 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA metal-oxide catalyst enhanced the desorption properties in complex metal hydrides
Tengfei
Zhang
*a,
Shigehito
Isobe
*ab,
Yongming
Wang
a,
Hiroshi
Oka
a,
Naoyuki
Hashimoto
a and
Somei
Ohnuki
a
aGraduate School of Engineering, Hokkaido University, N-13, W-8, Sapporo 060-8628, Japan. E-mail: isobe@eng.hokudai.ac.jp; zhangtengfei@eng.hokudai.ac.jp
bCreative Research Institution, Hokkaido University, N-21, W-10, Sapporo, 001-0021, Japan
First published on 6th January 2014
Abstract
In this study, LiTi2O4 was synthesized as a possible catalyst for complex metal hydrides. LiTi2O4 was stable in the sample after high-energy ball milling and heat treatment. LiTi2O4 exhibited a catalytic effect among the samples of MgH2, LiAlH4 and LiNH2. The desorption kinetics and the purity of the desorbed hydrogen gas have been improved by doping LiTi2O4. Furthermore, the catalytic mechanism of LiTi2O4 was discussed in accordance with the experimental results.
1. Introduction
Hydrogen is a kind of sustainable energy carrier compared to conventional ones such as fossil fuels. It has advantages such as being clean, is environmentally friendly and forms water as a non-polluting product during practical applications.1 Accordingly, the reduction of our dependence on fossil fuels and emissions of pollutants and greenhouse gases could be realized by using hydrogen as a primary fuel successfully. This would have a great influence on both changing energy structure and improving global warming. Recently hydrogen energy has been actively investigated in terms of production, storage and application (comprising combustion engines in vehicles and power application).2 Nowadays, compressed gaseous hydrogen is used in prototype and liquid form in advanced tank systems.3 However, storing hydrogen in solid-state materials has definite advantages on accessible hydrogen content and energy efficiency compared to complex system that use high-pressure gas or cryogenic liquid. A series of storage materials have been proposed, such as metal-hydrides, alanates and amides. While the hydrogen capacity of complex metal hydrides is reasonable (often in the 3–12 wt% range), relatively high thermal stability, slow kinetics of the pure hydrides, undesirable by-product gases (e.g. ammonia) and irreversibility on cycling are still limitations for on-board applications.4Extensive efforts are currently being made for the improvement of hydrogen storage properties by improving the absorption/desorption kinetics, reducing the operating temperatures and improving the purity of the desorbed hydrogen gas.5 One of the strategies for the improvement of hydrogen storage properties is the addition of catalysts. Ti compounds have been well known to catalyze the dehydrogenation of complex aluminium hydrides in solution. Wieberg et al. in 1951 observed that Ti catalysed the dehydrogenation of LiAlH4 in a diethyl ether suspension.6 A breakthrough came when Bogdanović and Schwickardi certificated that doping a few mol% Ti in the complex metal hydride NaAlH4 lowered the dehydrogenation temperature, improved the kinetics and allowed rehydrogenation of the decomposition products.7 This is an indication that Ti worked effectively for solid-state NaAlH4. Interest in using complex metal hydrides as hydrogen storage materials has dramatically reawakened. Meanwhile, the catalytic effect of Ti compounds was also found in a LiH/LiNH2 system which was reversible and had a high hydrogen capacity of 6.5 wt%.8 The absorption/desorption kinetics and the purity of the desorbed hydrogen gas had been improved. Among the metal hydrides, MgH2 exhibits a high hydrogen capacity up to 7.6 wt%. However, the absorption and desorption reactions of Mg/MgH2 itself are too slow. Some of the researchers reported that Ti compounds had a catalytic effect on the absorption and desorption.9–11 Subsequently, Hanada et al. reported the valence state of the Ti compounds that were doped in MgH2.12 However, the catalyst state of Ti is not the same as the original after ball-milling and/or heat treatment.12,13 In this case, the mechanism of the catalytic effect is difficult to understand clearly due to the instability of Ti compounds.
