per nmol depth for the porous 4500.4 thin films, and still acceptable noise for the 4500.04 depth profiles. For the dense 4500.04 thin film 20 nmol LSC would correspond to a 15.6 nm thick LSC layer. | ||
 | ||
| Fig. 7 Time-resolved data from spectra of Fig. 6. The detected amount of Sr for the 4500.4 (a) and 4500.04 (b) films of different thicknesses is plotted vs. measuring time. The total water-soluble amount of strontium for each sample is taken from the dashed line and summarized in Table 2. | ||
| Sample | Thickness [nm] | Amount of Sr [nmol] |
|---|---|---|
| 4500.4 | 167.5 | 3.09 ± 0.14 |
| 79.6 | 2.03 ± 0.19 | |
| 41.0 | 0.98 ± 0.11 | |
| 19.3 | 0.51 | |
| 4500.04 | 126.7 | 0.61 ± 0.01 |
| 68.5 | 0.42 ± 0.08 | |
| 36.4 | 0.38 ± 0.09 | |
| 19.4 | 0.39 ± 0.03 |
Assuming that the water soluble Sr-enriched phase is homogeneously distributed on the LSC surface it can be expected that the dissolved amount scales with the surface area accessible to the eluent. For films with a porous microstructure the accessible surface area increases with the thickness as long as pores are not closed. We therefore suggest that the much larger Sr amount indicates a six-fold larger surface area for the 167.5 nm thick film in comparison to the thinnest 4500.4 film. The almost constant Sr amount of 4500.04 layers, on the other hand, suggests dense films for lower deposition pressures. Hence, we conclude that ca. 0.4 nmol Sr exist on a thin film with the lateral dimensions of 0.5 × 0.5 cm2.
Based on these considerations it becomes possible to estimate the thickness of the Sr-rich surface phase, provided it homogeneously covers the entire LSC surface. Different heterogeneously or uniformly distributed Sr-rich phases (e.g. SrO, Sr(OH)2, SrCO3) were already proposed.6,7,12,13 In the case of a dense SrO layer covering the entire LSC surface, 0.4 nmol Sr would correspond to 0.32 nm SrO, i.e. 1.2 atomic layers. However, the result could also be interpreted as chemical dissolution of a terminating layer of pure SrO. Assuming the same amount of Sr segregates per area in the case of 4500.4 layers, our results suggest true surface areas (accessible by the solute) which are increased due to porosity by a factor of 1.2 for the 19 nm film to 8 for the thickest film, compared to the geometrical sample area. This, however, should also lead to significant differences of the polarization resistances and the corresponding measurements are shown in Section 3.3. As mentioned earlier, also traces of Co were found in the ultrapure H2O eluate, which suggests that Co is either part of the water soluble Sr-rich phase or forms a second water-soluble phase on top of LSC. Further experiments are planned involving an ICP-MS system, since the sensitivity of the ICP-OES system is not sufficient to make reliable quantitative statements about the low concentrations of Co (<0.5 mg l−1) in the H2O eluate.
Regarding the etching step in HCl, dense thin films suffered from noisy depth profiles, while porous thin films allowed qualitative insights into the first 10 nm of the LSC bulk due to the increased surface area – yielding more cations for detection. In Fig. 6, a slight depletion of strontium is found in the near surface region of LSC, removed by 0.006 mol l−1 HCl acid, which however by far does not correspond to the amount of strontium etched off in the beginning by H2O. Hence, larger parts of LSC have to be involved in Sr segregation during PLD preparation, which is possible due to sufficiently high cation diffusion coefficients in these thin films, particularly along grain boundaries.18 Another feature is Co depletion close to the surface accompanied by La enrichment thus suggesting a near-surface A-site enrichment (Sr + La)/Co in accordance with XPS measurements.13 However, it should be noted that for all thin films the expected LSC stoichiometry is observed after the first 20 nmol of the total cation amount.