Normally, the reactions that occur between the solid phases for complex metal hydrides are solid-state reactions. Diffusion plays an important role in the solid-state reactions since they require the coming together of reactive species. The rigid structure makes solids different from liquids and gases. The kinetics of solid-state reactions is therefore greatly dependent on the crystal structure and its defects. Based on these results, one proposal for improving the kinetics of the solid-state reaction is increasing the mobility of ions, such as Li+, Na+ and H− in the complex metal hydrides. This process would progress by doping the catalyst, especially with Ti compounds. Our previous study suggested that LiTi2O4 was discovered by transmission electron microscopy (TEM) in the Li–N–H sample which was doped with TiCl3 as the catalyst.13 The catalytic Ti compounds were active at the interface between LiH and LiNH2. Herein, we choose LiTi2O4 as the catalyst for hydrogen storage materials in lithium alanate, magnesium hydride and lithium amides. As a result of their investigation, hydrogen can be desorbed via the following reaction:
| LiAlH4 ↔ LiH + Al + 3/2H2 | (1) |
| MgH2 ↔ Mg + H2 | (2) |
| LiH + LiNH2 ↔ Li2NH + H2 | (3) |
For the case of LiAlH4, eqn (1) can be recast as a two step reaction as follows:
| LiAlH4 → 1/3Li3AlH6 + 2/3Al + H2 | (4) |
| 1/3Li3AlH6 → LiH + 1/3Al + 1/2H2 | (5) |
Consequently, the catalytic effect of LiTi2O4 will be discovered for samples with the aim of enhancing hydrogen desorption properties in this study.
2. Experimental
2.1. Sample preparation
Powders of LiAlH4 (95%), Li2CO3 (99.997%), TiO2 (99%), Ti2O3 (99.9%), LiH (95%) and LiNH2 (95%) were purchased from Sigma-Aldrich. MgH2 (98%) powder was purchased from Alfa Aesar. The single phase of LiTi2O4 was synthesized by a two-step solid-state reaction. Li2Ti2O5 forms as an intermediate compound.14 The reaction equations used here are:| Li2CO3 + 2TiO2 → Li2Ti2O5 + CO2 | (6) |
| Li2Ti2O5 + Ti2O3 → 2LiTi2O4 | (7) |
The synthesis method of LiTi2O4 according to Xu et al. has been used.15 Considering the volatility of Li, it was hard to get the exact composition of Li. Here an excess molar fraction 0.15 of Li2CO3 is added in eqn (6) for LiTi2O4. An additional factor is the sensitivity of LiTi2O4 to air. This issue was mentioned by several groups,16–18 regardless of the preparation method used. The aging reaction is the topotactic oxidation of LiTi2O4. LiTi2O4 reacts with oxygen or H2O in air and is progressively transformed into Li1−xTi2O4 (0 ≤ x ≤ 0.8). The polycrystalline sample is stored in Ar atmosphere in order to avoid the aging reaction with oxygen and moisture.
Typically, in order to ensure a homogeneous mixing between the starting materials and the additive, a ball-milling equipment (Fritsch P7) is used. Samples of LiAlH4 and 0.5 mol% LiTi2O4 were milled for 2 h. The samples, mixtures of MgH2 and 1 mol% LiTi2O4, were milled for 20 h. LiNH2 and LiH powders with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 molar ratio and 0.5 mol% LiTi2O4 were milled for 2 h. The total weight for each ball-milled sample was 300 mg. In the high energy ball-milling process, powder and 20 steel balls (SUJ-2) with a diameter of 7 mm were brought into a Cr steel pot (SKD-11) and milled at 400 rpm under a hydrogen gas (99.9999% purity) at a pressure of 1 MPa at room temperature. The ball-to-powder ratio was 100:1. The milling was interrupted every 1 h for 30 min in order to prevent frictional heat during the milling process.
:1.2 molar ratio and 0.5 mol% LiTi2O4 were milled for 2 h. The total weight for each ball-milled sample was 300 mg. In the high energy ball-milling process, powder and 20 steel balls (SUJ-2) with a diameter of 7 mm were brought into a Cr steel pot (SKD-11) and milled at 400 rpm under a hydrogen gas (99.9999% purity) at a pressure of 1 MPa at room temperature. The ball-to-powder ratio was 100:1. The milling was interrupted every 1 h for 30 min in order to prevent frictional heat during the milling process.
2.2. Characterization measurements
Structure properties were characterized by X-ray diffraction (XRD) measurement. A sample was firstly put on a glass plate in a glove box. Then the glass plate was covered with a plastic film to protect the sample from contact with air. The plastic film does not have any obvious influence on the obtained XRD patterns. The samples were measured with a diffraction 2θ angle from 10° to 90° under a speed of 0.04° per step. Furthermore, after the milling process, the mixtures were examined by thermogravimetry and differential thermal analysis (TG-DTA) equipment (HITACHI BRUKER TAPS3000S) combined with thermal gas desorption mass spectrometry (TDMS). TG-DTA equipment was installed in another glove box to avoid exposing the samples to air in their measurements.3. Results and discussion
LiTi2O4 was synthesized by sintering a mixture of Li2Ti2O5 and Ti2O3 at 880 °C under a pressure less than 10−4 Pa. Fig. 1 shows the powder X-ray diffraction (XRD) profile. There were no apparent impurities in the product of the single phase LiTi2O4. The color of this single phase was black, which is consistent with previous reports.19 The stability of LiTi2O4 is reflected by the XRD patterns in Fig. 2. Three samples after high-energy ball milling and dehydrogenation, 0.5 mol% LiTi2O4 + 99.5 mol% LiAlH4 mixture, 1 mol% LiTi2O4 + 99 mol% MgH2 mixture and the composite of LiH and LiNH2 with 0.5 mol% LiTi2O4, were prepared for XRD measurement. The results indicated that the structure of LiTi2O4 was the same as the single phase of the raw LiTi2O4 after high-energy ball milling and dehydrogenation.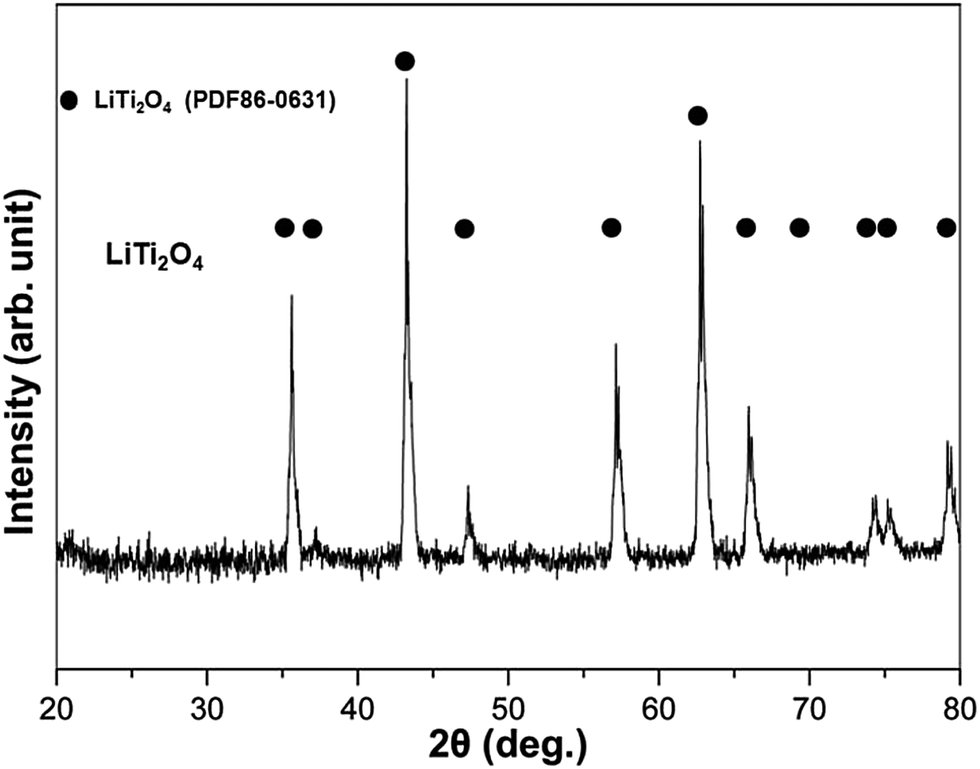 | ||
| Fig. 1 XRD pattern of the single phase LiTi2O4. | ||
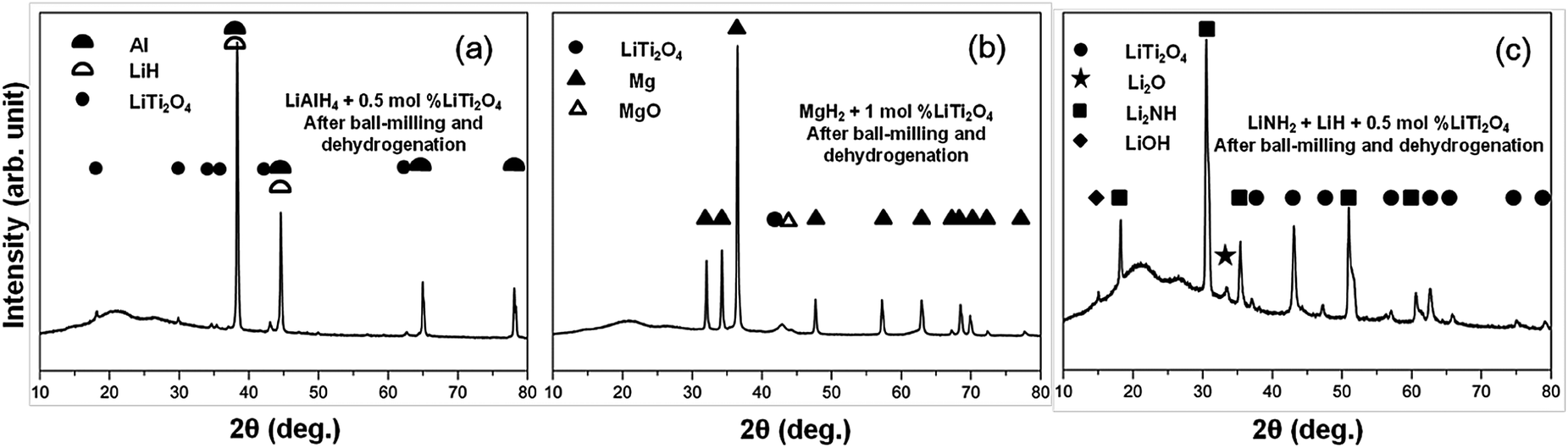 | ||
| Fig. 2 XRD patterns of the samples after high-energy ball milling and dehydrogenation: (a) 0.5 mol% LiTi2O4 + 99.5 mol% LiAlH4 mixture, (b) 1 mol% LiTi2O4 + 99 mol% MgH2 mixture, (c) the composite of LiH and LiNH2 with 0.5 mol% LiTi2O4. | ||