3.3 Electrochemical LSC performance investigated by impedance spectroscopy
AC impedance measurements were carried out on microelectrodes of 200 μm diameter, micro-structured by photo-lithography and chemical etching. Several impedance spectra were recorded for each sample to verify reproducibility. Representative spectra are shown in Fig. 8, normalized to the nominal (projected) electrode area. The high frequency intercept (>250 kHz) of ca. 3.5 Ω cm2 found for most impedance spectra can be attributed to oxide ion conduction in the YSZ bulk. Slight differences of the intercept most probably originate from temperature variations between measurements, since samples were heated only from the bottom and the set temperature deviates from the true electrode temperature. Using an Arrhenius-type equation it is rather possible to determine the actual temperature directly at the microelectrode from the YSZ bulk resistance, provided the ionic conductivity of YSZ is known as a function of temperature.28 The larger frequency intercept value of the 19.3 nm 4500.4 thin film can be attributed to an increased electronic sheet resistance in LSC. The position of the arc or plateau in the medium frequency range from 250 kHz to 1 kHz varied strongly between the different samples, similar to earlier studies on mixed conducting electrodes.29 As it is barely affected by long-term annealing experiments at 550 °C and not responding to LSC surface treatments,12 it is assumed to originate from the O2− transfer across the LSC/YSZ interface, possibly including a contribution of O2− transport through the LSC bulk.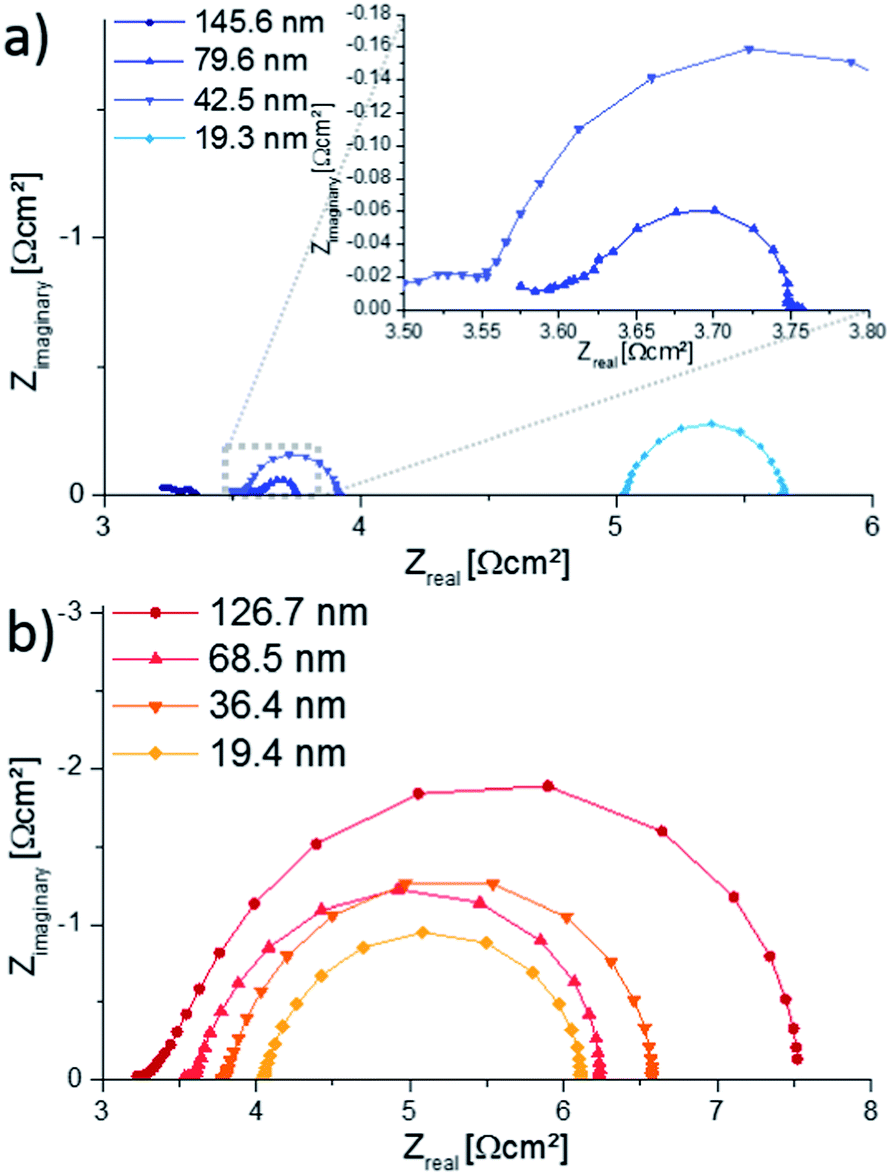 | ||
| Fig. 8 Nyquist plots showing impedance spectra of the 4500.4 (a) and 4500.04 (b) films, measured at approx. 550 °C electrode temperature (600 °C set temperature) on microelectrodes of 200 μm diameter. The increased intercept for the 19.3 nm thick film results from an increased electronic sheet resistance. | ||
At lower frequencies (1 kHz to 0.1 Hz) the shape of the impedance spectra became semicircle-like for all thin films and could be fitted to a parallel R-CPE-element (CPE = constant phase element with exponential fitting parameter n > 0.95). The resistive element R is attributed to the complex oxygen surface exchange reaction (Rsurface exchange) as it is sensitive to surface treatments and increases at low oxygen partial pressures, cf. also ref. 12 and 30. The capacitive element corresponds to the chemical capacitance of the film29,31 and depends on the volume of the microelectrode: The resulting values are plotted in Fig. 9 and the typical thickness dependence of bulk chemical capacitances can clearly be seen. This chemical capacitor is determined by the properties of the bulk material, since it virtually stores the charge carriers in the perovskite lattice of the LSC. Therefore crystallinity, strain, etc. can be assumed to play an important role and those may differ between the two thin film deposition routes.
 | ||
| Fig. 9 Relationship between the chemical capacitance and the thin film thickness of 4500.4 and 4500.04 thin films. | ||
This might explain the different absolute values for the same thickness of 4500.4 and 4500.04 films.
The size of the low-frequency semicircle strongly depends on the thickness of 4500.4 films. The increasing oxygen exchange resistance with decreasing 4500.4 film thickness is in accordance with the enlarged surface area of thicker films concluded from Sr amounts on the surface. However, all 4500.4 thin films exhibit similar peak frequencies due to a decreasing chemical capacitor for thinner films. The 4500.04 thin films, on the other hand, show only small variations of the surface resistance, particularly between 19.4 and 68.5 nm thicknesses but a change in the peak frequency. This is in accordance with little or no porosity concluded from ICP-OES measurements. A more detailed comparison of Sr amount and surface exchange resistance is given in the following.
3.4 Comparison of Sr amount and electrochemical properties
In Fig. 10, both the water soluble amount of Sr and 1/Rsurface exchange are plotted vs. film thickness of (a) LSC 4500.4 and (b) 4500.04 thin films. For 4500.04 films neither clear trends for different thicknesses nor a clear relationship between the (only moderate) variation of Sr amount and surface exchange resistance is found. This is in agreement with our assumption of dense 4500.04 films. However, both curves show a very clear trend for the 4500.4 thin films and both can be explained by porosity and thus a true surface area depending on the thickness: The larger the thickness (and the true surface area) the larger the inverse surface polarization resistance and the total amount of surface Sr. However, this does not necessarily mean that the entire 4500.4 films are porous. | ||
| Fig. 10 Dependence of the surface exchange resistance (normalized to the nominal surface area) and the water soluble Sr amount on the film thickness of (a) 4500.4 and (b) 4500.04 thin films. For the 4500.4 thin films both data sets clearly indicate an enhanced surface area. | ||
Rather, preliminary high-angle annular dark field (HAADF) measurements (Fig. 11) of the region close to the YSZ|LSC interface suggest density variations perpendicular to the interface.32 The intensity of the observed signal depends on the specimen thickness, atomic number and density. In our case, all factors except for the density are assumed to be rather constant over the film thickness. The data therefore indicate a denser film (<23 nm) close to the YSZ interface. High resolution images of our films (Fig. 12) and also of similar LSCF films22 revealed dense film regions close to the interface but open porosity for film parts on top of this dense layer.