Fig. 3 shows the TG-DTA results of the as received LiAlH4 and the 0.5 mol% LiTi2O4 + 99.5 mol% LiAlH4 mixture. It could be confirmed that during the examined temperature range, the dehydrogenation process showed rather different behavior from these two samples. In the case of the as received sample, two conspicuous peaks appeared around 165–180 °C. The endothermic peak (170 °C) resulted from the melting of LiAlH4. After the endothermic peak, there was an exothermic peak (179.5 °C) that indicated the decomposition of liquid LiAlH4. The reaction started at 160 °C and ended at 240 °C. The total weight loss was 6.4 wt%. In the case of the mixed sample, the hydrogen desorption started at 100 °C, and the whole decomposition of LiAlH4 was complete at around 220 °C. Two endothermic peaks could be seen from the DTA curve. The peak temperature of each peak was 160 °C and 210 °C. Accordingly, some authors have calculated in their reports that the enthalpy of the first dehydrogenation of LiAlH4 with an additive is in agreement with the enthalpy difference between the melting transition and the exothermic decomposition immediately following it for as received LiAlH4.20–23 Consequently, the composite’s endothermic–exothermic peak at 170–180 °C in the sample without the additive is in agreement with the absorption of latent heat of melting by LiAlH4 with fast decomposition of Li3AlH6 and desorption of H2, and immediate releasing the latent heat of fusion of Li3AlH6 with desorption of H2. Hence, the first dehydrogenation of LiAlH4 is intrinsically endothermic. Doping LiAlH4 with LiTi2O4 improved its thermal decomposition, bringing this dehydrogenation below the melting point of LiAlH4 and separating it from the complex melting–desorption–solidification event that appears at 170 °C for the undoped sample. This result indicated that LiTi2O4 has some catalytic effect on the lithium alanate.
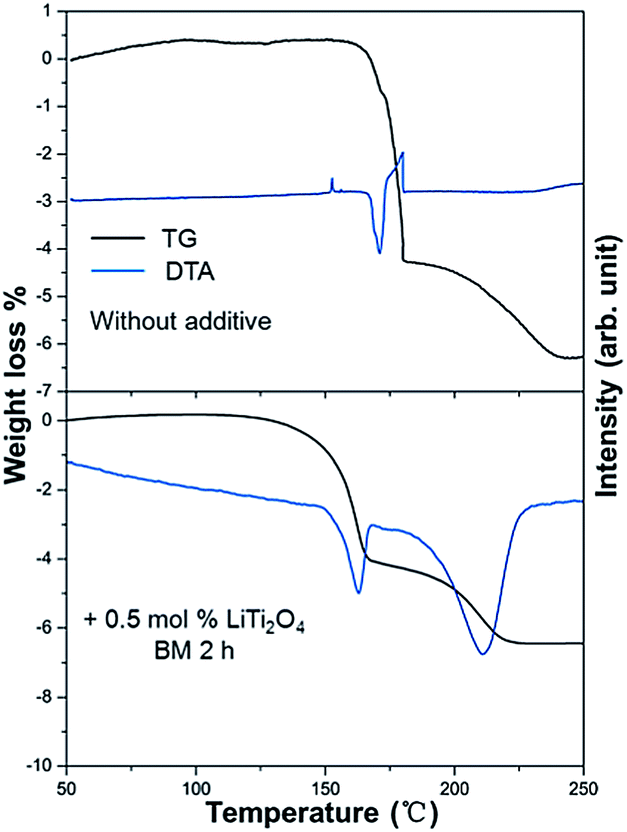 | ||
| Fig. 3 Differential thermal analysis and corresponding weight loss for the dehydrogenation of the as received LiAlH4 and the 0.5 mol% LiTi2O4 + 99.5 mol% LiAlH4 mixture. | ||
The TG-DTA results of the milled MgH2 and the 1 mol% LiTi2O4 + 99 mol% MgH2 mixture are shown in Fig. 4. There were two endothermic peaks for the milled MgH2 sample during the measurement. Dehydrogenation started around 260 °C and ended at 380 °C. The peak temperature for each peak was 290 °C and 340 °C. The weight loss of 5.1 wt% was much less than the theoretical value of 7.6 wt%, which indicated an incomplete decomposition of the milled sample under the conditions of heating up to 400 °C at a rate of 5 °C min−1. For the sample with an additive the DTA curve showed that hydrogen desorption started at 220 °C and finished at 330 °C. There was only one main endothermic peak for the mixture sample during the measurements. The peak temperature was at 260 °C. A weight loss of 6.6 wt% was achieved from the TG measurement compared with the theoretical value of 7.0 wt%. The difference between the experimental value and the theoretical value was caused by impurities in the original sample, such as MgO. The much better hydrogen desorption kinetics of the mixture sample was attributed to the catalytic effect of LiTi2O4. Recently, Nb2O5 doped MgH2 has been actively studied. The sluggish sorption kinetics of MgH2 can be improved dramatically to release 6 wt% of reversible hydrogen at 250 °C.8,24,25 Hanada et al. reported that MgH2 with 1 mol% Nb2O5 milled for 20 h, was able to release 4.5 wt% H2.26,27 After LiTi2O4 was introduced to this system, the kinetics of MgH2 has been accelerated at the same level compared with previous work.27
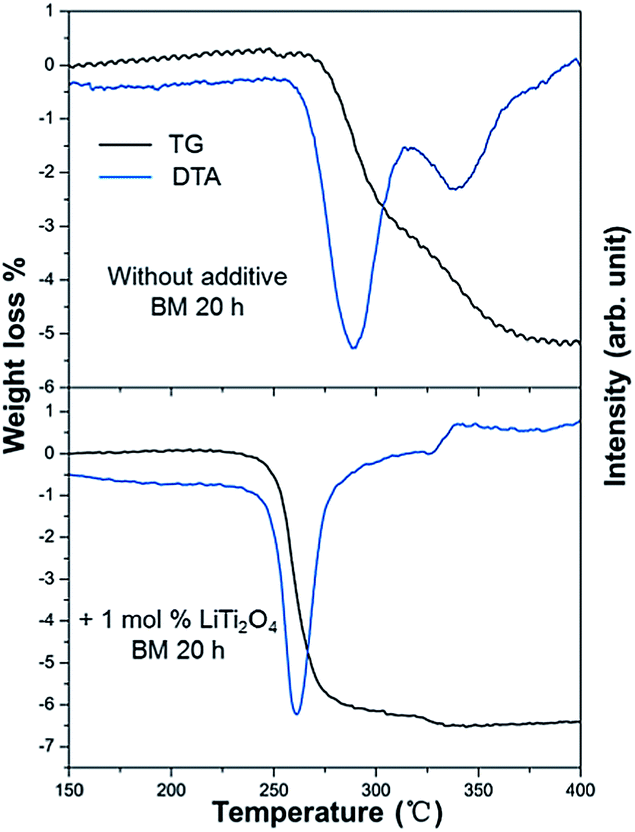 | ||
| Fig. 4 Differential thermal analysis and corresponding weight loss for the dehydrogenation of the milled MgH2 and the 1 mol% LiTi2O4 + 99 mol% MgH2 mixture. | ||
The dehydrogenation process of a composite of LiH and LiNH2 with 0.5 mol% LiTi2O4 additive was investigated. Fig. 5 presents the TDMS results. The sample of LiH and LiNH2 with LiTi2O4 showed a sharp H2 peak. The peak temperature was at 227 °C. No ammonia was detected during dehydrogenation. To the best of our knowledge, addition of LiTi2O4 to the system as a catalyst resulted in the lowest desorption temperature observed so far, compared with well known catalysts (BN, Si, TiCl3).28 However, hydrogen and ammonia were released from the sample without LiTi2O4. The broad hydrogen desorption curves and obvious ammonia emission can be seen in the temperature range from 200 °C to 400 °C. The results demonstrated clearly that the purity of the desorbed hydrogen gas was improved after doping with LiTi2O4. During heating, the decomposition of the LiNH2 + LiH mixture was observed to start at 200 °C and complete at 400 °C. By contrast, the addition of LiTi2O4 led to full desorption within the temperature range of 150–310 °C. The total weight loss for LiNH2 + LiH + 0.5 mol% LiTi2O4 was 5.7 wt% instead of the 7.5 wt% for LiNH2 + LiH. Accordingly, a lower dehydrogenation temperature indicated that desorption properties can be improved greatly by adding LiTi2O4 as the catalyst.
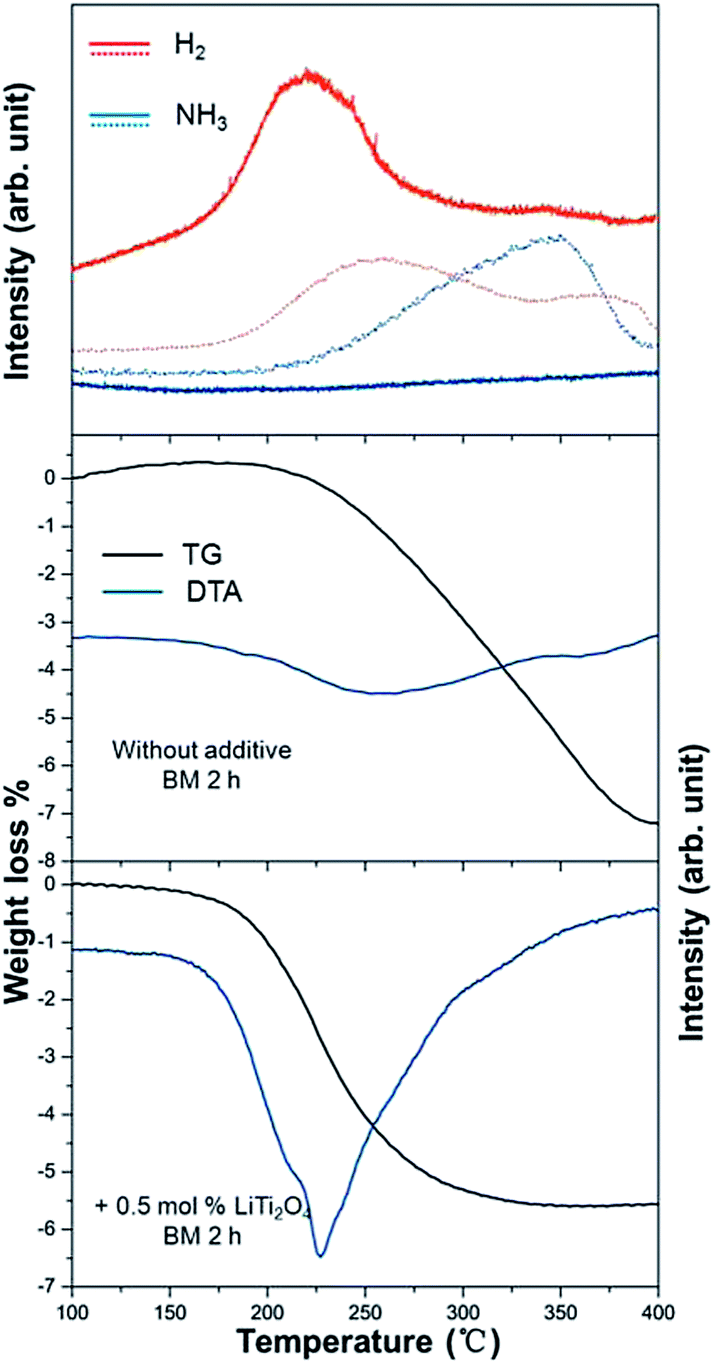 | ||
| Fig. 5 Mass spectra of gas desorption for the dehydrogenation of LiNH2 + LiH mixture without any additive (dot line) and with 0.5 mol% LiTi2O4 (solid line); corresponding weight loss and differential thermal analysis for each sample. | ||
On the basis of these experimental results, it can be figured out that LiTi2O4 worked catalytically for the reactions of lithium alanate, magnesium hydride and lithium amides on hydrogen desorption. As far as the catalytic mechanism of a Ti compound is concerned, some investigations have been performed. For the situations of lithium alanate and magnesium hydride, the explanation of how Ti compounds might work as a catalyst is that it destabilizes the metal–H bonds of the complex metal hydrides, such as the bonding between Mg–H or Al–H. Sandrock et al. suggested that the Ti compound aided the “breaking and re-forming of covalent metal–H bonds”.29 Subsequently, starting from 2005, a number of researchers have investigated similar ideas.30–35 Additionally, Seong Mu Jo certificated that LiTi2O4 nanofibers could absorb/desorb hydrogen at ambient temperature.36 That means LiTi2O4 could probably have a similar effect on helping to weaken the metal–H bond, and suggests the increasing lability of the hydride. The catalytic effect is generated by nanocontacts between the sample and LiTi2O4 in the mechanical milling process.
For the case of a lithium–nitrogen–hydrogen system, David et al. proposed in their report that the reaction mechanism was based on Li+ migration across reactive interfaces between LiH and LiNH2.37 Additionally, our previous results revealed that the catalytic effect is generated by nanocontacts between the sample and the Ti compound in the interface.13 In the present studies, LiTi2O4 has been experimentally proven to have a catalytic effect on the Li–N–H system. The catalytic mechanism might be related to the crystal structure of LiTi2O4. LiTi2O4 has a spinel-structure, where Li+ ions could go through into/out of LiTi2O4 without high energy barriers at ambient temperature.38 Because of these characteristics of the crystal structure, LiTi2O4 has a high diffusion rate of Li+ which is reported to be 10−8 cm2 s−1.39 All of these could help increase Li+ ion mobility in the Li–N–H system. Accordingly, the catalytic effect of LiTi2O4 probably results from the increase of the Li+ ion mobility to improve the reaction kinetics. Meanwhile, the improvement of desorption kinetics could lead to the consumption of LiNH2 and therefore the ammonia emission at high temperature could be restricted.
4. Conclusions
In conclusion, we performed a study on the catalytic effect of LiTi2O4 in complex metal hydrides, such as magnesium hydride, lithium alanate and lithium amide. The purity and stability of LiTi2O4 were confirmed by XRD results. LiTi2O4 was stable after high energy ball milling and heat treatment. After adding LiTi2O4, the dehydrogenation temperature was decreased effectively among the three kinds of materials. The desorption kinetics and the purity of the desorbed hydrogen gas have been improved by doping with LiTi2O4. The catalytic mechanism for complex metal hydrides was also discussed in this study. This is the first time these results have appeared in the literature. It is hoped that they are helpful for hydrogen storage materials as well as fuel cell industries.Acknowledgements
This work was partially supported by L-station (Hokkaido University) and the “Nanotechnology Platform” program of MEXT, Japan. I would like to thank my colleagues, Dr Tao Ma and Mr Yudai Ikarashi, for their cooperation on my work.References
- A. Züttel and L. Schlapbach, Nature, 2001, 414, 353 CrossRef PubMed.
- http://www.hydrogen.energy.gov/ .
- R. von Helmolt and U. Eberle, J. Power Sources, 2007, 165, 833 CrossRef CAS PubMed.
- T. Frankcombe, Chem. Rev., 2012, 112, 2164 CrossRef CAS PubMed.
- H. Wu, ChemPhysChem, 2008, 9, 2157 CrossRef CAS PubMed.
- E. Wieberg, R. Bauer, M. Schmidt and R. Z. Uson, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1951, 6, 393 Search PubMed.
- B. Bogdanović and M. Schwickardi, J. Alloys Compd., 1997, 253, 1 CrossRef.
- T. Ichikawa, N. Hanada, S. Isobe, H. Y. Leng and H. Fujii, J. Alloys Compd., 2005, 404–406, 435 CrossRef CAS PubMed.
- G. Barkhordarian, T. Klassen and R. Bormann, Scr. Mater., 2003, 49, 213 CrossRef CAS.
- W. Oelerich, T. Klassen and R. Bormann, J. Alloys Compd., 2001, 315, 237 CrossRef CAS.
- G. Barkhordarian, T. Klassen and R. Bormann, J. Phys. Chem. B, 2006, 110, 11020 CrossRef CAS PubMed.
- N. Hanada, T. Ichikawa, S. Isobe, T. Nakagawa, K. Tokoyoda, T. Honma, H. Fujii and Y. Kojima, J. Phys. Chem. C, 2009, 113, 13450 CAS.
- T. Zhang, S. Isobe, Y. Wang, N. Hashimoto and S. Ohnuki, RSC Adv., 2013, 3, 6311 RSC.
- P. Kichambare, N. Kijima, S. Honma, S. Ebisu and S. Nagata, J. Phys. Chem. Solids, 1996, 57, 1615 CrossRef CAS.
- F. Xu, Y. C. Liao, M. J. Wang, C. T. Wu, K. F. Chiu and M. K. Wu, J. Low Temp. Phys., 2003, 131, 569 CrossRef CAS.
- M. Rygula, S. Kemmler-Sack, T. Nissel and R. P. Hubener, Ann. Phys., 1993, 2, 685 CrossRef CAS.
- D. W. Murphy, M. Greenblatt, S. M. Zahurak, R. J. Cava, J. V. Waszczak, G. W. Hull, Jr and R. S. Hutton, Rev. Chim. Miner., 1982, 19, 441 CAS.
- E. Moshopoulou, Ph.D. Dissertation, University Joseph Fourier-Grenoble I, Grenoble-France, March 1995.
- L. H. Yang, C. Dong, H. H. Song, J. Guo and G. C. Fu, Chin. Phys. Lett., 2005, 22, 243 CrossRef CAS.
- B. Bogdanović, R. A. Brand, A. Marjanović, M. Schwickardi and J. Tölle, J. Alloys Compd., 2000, 302, 36 CrossRef.
- F. Schüth, B. Bogdanović and M. Felderhoff, Chem. Commun., 2004, 2249 RSC.
- B. Bogdanović, M. Felderhoff and G. Streukens, J. Serb. Chem. Soc., 2009, 74(2), 183 CrossRef.
- H. W. Langmi, G. S. McGrady, X. Liu and C. M. Jensen, J. Phys. Chem. C, 2010, 114, 10666 CAS.
- G. Barkhordarian, T. Klassen and R. Bormann, J. Alloys Compd., 2004, 364, 242 CrossRef CAS.
- S. T. Sabitu and A. J. Goudy, Metals, 2012, 2, 219 CrossRef CAS.
- N. Hanada, T. Ichikawa, S. Hino and H. Fujii, J. Alloys Compd., 2006, 420, 46 CrossRef CAS PubMed.
- N. Hanada, T. Ichikawa and H. Fujii, J. Alloys Compd., 2005, 404, 716 CrossRef PubMed.
- S. Nayebossadri and K. F. Aguey-Zinsou, Phys. Chem. Chem. Phys., 2011, 13, 17683 RSC.
- G. Sandrock, K. Gross and G. Thomas, J. Alloys Compd., 2002, 339, 299 CrossRef CAS.
- P. Wang, X. D. Kang and H. M. Cheng, J. Phys. Chem. B, 2005, 109, 20131 CrossRef CAS PubMed.
- A. Blomqvist, C. M. Araújo, Jena and R. Ahuja, Appl. Phys. Lett., 2007, 90, 141904 CrossRef PubMed.
- T. Vegge, Phys. Chem. Chem. Phys., 2006, 8, 4853 RSC.
- T. Ma, S. Isobe, E. Morita, Y. Wang, N. Hashimoto, S. Ohnuki, T. Kimura, T. Ichikawa and Y. Kojima, Int. J. Hydrogen Energy, 2011, 36, 12319 CrossRef CAS PubMed.
- J. F. Pelletier, J. Huot, M. Sutton and R. Schulz, J. Alloys Compd., 2003, 384, 319 Search PubMed.
- T. Ma, S. Isobe, Y. Wang, N. Hashimoto and S. Ohnuki, J. Phys. Chem. C, 2013, 117, 10302 CAS.
- S. M. Jo, Electrospun Nanofibrous Materials and Their Hydrogen Storage, ed. J. Liu, InTech, 2012, pp. 181–210 Search PubMed.
- W. I. F. David, M. O. Jones, D. H. Gregory, C. M. Jewell, S. R. Johnson, A. Walton and P. P. Edwards, J. Am. Chem. Soc., 2007, 129, 1594 CrossRef CAS PubMed.
- R. J. Cava, D. W. Murphy and S. Zahurak, J. Solid State Chem., 1984, 53, 64 CrossRef CAS.
- O. W. Johnson, Phys. Rev., 1964, 136, A284 CrossRef.
| This journal is © The Royal Society of Chemistry 2014 |