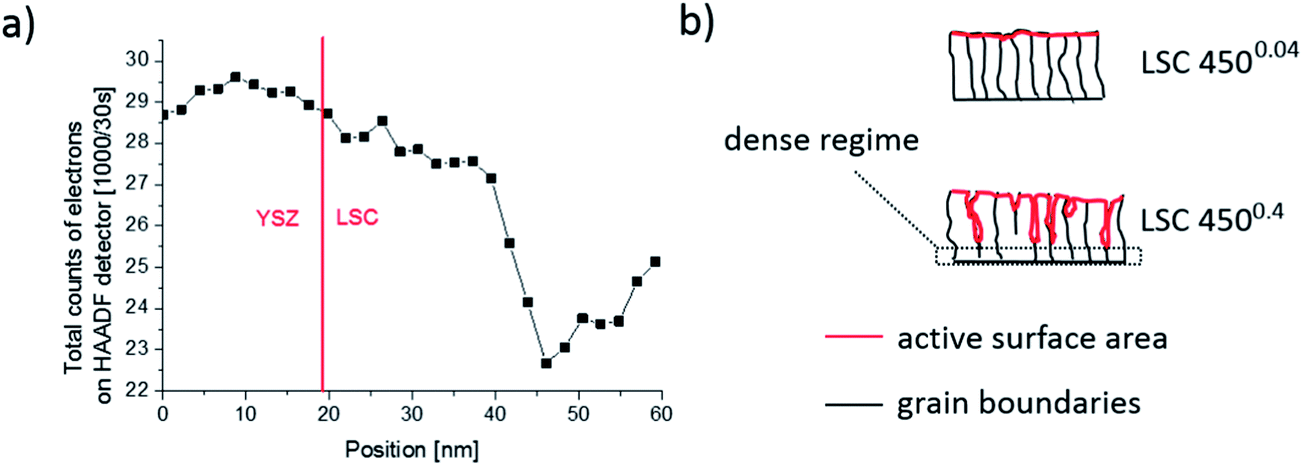 | ||
| Fig. 11 (a) Total counts of electrons detected by HAADF STEM close to the YSZ|LSC interface (dotted line). A decrease, most probably related to pores, is observed after 20 nm of thin film growth. (b) Sketch of LSC thin film microstructure with columnar grains in both cases but pores only for 4500.4. | ||
 | ||
| Fig. 12 TEM bright field image of 4500.4 (l.h.s.) and 4500.04 (r.h.s.) film. | ||
From this we suggest that the first ca. 20 nm of 4500.4 films are still dense which is also in accordance with similar Sr amounts found for all dense 4500.04 films and the 19.3 nm thin 4500.4 film. A sketch of the microstructure of both film types, highlighting the active surface area (red), is given in Fig. 11b. For a rough estimate of the true surface area in porous 4500.4 films we fit the Sr amount β and film thickness y to eqn (1)
| β = β0 + α(y − 20 nm) | (1) |
Microstructural differences of the 4500.4 and 4500.04 films can be explained in terms of Thornton's well known structure zone model (SZM)33 and the so-called “shadow effect”34 during deposition. The 4500.4 thin film shows characteristics of zone 1 in the SZM, where only limited surface diffusion of deposited particles takes place.22 At higher oxygen partial pressures, more particle collisions occur during deposition, causing an increased angle distribution of decelerated incoming particles, which amplifies shadowing effects for the 4500.4 thin films and thus results in an increased porosity.35 Pores can be expected to form at a certain roughness of the film but not from the very beginning of the film growth, see Fig. 12.
Based on these results we can also further interpret the very low polarization resistance of low temperature deposited LSC films reported in ref. 27 (0.1 Ω cm2 for a 200 nm thin film at 600 °C set temperature). In contrast to the first impression, those films were most probably porous since the same deposition parameters were applied and the same electrochemical properties were observed as in this study. Such an assumption is also in accordance with the systematic investigation of LSCF thin film growth on YSZ substrates at different substrate temperatures and gas pressures.22 Hence, differences in electrode resistance between 40 and 200 nm thin films in ref. 27 can rather be attributed to different electrode surface areas than to surface chemistry variations. In accordance with the estimated surface area increase of a 200 nm film, the polarization resistance related to the true surface area has to be corrected to ca. 0.5 Ω cm2 at 600 °C set temperature (instead of 0.1 Ω cm2).
It is finally worth mentioning that the polarization resistance of a LSC film measured at 600 °C is not altered when removing the surface Sr by ultrapure water. This, however, is simply caused by the fact that during heating and thermal equilibration (1 h at 600 °C), before the impedance measurements are started, water soluble Sr is again formed. Quantification by ICP-OES even showed that this newly formed amount of Sr is almost identical to that found for freshly prepared PLD layers. This suggests that already at rather low temperatures and short annealing times a surface reconstruction takes place which leads to the water soluble Sr surface phase.
4 Conclusions
By means of an improved on-line in situ etching procedure with subsequent ICP-OES analysis using different eluents, a water soluble Sr-rich surface phase was found on top of La0.6Sr0.4CoO3−δ (LSC) thin films, together with traces of Co. Assuming a dense SrO layer, the ca. 0.4 nmol Sr per 0.25 cm2 found for low pressure (0.04 mbar) deposited films corresponds to 1.2 atomic layers covering the whole LSC surface. For thin films deposited at higher oxygen partial pressures (0.4 mbar) the water soluble Sr amount strongly increased with increasing film thickness. This was related to the effective porosity and thus to an increased surface area of thicker films. The influence of this porosity on the surface exchange resistance could be quantified by combining impedance spectroscopy measurements and ICP-OES. It was possible to relate the results of the electrochemical measurements to the true surface area. The lowest surface exchange resistance of LSC seems to be approximately 0.5 Ω cm2 at 550 °C. The dependence of the electrochemical performance of LSC thin films on the oxygen partial pressure during deposition was thus shown to be partly caused by microstructural differences, i.e. effective porosity.Acknowledgements
The authors gratefully acknowledge funding by Austrian Science Fund (FWF) projects P21960-N17 and W1243.Notes and references
- T. Yamaguchi, S. Shimizu, T. Suzuki, Y. Fujishiro and M. Awano, Electrochem. Commun., 2008, 10, 1381–1383 CrossRef CAS PubMed.
- J. P. P. Huijsmans, F. P. F. van Berkel and G. M. Christie, J. Power Sources, 1998, 71, 107–110 CrossRef CAS.
- S. B. Adler, Chem. Rev., 2004, 104, 4791–4844 CrossRef CAS.
- F. Zhao, R. Peng and C. Xia, Fuel Cells Bulletin, 2008, 2008, 12–16 CrossRef.
- W. Lee, Z. Cai and B. Yildiz, ECS Trans., 2012, 45, 405–412 CrossRef CAS PubMed.
- A. Huber, M. Falk, M. Rohnke, B. Luerssen, M. Amati, L. Gregoratti, D. Hesse and J. Janek, J. Catal., 2012, 294, 79–88 CrossRef CAS PubMed.
- Q.-H. Wu, M. Liu and W. Jaegermann, Mater. Lett., 2005, 59, 1980–1983 CrossRef CAS PubMed.
- E. Bucher, W. Sitte, F. Klauser and E. Bertel, Solid State Ionics, 2012, 208, 43–51 CrossRef CAS PubMed.
- E. Bucher, C. Gspan, F. Hofer and W. Sitte, Solid State Ionics, 2013, 238, 15–23 CrossRef CAS PubMed.
- S. P. Simner, M. D. Anderson, M. H. Engelhard and J. W. Stevenson, Electrochem. Solid-State Lett., 2006, 9, A478–A481 CrossRef CAS PubMed.
- F. S. Baumann, J. Fleig, M. Konuma, U. Starke, H. Habermeier and J. Maier, J. Electrochem. Soc., 2005, 152, A2074–A2079 CrossRef CAS PubMed.
- M. Kubicek, A. Limbeck, T. Fromling, H. Hutter and J. Fleig, J. Electrochem. Soc., 2011, 158, B727–B734 CrossRef CAS PubMed.
- Z. Cai, M. Kubicek, J. Fleig and B. Yildiz, Chem. Mater., 2012, 24, 1116–1127 CrossRef CAS.
- K. Szot, M. Pawelczyk, J. Herion, C. Freiburg, J. Albers, R. Waser, J. Hulliger, J. Kwapulinski and J. Dec, Appl. Phys. A: Mater. Sci. Process., 1996, 62, 335–343 Search PubMed.
- Y. Chen, W. Jung, Z. Cai, J. J. Kim, H. L. Tuller and B. Yildiz, Energy Environ. Sci., 2012, 5, 7979–7988 CAS.
- P. Hjalmarsson, M. Søgaard and M. Mogensen, Solid State Ionics, 2008, 179, 1422–1426 CrossRef CAS PubMed.
- W. Lee, J. W. Han, Y. Chen, Z. Cai and B. Yildiz, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS PubMed.
- M. Kubicek, G. M. Rupp, S. Huber, A. Penn, A. K. Opitz, J. Bernardi, M. Stoger-Pollach, H. Hutter and J. Fleig, Phys. Chem. Chem. Phys., 2014, 16, 2715–2726 RSC.
- M. R. Baklanov, K. P. Mogilnikov, V. G. Polovinkin and F. N. Dultsev, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 2000, 18, 1385–1391 CrossRef CAS.
- P. Gilbert, J. Theor. Biol., 1972, 36, 105–117 CrossRef CAS.
- L. Dieterle, P. Bockstaller, D. Gerthsen, J. Hayd, E. Ivers-Tiffée and U. Guntow, Adv. Energy Mater., 2011, 1, 249–258 CrossRef CAS PubMed.
- P. Plonczak, A. Bieberle-Hütter, M. Søgaard, T. Ryll, J. Martynczuk, P. V. Hendriksen and L. J. Gauckler, Adv. Funct. Mater., 2011, 21, 2764–2775 CrossRef CAS PubMed.
- J. Fleig, F. S. Baumann, V. Brichzin, H. R. Kim, J. Jamnik, G. Cristiani, H. U. Habermeier and J. Maier, Fuel Cells, 2006, 6, 284–292 CrossRef CAS PubMed.
- M. P. Pechini, US Pat., 3330697, 1967.
- F. S. Baumann, J. Fleig, G. Cristiani, B. Stuhlhofer, H.-U. Habermeier and J. Maier, J. Electrochem. Soc., 2007, 154, B931–B941 CrossRef CAS PubMed.
- G. M. Rupp, 2012, Master’s Thesis, Vienna University of Technology.
- J. Januschewsky, M. Ahrens, A. K. Opitz, F. Kubel and J. Fleig, Adv. Funct. Mater., 2009, 19, 3151–3156 CrossRef CAS PubMed.
- A. K. Opitz and J. Fleig, Solid State Ionics, 2010, 181, 684–693 CrossRef CAS PubMed.
- F. S. Baumann, J. Fleig, H. Habermeier and J. Maier, Solid State Ionics, 2006, 177, 1071–1081 CrossRef CAS PubMed.
- A. Ringuedé and J. Fouletier, Solid State Ionics, 2001, 139, 167–177 CrossRef.
- J. Jamnik and J. Maier, Phys. Chem. Chem. Phys., 2001, 3, 1668–1678 RSC.
- A. Penn, Master thesis, Vienna University of Technology, 2012.
- J. A. Thornton, J. Vac. Sci. Technol., 1974, 11, 666–670 CrossRef CAS.
- A. Infortuna, A. S. Harvey and L. J. Gauckler, Adv. Funct. Mater., 2008, 18, 127–135 CrossRef CAS PubMed.
- L. Dong, R. W. Smith and D. J. Srolovitz, J. Appl. Phys., 1996, 80, 5682–5690 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |