 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A study of dye anchoring points in half-squarylium dyes for dye-sensitized solar cells†
Arthur
Connell
a,
Peter J.
Holliman
*a,
Matthew L.
Davies
a,
Christopher D.
Gwenin
a,
Sophie
Weiss
a,
Mateusz B.
Pitak
b,
Peter N.
Horton
b,
Simon J.
Coles
b and
Graeme
Cooke
c
aSchool of Chemistry, Bangor University, Gwynedd LL57 2UW, UK. E-mail: p.j.holliman@bangor.ac.uk; Fax: +44 (0)1248 370528; Tel: +44 (0)1248 382375
bUK National Crystallography Service, Chemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Highfield Campus, Southampton, SO17 1BJ, UK
cWestCHEM, School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK
First published on 29th January 2014
Abstract
This paper reports the synthesis of a series of new half-squaraine dyes (Hf-SQ) based around a common chromophoric unit consisting of linked indoline and squaric acid moieties. Carboxylate groups have been incorporated onto this core structure at four different points to study the influence of the anchoring group position on dye-sensitized solar cell (DSC) device performance. Dyes have been linked to TiO2 directly through the squaric acid moiety, through a modified squaric acid unit where a vinyl dicyano group has replaced one carbonyl, via an alkyl carboxylate attached to the indole N or through a carboxylate attached to the 4 position of a benzyl indole. Contact angle measurements have been studied to investigate the hydrophobic/hydrophilic properties of the dyes and the results have been compared to N719 and Z907. Full characterization data of all the dyes and synthetic intermediates are reported including single-crystal X-ray structural analysis for dye precursors; the indole (2a) and the half-squarylium esters (3a) and (6b), as well as the dyes (4c), (8) and (12). Dye colours range from yellow to red/brown in solution (λmax range from 430 to 476 nm) with ε ranging from 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 133100 M−1 cm−1. The performance of the dyes in DSCs shows the highest efficiency yet reported for a Hf-SQ dye (η = 5.0%) for 1 cm2 devices with a spectral response ranging from 400 to 700 nm depending on the dye substituents. Co-sensitization of half-squarylium dye (7b) with squaraine dye (SQ2) resulted in a broader spectral response and an improved device efficiency (η = 6.1%). Density functional theory (DFT) calculations and cyclic voltammetry have been used to study the influence of linker position on dye HOMO–LUMO levels and the data has been correlated with I–V and EQE data.
000 to 133100 M−1 cm−1. The performance of the dyes in DSCs shows the highest efficiency yet reported for a Hf-SQ dye (η = 5.0%) for 1 cm2 devices with a spectral response ranging from 400 to 700 nm depending on the dye substituents. Co-sensitization of half-squarylium dye (7b) with squaraine dye (SQ2) resulted in a broader spectral response and an improved device efficiency (η = 6.1%). Density functional theory (DFT) calculations and cyclic voltammetry have been used to study the influence of linker position on dye HOMO–LUMO levels and the data has been correlated with I–V and EQE data.
O'Regan and Grätzel's breakthrough in DSC devices used Ru bipyridyl dye N3 to sensitize nanoparticulate TiO2.1 Since 1991, Ru-bipy dyes have remained a widely used family of DSC dyes.2 However, these dyes are expensive which is partly due to raw material costs but also due to lengthy purification procedures which hinder scaling and they have relatively low molar extinction coefficients (ε) and generally poor spectral response above 600 nm leading to DSC efficiencies of 11.1% for N719 (ref. 3) and 11.4% for C101.4 This has led to the development of “Ru-free” organic DSC dyes (e.g. triphenylamines,5 coumarins6 and indolines7) which absorb in the same region as Ru-bipy dyes (450–600 nm) where AM1.5 solar intensity is highest. Generally, these organic dyes are simpler to purify than Ru-bipy complexes and have significantly higher ε. This allows thinner photo-electrodes to be used which can reduce recombination losses and improve device voltages. High ε is also advantageous when co-sensitizing the TiO2 electrode to broaden spectral response because if fewer dye sorption sites utilized for the dye harvesting light at 400–650 nm, this leaves more space for near infrared (NIR) dyes which absorb at λ > 650 nm.
Extending DSC spectral response remains key to increasing device efficiency. This has led to the development of panchromatic dyes (e.g. “Black dye”,8 squaraine9–12). However, the synthetic and purification procedures associated with such dyes make them expensive and ensuring optimized spectral response across 400–900 nm in a single dye has proved very difficult. Co-sensitizing DSC photo-anodes with combinations of two or more dyes is another approach to increasing light harvesting13–16 which has led to the first reports of ultra-fast co-sensitization17,18 and η > 12% for a combination of porphyrin and triphenylamine dyes.19
Co-sensitization requires dyes which can harvest longer wavelength photons. In this context, squaraines have been studied as DSC dyes20–22 because of their high ε and λmax ≥ 650 nm where Ru-bipy dyes are less effective sensitizers. Half-squarylium (Hf-SQ) dyes are produced as an intermediate during the synthesis of unsymmetrical squaraine dyes.23,24 Hf-SQ dyes are known to be fluorescent25 and have been tested in ZnO giving η = 0.27% (ref. 26) 0.53%,27 and, more recently, in TiO2 DSC devices giving η = 3.54%.28 The Hf-SQ unit is also interesting because it offers a number of locations where a carboxylate anchoring group can be positioned. It is widely believed that the most efficient DSC dyes possess a donor–π bridge–acceptor (D–π–A) configuration.29 In this paper, we have used the synthetic flexibility of the Hf-SQ moiety to investigate the influence of anchor position on DSC performance using a common chromophoric unit. Scheme 1 shows the anchor points tested (A/B = indole aromatic moiety, C = alkyl carboxylate on the indole N, D = carbonyl or vinyl dicyano moiety and E = H+ for an acid or an alkyl group for an ester). We believe this the first report of this type. We have also investigated the effect of varying Hf-SQ side chain on dye/TiO2 surface hydrophobicity. As such, this paper reports the synthesis of new Hf-SQ dyes along with detailed characterization, calculations of HOMO–LUMO energy levels and electron density distributions which have been correlated with DSC device testing. The co-sensitization of Hf-SQ and squaraine dyes has also been studied which offers the potential to reduce dye cost whilst also improving spectral response and device efficiency due to both sensitizers being prepared from the same synthetic pathway.
 | ||
| Scheme 1 Dye anchoring points on half-squaraine chromophore. | ||
Results and discussion
Dye synthesis
The synthetic routes used to produce the Hf-SQ used in this work are shown in Schemes 1 and 2. The first step is the esterification of squaric acid in ethanol to give (1) which was prepared according to the method described by Terpetschnig et al. in good yield with spectroscopic data in line with the literature.30 Compounds (2a–c) and (5a–b) were prepared using a general N-alkyl substitution reaction at the indole nitrogen with various iodo-alkanes.31 All of the materials were prepared in high yield (>90%) with spectroscopic data in line with the literature.31 The molecular structures of (2a), (2b) and (5a) have been confirmed by single crystal X-ray analysis (Fig. 1). Compounds (2a–c) and (5) were reacted with (1) to prepare (3a–c) and (6), respectively. In addition, single crystal analysis of (3a) shows the expected configuration (Fig. 1). Finally, hydrolysis of (3a–c) and (6a–c) using NaOH as described elsewhere20,30 gives the Hf-SQ dyes (4a–c) and (7a–b), respectively. The target compounds (4a–c) and (7a–b) were analyzed using 1H and 13C NMR, high resolution mass spectrometry, UV-visible spectroscopy, I.R. spectroscopy and melting points. In addition, single crystal analysis of (4b) and (4c) sodium salts show the expected molecular configuration as shown in (Fig. 3 and 4, ESI†). The synthetic routes to (8), (10) and (12) are shown in Scheme 2. In situ hydrolysis of (6c) together with nucleophilic substitution of a squaric acid carbonyl with malononitrile was used to prepare compound (8). These reaction conditions have been reported elsewhere in the literature for the preparation of related vinyl dicyano substituted Hf-SQ molecules.23,32 Dye (10) and (12) were prepared by reacting (9)33 and (11)34 with (1), respectively. All of the dyes were isolated in high yield (>75%) and characterised using 1H, 13C NMR, high resolution mass spectrometry, melting points, infrared and UV-visible spectroscopy. The expected molecular configuration (8) (sodium salt) and (12) with the carboxylate linker groups sited on a vinyl dicyano-derivatized squaraine unit and on the benzyl of the indole, respectively have been confirmed by, single crystal X-ray analysis (Fig. 1) and (Fig. 6 ESI†). Whilst some structurally-related Hf-SQ dyes and their precursors have been reported previously,20,30 (5), (6b), (7b) and (8) are variants (see ESI†) with a dodecyl chain attached to the nitrogen of the indole. The spectroscopic data for these new compounds are in line with the previously reported data (see ESI for references†). Dye (10) is significantly different to previously reported Hf-SQ dyes due to the linker being attached to the nitrogen of the indole. However, the 1H and 13C NMR of (10) show resonances in the expected regions for the various functional groups in the molecule and high resolution mass spectrometry has identified the expected mass for this compound. In addition, the expected functional groups, including the carboxylate moiety, have been identified using infrared spectroscopy and (10) has a λmax of 440 nm and an extinction coefficient of 116000 M−1 cm−1 in ethanol measured using UV-visible spectroscopy.
 | ||
| Scheme 2 Synthetic pathways to half-squaraine dyes. | ||
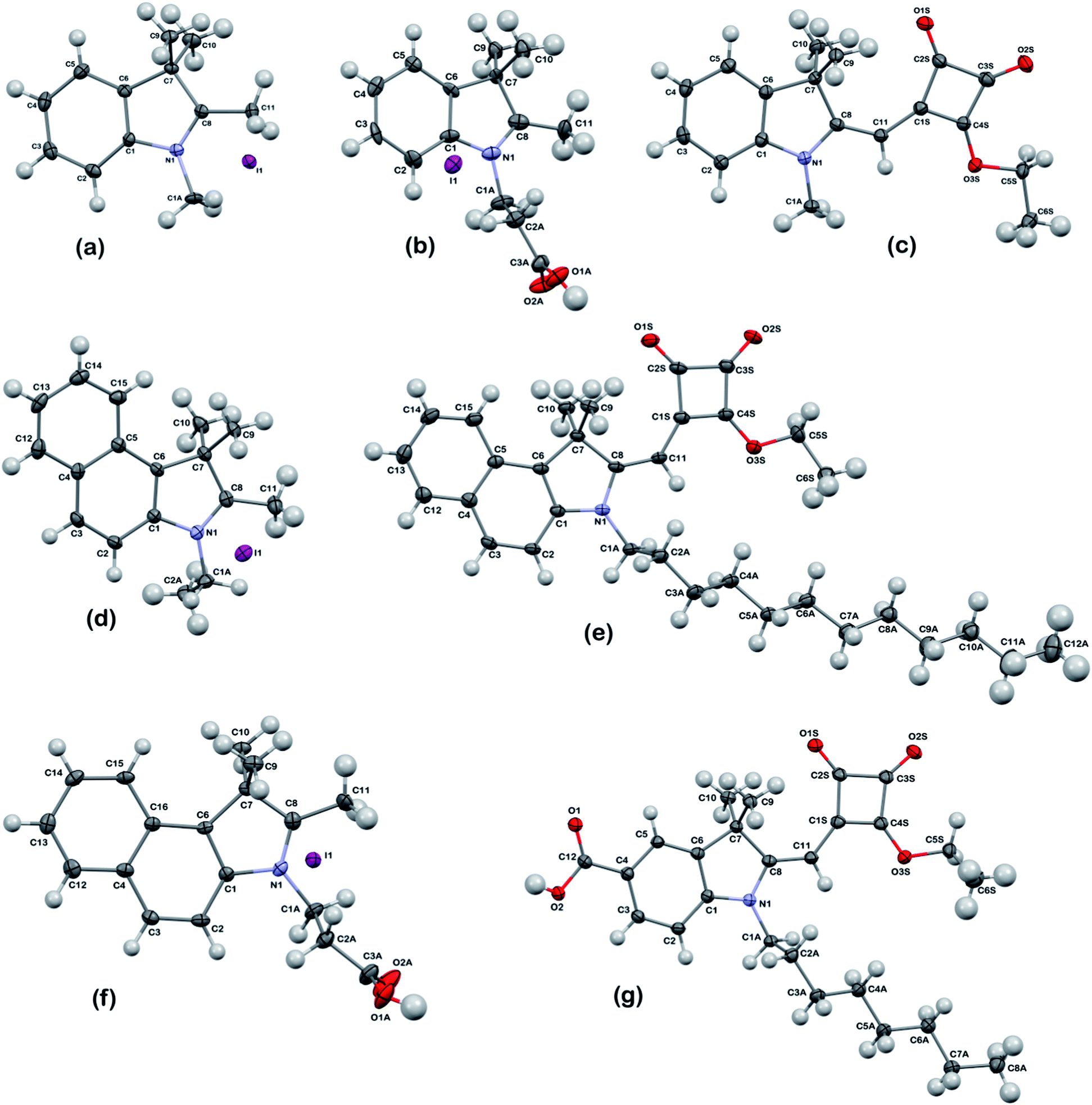 | ||
| Fig. 1 Crystal structures of dye precursors and dyes (a) (2a), (b) (2b), (c) (3a), (d) (5a), (e) (6b), (f) (9) and (g) (12). Displacement ellipsoids – 50% probability. | ||
The new dyes in this paper have been designed with bulky side groups to try to minimize inter-molecular interactions which can give rise to dye aggregation and increased recombination processes between adsorbed dye molecules on the TiO2 surface. This is an established approach to DSC dye design, which has been described previously.2,3 However, this can make it more difficult to grow single crystals for X-ray analysis which hinders structural studies of DSC dyes. Using careful purification and slow evaporation, it was possible to grow suitable crystals of several HF-SQ dyes and precursors reported including; (4c), the esterified precursor of (7b) i.e. (6b), (8) and (12). Analysis of the crystal structure obtained from (4c) i.e. the molecule containing the longest and potentially most hydrophobic alkyl unit (Fig. 4, ESI†) shows a complex structure where the negatively charged squaric acid moiety is balanced by sodium ions but with several molecules H-bonded together through the carbonyl and C–OH of this acyloin moiety (ESI, Fig. 5 and Table 4†). The hydrophobic hexadecyl alkyl chain is then arranged pointing away from the hydrophilic, electrostatically charged squaric acid linker group in the same way that might be observed in a surfactant. Interestingly, this localized arrangement is then extended to the wider crystal packing arrangement so that alternating layers of hydrophilic squaric acid and hydrophobic alkyl chains can be observed (Fig. 5, ESI†). This structural arrangement is important in the context of DSC devices because these dyes were designed with the hope that the charged, hydro-philic squaric acid moiety of the Hf-SQ dyes (4a–c), (7a–b) would chemisorb to the TiO2 surface through a bi-dentate ester-like linkage whilst the long chain alkyl moiety might point away from the surface (Fig. 3). It was also hoped that this localized structure might extend to the inter-molecular packing arrangement of dyes on the TiO2 surface creating a hydrophobic surface and that this might affect electrolyte–dye interactions and device stability in a similar manner to that which occurs for Z907 versus N719.35 To test this hypothesis, contact angles have been measured for TiO2 electrodes dyed either with N719, Z907, (7b), (8), (10) and (12) are shown in Fig. 2 and Table 1. The data show that films dyed with N719 have a lower contact angle and are relatively hydrophilic by comparison to films dyed with Z907 in line with literature reports35,36 for these and other dyes incorporating various hydrophobic molecular entities such as the alkyl chains on Z907.35 Interestingly, although (7b) and (8) both contain dodecyl alkyl chains attached to the nitrogen of the indole moiety, neither dye displays the expected hydrophobic properties that Z907 does with lower contact angles observed than for N719. Neither do (7b) or (8) show enhanced hydrophobicity relative to (10), which does not contain a long chain alkyl unit. By comparison, (12) does show a contact angle similar to Z907 despite possessing a shorter (octyl) alkyl chain compared to the longer, dodecyl unit in (7b) and (8). This suggests that hydrophobicity is also influenced by other factors as well as alkyl chain length. These are believed to include the orientation of individual dye molecules on the surface which will, in turn, influence the orientation of the alkyl chain to the surface and, hence, any inter-molecular dye interactions and wider surface arrangement of dyes. All of these factors are important because contact angles only measure average surface hydrophobicity which results from the collective action of many molecules across the surface. For DSC dyes, such as Z907, where a dye monolayer is believed to form, it is reported that the alkyl chains are orientated towards the periphery of the adsorbed dye such that these groups will be the first part of the molecule in contact with any species approaching the TiO2 surface. For the Hf-SQ dyes, the poor hydrophobicity of (7b) and (8) by comparison to (10) could be due to the alkyl chains being orientated in a way that does not point them towards the periphery of the molecule, or because a complete monolayer has not formed and/or because the alkyl chains do not form a collective surface. This is clearly not the case for (12) and therefore the position of the linker group and alkyl chains result in an orientation of the alkyl chain to repel hydrophilic species. Equilibrium dye loading measurements show that 81.7 μg cm−2 of (7b) sorbs to titania whilst (10) and (12) show comparable and much lower dye loadings at 33.9 and 34.0 μg cm−2. It was not possible to measure the dye loading of (8) because this dye bleached rapidly and completely on exposure to alkali. Dye loading correlates with Jsc for (7b), (10) and (12) with the highest dye loading and Jsc for (7b) whilst lower dye loadings and Jsc are observed for (10) and (12). In addition, the broader spectral response of (10) relative to (12) explains the increased Jsc of (10) compared to (12). The dye loading data further suggest that dye orientation is important and appears to be more influential than dye loading when influencing contact angles (assuming dye uptake is complete). Interestingly, very low contact angles were observed for all dyes when measurements were made using the electrolyte solvent methoxypropionitrile instead of water suggesting complete wetting of the surface should occur in DSC devices. This should ensure the redox couple (I3−/I−) is able to interact with the dye-oxide surface avoiding any dye regeneration limitations on device performance.
 | ||
| Fig. 2 Contact angle measurements and pictures for TiO2 surfaces dyed with (a) N719, (b) Z907, (c) (7b), (d) (8), (e) (10) and (f) (12). | ||
| Surface | Contact angle/° |
|---|---|
| N719 | 45.8 ± 3.0 |
| Z907 | 110.1 ± 5.0 |
| (7b) | 42.3 ± 3.0 |
| (8) | 25.5 ± 3.0 |
| (10) | 38.7 ± 3.0 |
| (12) | 106.2 ± 4.0 |
The optical properties of the dyes (7b), (8), (10) and (12) have been studied both in solution and adsorbed to translucent TiO2 films (Fig. 3). (12) has a λmax at 428 nm (133100 M−1 cm−1) in solution which red shifts to 444 nm when adsorbed to TiO2. Bathochromic shifts are observed for (7b) (λmax 442 nm, 38000 M−1 cm−1) and (10) (λmax 440 nm, 116000 M−1 cm−1) in solution which are attributed to the increased conjugation from the naphthyl indole by comparison to the benzyl indole in (12). (10) also exhibits narrower absorption peaks than (7b) and the solution appears bright orange by comparison to the more diffuse yellow hue of (7b) and (12). Furthermore, a decrease in the extinction coefficient is observed for (7b) compared to (6b) which is similar to observations reported in the literature for other de-esterified Hf-SQ molecules.26,28 A relatively large bathochromic shift and significantly broadened absorption peaks are observed for (7b) and (10) when adsorbed to TiO2 (Fig. 3). To help understand these effects, the HOMO and LUMO maps for molecules (7a), (8), (10) and (12) have been calculated (Fig. 4). All of the dyes show that both the HOMO and LUMO are delocalized throughout the π-framework although there is significantly greater electron density for the LUMO around the squaric acid unit and at the nitrogen of the indole group. This should result in greater electronic interaction between the anchoring unit and TiO2 surface which contributes to the broader absorption spectra for (7a), (8) and (10) which link via these groups by comparison to (12) which links via a benzyl carboxylate. Interestingly, the Hf-SQ–TiO2 interface has also been investigated using DFT calculations by Cicero et al.28 which also shows that the HOMO is delocalized through the π-framework of the whole structure while the LUMO contains a notable electron density contribution arising from the squaric acid group.28 These workers reported that the HOMO–LUMO transition should therefore move electron density to the anchoring group resulting in directional electron transfer to the TiO2 surface.28 A similar phenomenon should also occur in the dyes reported here. Finally, incorporating the vinyl dicyano entity into the Hf-SQ central unit in (8) results in the largest peak broadening and bathochromic shift both in solution (λmax 476 nm, 57700 M−1 cm−1) and when adsorbed to TiO2 films. Whilst this influence of the electron donating vinyl dicyano group is similar to that reported for squaraines,23,32 to the best of our knowledge, this effect has not yet been reported for half-squaraines.
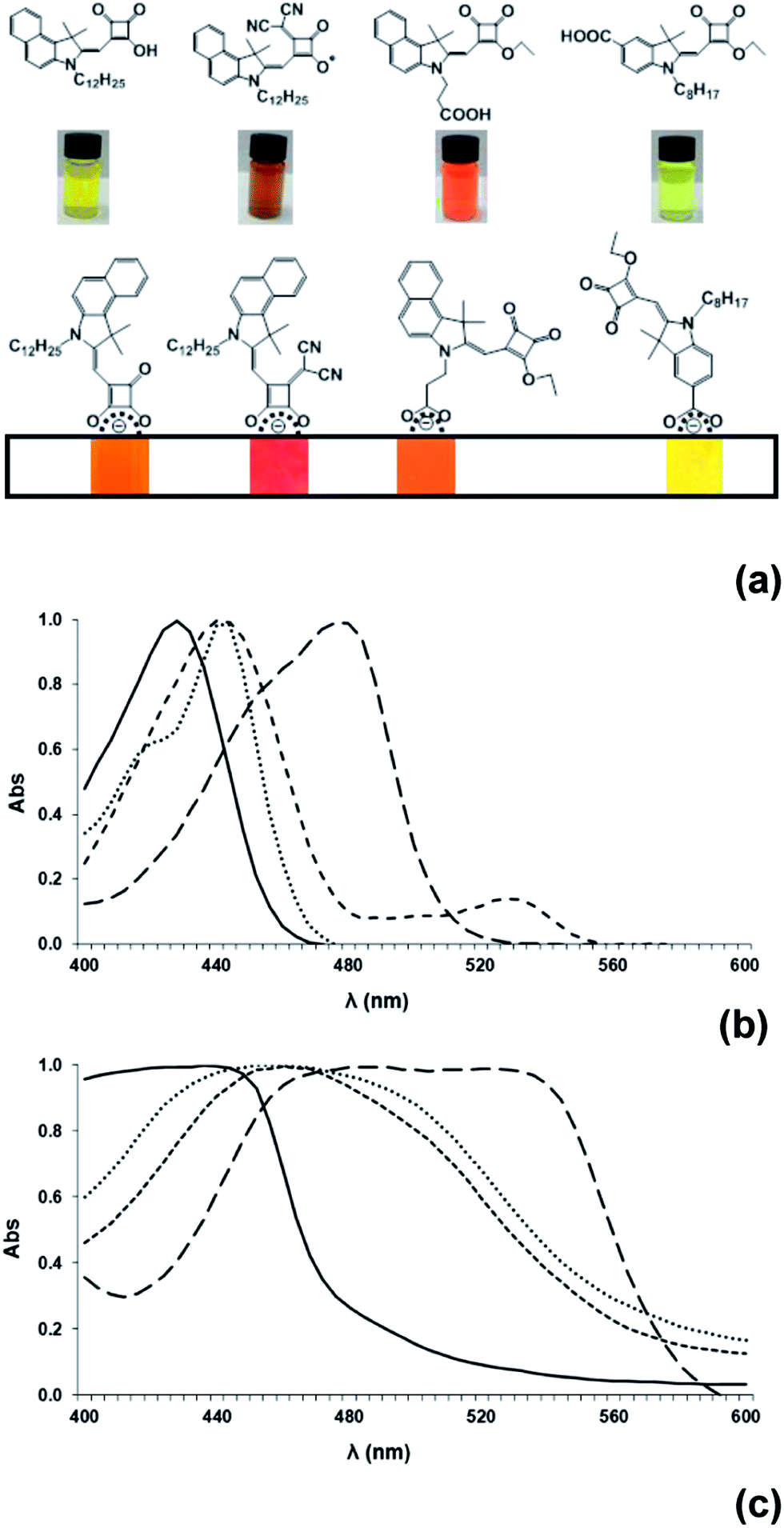 | ||
| Fig. 3 (a) Colours and molecular configurations of selected dyes in solution and sorbed to TiO2 and normalized UV-visible spectra of (b) dyes in solution and (c) dyes adsorbed onto TiO2 films. Data show (7b) dotted line, (8) long dashes, (10) short dashes and (12) solid line. | ||
 | ||
| Fig. 4 Optimized geometries and HOMO and LUMO maps of the methyl indoline derivatives of selected dyes calculated using DFT. | ||
DSC device testing
Table 2 shows I–V data for selected Hf-SQ devices. Initially, DSC devices were prepared with and without 5 mM the additive chenodeoxycholic acid (CDCA). However, the power conversion efficiencies of devices without CDCA were always at least 25% lower than those with CDCA; see e.g. (10) or (12) due to slight improvements in Jsc, Voc and FF. This is in line with published reports that CDCA suppresses aggregation28,32,37 and affects the position of the conduction band resulting in improved electron injection.32,37| Dye | η/% | FF | V oc/V | J sc/mA cm−2 |
|---|---|---|---|---|
| Single dye devices | ||||
| (4a) + CDCA | 3.3 | 0.56 | 0.64 | 9.20 |
| (4b) + CDCA | 3.8 | 0.62 | 0.66 | 9.22 |
| (4c) + CDCA | 3.5 | 0.67 | 0.63 | 7.92 |
| (7a) + CDCA | 4.0 | 0.61 | 0.60 | 10.90 |
| (7b) | 4.5 | 0.66 | 0.65 | 10.40 |
| (7b) + CDCA | 5.0 | 0.68 | 0.71 | 10.25 |
| (8) | 2.8 | 0.47 | 0.53 | 11.11 |
| (10) | 3.5 | 0.65 | 0.67 | 8.12 |
| (10) + CDCA | 3.9 | 0.68 | 0.68 | 8.38 |
| (12) | 2.4 | 0.74 | 0.61 | 5.31 |
| (12) + CDCA | 2.7 | 0.72 | 0.68 | 5.44 |
| N719 only | 6.4 | 0.60 | 0.73 | 14.69 |
| Co-sensitized devices | ||||
| SQ2 + CDCA | 3.4 | 0.67 | 0.62 | 8.25 |
| (7) + SQ2 + CDCA | 6.1 | 0.66 | 0.63 | 14.61 |
Table 2 shows device data for TiO2 devices sensitized with Hf-SQs containing either benzyl or napthyl indoles with alkyl chains of varying in length (ethyl to octadecyl). Comparing the benzyl indole dyes (4a–c) shows very similar device performance data for all three dyes with η ranging from 3.3–3.8%. The variance was mainly due to differences in Jsc which ranges from 7.9–9.2 mA cm−2 with very similar Voc values observed for all three dyes (ca. 0.65 V). The slightly lower Jsc value was observed for the indole with the longest alkyl chain (hexadecyl) may reflect slightly poorer electron injection or dye regeneration from the electrolyte but this is only has a relatively small effect on η. However, after synthesis, this dye was also the most difficult to purify from the iodohexadecane precursor by comparison to the shorter chain iodoalkane dyes and precursors which may also have had a slight negative effect on Jsc for (4c).
Studying the effects of alkyl chain length on the naphthyl indoles of dyes (7a) and (7b) shows two main differences. Firstly, a significant uplift in η is observed for the napthyl indoles (4.0–5.0%) compared to their benzyl indoles which mainly results from increased Jsc (10.2–10.9 mA cm−2). This Jsc increase is attributed to a slight broadening of the absorption spectra due to the extended conjugation of the napthyl indole. This is in line with literature data for squaraines which report improved η for the napthyl indole squaraine, SQ2 versus the benzyl indole, SQ1.20,21 Interestingly, for the napthyl indoles, increasing the length of the alkyl chain attached to the indole N from an ethyl group in (7a) to a dodecyl chain in (7b) increases Voc from 0.61 to 0.71 V respectively. This is the highest Voc observed for the Hf-SQ dyes reported here which, along with an improved FF, gives the highest single dye efficiency (η = 5.0%). In addition, the Voc of 0.61 V observed for (7a) is close to the literature value of 0.64 V reported elsewhere for this dye.28 We believe this to be the highest efficiency Hf-SQ reported to date. The improved Voc and FF may be due to decreased dye aggregation on the TiO2 photo-electrode which could be caused by the longer alkyl chain in (7b) sterically reducing inter-dye interactions. Due to the improved device performance of (7b) by comparison to (4a–c) or (7a), the dye (7b) was taken forward for further study using co-sensitization studies and for comparison with dyes (8), (10) and (12).
Cyclic voltammetry (CV) measurements of (7b), (8), (10) and (12) in solution (10 mM in degassed THF) have been carried out to compare dye oxidation and reduction processes with spectral and DSC device data along with theoretical DFT calculations to try to further examine any structure–activity relationships arising from changes to the dye linker position. The CV data for (7b) show three anodic waves (Fig. 5) similar to that reported in the literature.27
 | ||
| Fig. 5 Cyclic voltammetry data for 5 mM of (8) in degassed THF with [Bu4N] [PF6] as supporting electrolyte (solid line). Dashed line shows CV of supporting electrolyte only. | ||
Shifts in the anodic and cathodic waves were observed for (8) and (10) which were expected from the changes in the electronic spectra of these dyes (Fig. 3). The CV data have been used to calculate the HOMO–LUMO potentials and band gaps versus the NHE and the band gap energies obtained correlate well with the onset of the absorption measured from the solution UV-Visible spectroscopy (Table 3). For example, the bandgap calculated from the absorption onset obtained for (7b), (8) and (10) is 2.20, 2.35 and 2.60 (eV) respectively which correlates with the bandgap energies of 2.23, 2.4 and 2.6 (eV) calculated from the CV data. By comparison, the anodic and cathodic waves measured for (12) are not as clear as for the other dyes (see ESI†). However, a sensible estimation of the HOMO–LUMO energy gap i.e. 2.62 (eV) could still be made by using the bandgap of 2.6 (eV) calculated from absorption onset obtained from solution UV-visible spectroscopy. Furthermore, both the DFT and CV data (Fig. 4 and 5) suggest that the LUMO levels of (7b), (8), (10) and (12) should be at the correct energy for successful electron injection from the dye into the TiO2 conduction band and that the energy of the HOMO levels should enable successful regeneration of the dye using an I−/I3− redox couple in the electrolyte.
| Dye | λ onset/nm | E B calc. from λ onset/eV | E B calc. from CV/eV |
|---|---|---|---|
| (7b) | 556 | 2.20 | 2.23 |
| (8) | 526 | 2.35 | 2.40 |
| (10) | 476 | 2.60 | 2.60 |
| (12) | 472 | 2.62 | 2.5 |
To study the influence of linker position, the performance of (7b), (8), (10) and (12) were evaluated in DSC devices with and without CDCA (Table 2). Interestingly, it was not possible to include CDCA with (8) as this caused desorption of the sensitizer from the TiO2 surface. However, where CDCA was present, device efficiency was consistently improved due to increased Voc as has been widely reported in the literature for various dye systems.38,39 Further desorption of (8) was also observed when an electrolyte was introduced into the device which meant that the I–V data for (8) was measured immediately after electrolyte addition. The desorption problems of (8) meant that, despite having the highest photocurrent of all dyes reported in this study (Jsc > 11 mA cm−2), the overall efficiency was limited to η = 2.8% as a result of much lower Voc and FF which must reflect the significant changes to the photo-electrode surface during desorption. These problems are ascribed to the addition of the vinyl dicyano group to one carbonyl of the squaric acid unit which in (8) must act as the linker group to the TiO2 surface. This new group, rich in π-related electron density, presumably increases steric hindrance at the dye sorption site. Whilst this does not prevent the formation of an ester link between the dye and TiO2 surface, it does appear to weaken this interaction to the extent that dye desorption occurs more easily. Indeed, desorption in the presence of the electrolyte suggests a dye binding energy of the same order as iodide which means the dye and electrolyte anions are competing for surface sorption sites. The instability of (8) is further evidenced by the fact that it bleaches on exposure to alkali (see earlier dye loading discussion).
Fig. 6 shows that the spectral responses of (7b), (8), (10) and (12) are all slightly red shifted by comparison to the corresponding absorption spectra of TiO2 dyed films (Fig. 3). The narrowest spectral response and absorption data are observed for (12) which correlate with the lowest Jsc and η of these four dyes. Interestingly, the carboxylate linker in (12) is placed on the benzene ring and so is expected to lie furthest from greatest electron density of the LUMO on the squaraine moiety. In addition, (12) has the least conjugation of the 4 dyes tested here which limits the extent of spectral response. This is despite this dye having the highest ε of the four dyes tested here (133100 M−1 cm−1) which reflects the importance of electron injection over simply light harvesting. By comparison, the broadest spectral response (out to ca. 650 nm) and absorption data are observed for (8) which reflect the much greater conjugation in this molecule arising from the addition of the napthyl indole and vinyl dicyano moieties. The broader light harvesting gives rise to the highest Jsc despite this dye having a moderate ε of (57700 M−1 cm−1). Dye (10) shows a slightly broader spectral response compared to (12) at ca. 400 nm along with a shoulder at ca. 575 nm which is reflected in the higher Jsc; 8.12 versus 5.44 mA cm−2. These two dyes have almost identically high ε relative to (7b) and (8) and although Jsc is improved for (10), it is lower than for (7b) which presumably is related the fact that the linker group is attached to the N of the indole. This means the linker is closer to the LUMO electron density on the squaraine ester but that electron injection must occur through the N of the indole. In squaraine dyes, it has been reported that charge generation occurs around the squaraine moiety during photo-excitation40,41 but that electron transfer through the indole nitrogen such as that which occurs in the dye B1 is less efficient than through a pathway composed entirely of carbon and hydrogen atoms as occurs in SQ1.20,33 The highest spectral response is observed for (7b) which approaches 75% between ca. 410 and 550 nm. This is despite (7b) having the lowest ε of these 4 dyes (38000 M−1 cm−1). This is ascribed to the linker group being the squaric acid moiety where DFT calculations (Fig. 4) show that the greatest LUMO electron density is found. Hence, electron injection should be enhanced by the proximity of the LUMO to the TiO2 surface resulting in increased Jsc (over 10 mA cm−2) despite significantly narrower spectral response than (8). To the best of our knowledge (7b) gives rise to the highest efficiency reported for a Hf-SQ DSC dye to date.
 | ||
| Fig. 6 EQE of (a) (7b) dotted line, (8) long dashes, (10) short dashes and (12) solid line and (b) (7b) dotted line and (7b) + SQ2. | ||
In an attempt to improve device and broaden the spectral response further towards the NIR, (7b) was co-sensitized with squaraine dye SQ2 using a fast pump dyeing method reported elsewhere.17 The spectral response for a co-dyed (7b)/SQ2 device shows significantly greater light harvesting out to ca. 700 nm leading to a significant increase in Jsc to 14.61 mA cm−2 and η = 6.1% (Table 2). By comparison, an efficiency of 6.4% was obtained for a device sensitized using N719. In addition, the performance of this co-dyed device is very similar to many panchromatic SQ dyes reported9–11 but has the advantage that a complicated synthesis is not required to incorporate the molecular entities required for higher energy photon harvesting across the solar spectrum. This could encourage research efforts to be directed towards incorporating molecular entities into dye systems that broaden the spectral response further into the near infrared as another approach alongside the development of panchromatic responses from individual dye systems.
Experimental
Instrumentation and chemicals
All chemicals were purchased from Aldrich and used as supplied unless otherwise stated. Anhydrous solvents were used as purchased except THF which was dried with Na wire. NMR spectra were recorded on a Bruker AC500 instrument operating at 500 MHz for 1H and 125 MHz for 13C. Chemical shifts (δ) are given in ppm relative to tetramethylsilane (for 1H and 13C). J values are given in Hz and refer to JH,H unless otherwise indicated. Mass spectra were recorded using electron impact, chemical ionisation (NH3), fast atom bombardment, electrospray, matrix-assisted laser desorption ionisation and laser desorption ionisation at the EPSRC National Mass Spectrometry Service at the University of Swansea. Infrared spectra were recorded on a Perkin-Elmer 1600 series FTIR spectrometer. Elemental analysis was performed on a Carlo Erba EA1108 elemental analyser. Dye loadings were measured by desorption from 1 cm2 TiO2 electrodes using 40 mM Bu4NOH in 1:1 v/v ethanol–water according the method described previously.42 The concentration of dye in solution was measured using UV/Visible spectroscopy and dye loadings were calculated (μg cm−2) using the appropriate ε (see ESI†) and the Beer–Lambert Law.
Cyclic voltammetry was measured on dyes (5 mM) at room temperature in N2-saturated anhydrous tetrahydrofuran (THF) using 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) as the supporting electrolyte. Measurements were performed using an Autolab PGSTAT 30 computer-controlled electrochemical measurement system (Eco Chemie, Holland). with a sweep rate of 0.05 V s−1 in a three-electrode cell with a glassy carbon (1 cm2) working electrode, a silver/silver chloride (Ag/AgCl) reference electrode and a Pt sheet (1 cm2) counter electrode. All voltammetric potentials were re-calculated and are reported versus NHE.
X-ray crystallography
Single-crystal X-ray diffraction analyses of 3a, 4c_salt and 5a were performed at 120 K using a Bruker APEXII CCD diffractometer mounted at the window of a Bruker FR591 rotating anode (Mo Kα, λ = 0.71073 Å) and equipped with an Oxford Cryosystems Cryostream device. Data were processed using the COLLECT package.43The X-ray data for compounds 2a, 2b, 4b_salt, 6b, 6c, 9, 12(I) and 12(II) were collected at 100 K on Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn 724+ detector mounted at the window of an FR-E+ Superbright MoKα rotating anode generator with HF Varimax optics.44
The X-ray data for compound 8_salt were collected with the use of synchrotron radiation at Diamond Light Source UK, beamline I19 (λ = 0.6889 Å) on a Crystal Logic diffractometer and Rigaku Saturn 724+ detector.
Unit cell parameters were refined against all data. For 3a, 4c_salt, 5a an empirical absorption correction was carried out using SADABS,45 whereas for 2a, 2b, 4b_salt, 6b, 6c, 9, 12(I) and 12(II) CrystalClear46 software was used.
All structures were solved by direct methods and refined on Fo2 by full-matrix least-squares refinements using programs of the SHELX-97/SHELX-2013 software.47 All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were added at calculated positions and refined using a riding model with isotropic displacement parameters based on the equivalent isotropic displacement parameter (Ueq) of the parent atom.
The crystal structure of 4c_salt is non-merohedraly twinned (180° rotation around 001 reciprocal vector). EvalCCD48 was used to integrate the domains and the structure was refined using the HKLF 5 instruction in Shelxl.47 The refined percentage ratio of the twin domains was 25:75 (refined BASF = 0.25714).
The crystal data for 8_salt is not of the highest quality, but is sufficient to prove structural connectivity.
The long aliphatic chains present in the crystal structures of 8_salt and 12(II) are disordered and modelled over two sites.
For disordered components vibrational restraints (SIMU/DELU), similar displacement restraints (EADP) and distance/angle restraints DFIX/DANG used to maintain sensible geometries and atomic displacement ellipsoids. Some atoms required ISOR restraint to approximate isotropic behaviour. Crystal structure of 4b_salt exhibits positional disorder which has been modelled over two sites. Additionally, in 4b_salt highly diffused solvent electron density has been removed with PLATON/SQUEEZE routine.49
2a: a = 6.944(2) Å, b = 11.434(4) Å, c = 15.743(6) Å, α = β = γ = 90°; V = 1249.9(8) Å3, orthorhombic, P212121, Z = 4, ρcalc = 1.600 Mg m−3; μ = 2.527 mm−1; T = 100 K; θmax = 27.48°, 8462 measured reflections, 2810 unique reflections [Rint = 0.0230], 2696 with F2 > 2σ, R(F, F2 > 2σ) = 0.0151; Rw(F2, all data) = 0.0385; Δρmin/max = 0.370/−0.230 e Å−3; CCDC: 977417.
2b: a = 7.4137(5) Å, b = 13.6430(12) Å, c = 29.756(3) Å, α = β = γ = 90°; V = 3009.7(5) Å3, orthorhombic, Pbca, Z = 8, ρcalc = 1.585 Mg m−3; μ = 2.123 mm−1; T = 100 K; θmax = 25.02°, 12057 measured reflections, 2644 unique reflections [Rint = 0.1625], 1333 with F2 > 2σ, R(F, F2 > 2σ) = 0.0486; Rw(F2, all data) = 0.0690; Δρmin/max = 0.974/−0.595 e Å−3; CCDC: 977418.
3a: a = 7.2314(2) Å, b = 9.4760(3) Å, c = 11.6165(4) Å, α = 82.094(2)°, β = 82.082(2)°, γ = 78.631(2)°; V = 767.95(4) Å3, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, ρcalc = 1.286 Mg m−3; μ = 0.087 mm−1; T = 120 K; θmax = 27.48°, 13277 measured reflections, 3509 unique reflections [Rint = 0.0435], 2951 with F2 > 2σ, R(F, F2 > 2σ) = 0.0542; Rw(F2, all data) = 0.1243; Δρmin/max = 0.261/−0.212 e Å−3; CCDC: 977419.
, Z = 2, ρcalc = 1.286 Mg m−3; μ = 0.087 mm−1; T = 120 K; θmax = 27.48°, 13277 measured reflections, 3509 unique reflections [Rint = 0.0435], 2951 with F2 > 2σ, R(F, F2 > 2σ) = 0.0542; Rw(F2, all data) = 0.1243; Δρmin/max = 0.261/−0.212 e Å−3; CCDC: 977419.
4b_salt: a = 8.810(4) Å, b = 12.003(5) Å, c = 17.977(8) Å, α = 82.678(14)°, β = 86.660(17)°, γ = 74.635(14)°; V = 1817.5(14) Å3, triclinic, P, Z = 2, ρcalc = 1.211 Mg m−3; μ = 0.108 mm−1; T = 100 K; θmax = 25.06°, 26587 measured reflections, 6434 unique reflections [Rint = 0.0518], 4237 with F2 > 2σ, R(F, F2 > 2σ) = 0.0751; Rw(F2, all data) = 0.2457; Δρmin/max = 0.747/−0.525 e Å−3; CCDC: 977420.
4c_salt: a = 8.6755(7) Å, b = 10.3795(8) Å, c = 35.240(3) Å, α = 92.172(2)°, β = 91.370(2)°, γ = 99.956(3)°; V = 3121.7(4) Å3, triclinic, P, Z = 2, ρcalc = 1.155 Mg m−3; μ = 0.087 mm−1; T = 120 K; θmax = 24.71°, 10081 measured reflections, 10081 unique reflections [Rint = 0.000], 8831 with F2 > 2σ, R(F, F2 > 2σ) = 0.0958; Rw(F2, all data) = 0.2473; Δρmin/max = 0.926/−0.404 e Å−3; CCDC: 977421.
5a: a = 15.3349(3) Å, b = 7.5158(2) Å, c = 13.7564(2) Å, α = 90°, β = 92.5450(10)°, γ = 90°; V = 1583.92(6) Å3, monoclinic, P21/c, Z = 4, ρcalc = 1.532 Mg m−3; μ = 2.010 mm−1; T = 120 K; θmax = 27.48°, 14035 measured reflections, 2792 unique reflections [Rint = 0.0390], 2592 with F2 > 2σ, R(F, F2 > 2σ) = 0.0217; Rw(F2, all data) = 0.0528; Δρmin/max = 0.366/−0.367 e Å−3; CCDC: 977422.
6b: a = 19.2363(14) Å, b = 10.2830(7) Å, c = 29.048(2) Å, α = 90°, β = 92.704(2)°, γ = 90°; V = 5739.5(7) Å3, monoclinic, C2/c, Z = 8, ρcalc = 1.161 Mg m−3; μ = 0.073 mm−1; T = 100 K; θmax = 25.02°, 15920 measured reflections, 5046 unique reflections [Rint = 0.0461], 4129 with F2 > 2σ, R(F, F2 > 2σ) = 0.0401; Rw(F2, all data) = 0.1077; Δρmin/max = 0.194/−0.182 e Å−3; CCDC: 977423.
6c: a = 9.802(13) Å, b = 9.8259(10) Å, c = 15.69(2) Å, α = 83.509(10)°, β = 89.40(9)°, γ = 66.40(6)°; V = 1375(3) Å3, triclinic, P, Z = 2, ρcalc = 1.178 Mg m−3; μ = 0.074 mm−1; T = 100 K; θmax = 25.03°, 15659 measured reflections, 4847 unique reflections [Rint = 0.0554], 3510 with F2 > 2σ, R(F, F2 > 2σ) = 0.0848; Rw(F2, all data) = 0.2128; Δρmin/max = 0.593/−0.330 e Å−3; CCDC: 977424.
8_salt: a = 11.9454(11) Å, b = 12.4652(11) Å, c = 24.378(2) Å, α = 90.54(2)°, β = 101.50(2)°, γ = 118.10(3)°; V = 3115.3(5) Å3, triclinic, P, Z = 2, ρcalc = 1.210 Mg m−3; μ = 0.084 mm−1; T = 100 K; θmax = 24.21°, 22317 measured reflections, 10395 unique reflections [Rint = 0.2525], 2921 with F2 > 2σ, R(F, F2 > 2σ) = 0.1277; Rw(F2, all data) = 0.4276; Δρmin/max = 0.459/−0.422 e Å−3; CCDC: 977425.
9: a = 7.530(2) Å, b = 13.635(3) Å, c = 34.164(8) Å, α = β = γ = 90°; V = 3507.9(15) Å3, orthorhombic, Pbca, Z = 8, ρcalc = 1.550 Mg m−3; μ = 1.832 mm−1; T = 100 K; θmax = 27.48°, 21886 measured reflections, 3992 unique reflections [Rint = 0.0666], 3503 with F2 > 2σ, R(F, F2 > 2σ) = 0.0595; Rw(F2, all data) = 0.0896; Δρmin/max = 0.908/−0.631 e Å−3; CCDC: 977426.
12 (polymorph I): a = 12.7035(9) Å, b = 18.2291(13) Å, c = 11.1693(8) Å, α = 90.0°, β = 111.529(2)°, γ = 90.0°; V = 2406.1(3) Å3, monoclinic, P21/c, Z = 4, ρcalc = 1.213 Mg m−3; μ = 0.083 mm−1; T = 100 K; θmax = 27.48°, 30172 measured reflections, 5504 unique reflections [Rint = 0.0562], 4195 with F2 > 2σ, R(F, F2 > 2σ) = 0.0406; Rw(F2, all data) = 0.1137; Δρmin/max = 0.305/−0.199 e Å−3; CCDC: 977427.
12 (polymorph II): a = 10.8885(8) Å, b = 17.3070(11) Å, c = 27.892(2) Å, α = 78.372(3)°, β = 80.234(3)°, γ = 77.874(3)°; V = 4988.8(6) Å3, triclinic, P, Z = 2, ρcalc = 1.178 Mg m−3; μ = 0.081 mm 1; T = 100 K; θmax = 25.028°, 47382 measured reflections, 17294 unique reflections [Rint = 0.0964], 9296 with F2 > 2σ, R(F, F2 > 2σ) = 0.1066; Rw (F2, all data) = 0.3071; min/max = 1.222/−0.468 e Å−3; CCDC: 977428.
Theoretical calculations
All calculations were performed using Spartan '08.50 Geometries were first optimized semi-empirically (AM1) and then re-optimized using DFT (B3LYP/6-31G*) level of theory. The optimized structures were shown to be local minima on their respective potential energy surfaces, as none of the vibrational frequencies in the optimized geometries generated imaginary frequencies.Preparation of 1,1,2-trimethyl-1-dodecyl-1H-benzo[e]indole (5b)
A mixture of 1,1,2-trimethyl-1H-benzo[e]indole (8 g, 38 mmol) and dodecyl iodide (25 g, 87 mmol) was heated under reflux overnight under nitrogen in anhydrous acetonitrile (80 ml). After cooling, the solvent was removed in vacuo and the product was re-crystallized from ethyl acetate to give a green solid. (Yield 17.3 g, 90%.)1H NMR (400 MHz, CDCl3) δ: 0.86 (q, J = 6.7 Hz, 2H), 1.03–1.41 (m, 14H), 1.41–1.60 (m, 1H), 1.73–1.91 (m, 4H), 1.97 (dd, J = 15.2 Hz, 7.8, 1H), 3.13–3.22 (m, 2H), 4.77 (t, J = 7.7 Hz, 1H), 7.72 (ddd, J = 29.6 Hz, 17.8 Hz, 8.1 Hz, 2H), 7.99–8.15 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 7.41, 14.10, 15.27, 16.90, 22.64, 22.66, 22.73, 26.80, 28.18, 28.52, 29.13, 29.27, 29.31, 29.39, 29.44, 29.53, 29.59, 30.48, 31.85, 31.88, 33.55, 50.37, 55.30, 55.95, 76.83, 77.15, 77.36, 77.46, 112.53, 119.75, 122.49, 122.89, 124.41, 126.33, 127.65, 127.84, 128.69, 130.06, 131.47, 133.69, 137.17, 138.21, 195.17. MS (FTMS+) M+ calculated = 378, M+ observed = 378. m/z accurate mass (FTMS+), M+ calculated = 378.1355, M+ observed = 378.1359.
Preparation of 3-ethoxy-4-[(1-dodecyl-1,3-dihydro-3,3-dimethyl-2H-benzo[e]indol-2-ylidene)methyl]-3-cyclobutene-1,2-dione (6b)
(Yield 3 g, 65%). 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.27 (s, 16H), 1.38–1.54 (m, 4H), 1.58 (t, J = 7.1 Hz, 3H), 1.96–1.60 (m, 9H), 3.95 (t, J = 7.5 Hz, 2H), 4.95 (q, J = 7.1 Hz, 2H), 5.48 (s, 1H), 7.20–7.31 (m, 1H), 7.39 (t, J = 7.4 Hz, 1H), 7.55 (t, J = 7.3 Hz, 1H), 7.87 (dd, J = 13.4 Hz, 8.5, 2H), 8.12 (d, J = 8.5 Hz, 1H), 13C NMR (101 MHz, CDCl3) δ 14.13, 16.01, 22.68, 26.71, 26.99, 28.05, 29.32, 29.47, 29.54, 29.61, 31.91, 43.12, 49.84, 69.86, 76.78, 77.10, 77.30, 77.42, 80.90, 109.80, 122.22, 123.77, 127.19, 128.59, 129.58, 129.74, 130.82, 132.51, 139.82, 170.33, 173.30, 187.09, 187.14, 192.71. MS (FTMS+) [M + H]+ calculated = 488, [M + H]+ observed = 488, m/z accurate mass (FTMS+), reference compound: NH4OAc, [M + H]+ calculated = 488.3159, [M + H]+ observed = 488.3153. FT-IR (KBr) ν/cm−1 2988 (w), 1736 (m), 1707 (s).Preparation of 4-[(1-dodecyl-1,3-dihydro-3,3-dimethyl-2H-benzo[e]indol-2-ylidene)methyl]-3-cyclobutene-1,2-dione (7b)
(Yield 2.5 g, 70%). 1H NMR (400 MHz, MeOD) δ 0.89 (t, J = 6.8 Hz, 4H), 0.99–1.52 (m, 57H), 1.42 (dq, J = 14.6 Hz, 7.3, 19H), 1.56–1.91 (m, 18H), 1.95 (d, J = 5.9 Hz, 10H), 3.24 (dd, J = 10.0 Hz, 7.1 Hz, 14H), 3.33 (dt, J = 3.2 Hz, 1.6, 2H), 3.94 (t, J = 7.3 Hz, 2H), 4.90 (s, 2H), 5.73 (s, 1H), 7.28 (dd, J = 7.9 Hz, 5.0 Hz, 2H), 7.47 (s, 1H), 7.76–7.89 (m, 2H), 8.12 (d, J = 8.5 Hz, 1H). 13C NMR (101 MHz, MeOD) δ 12.58, 13.08, 19.31, 19.32, 19.34, 21.36, 22.32, 23.39, 26.34, 26.61, 27.93, 29.02, 29.17, 29.19, 29.26, 29.32, 31.66, 42.11, 46.98, 47.20, 47.41, 47.62, 47.84, 48.05, 48.26, 48.72, 48.75, 49.25, 58.08, 58.11, 58.14, 82.60, 109.52, 121.66, 122.32, 126.27, 128.93, 128.96, 129.39, 130.29, 130.73, 140.97, 164.76, 176.55, 179.71, 196.59, 203.63. MS (FTMS+) [M + H]+ calculated = 474, [M + H]+ observed = 474, m/z accurate mass (FTMS+), reference compound: NH4OAc, [M + H]+ calculated = 474.3003, [M + H]+ observed = 474.2998. FT-IR (KBr) ν/cm−1 3473–3010 (m), 2960 (m), 1740 (s), 1600 (s). UV-Visible λmax 442 nm (38000 M−1 cm−1) in ethanol.
Preparation of triethylammonium-2-[(1-dodecyl-3,3-dimethyl-1,3-dihydro-2H-indol-2-ylidene)-3-(dicyanomethylidene)-4-oxocyclobut-1-en-1-olate (8)
Triethylamine (1 ml, 7.14 mmol) was added dropwise to a mixture of (6b) (2 g, 6.15 mmol) and malononitrile (440 mg, 6.66 mmol) in ethanol (35 ml) and stirred for 2 h at room temperature. The solvent was removed in vacuo and the product was obtained as an orange solid after purification by column chromatography (SiO2) with chloroform and methanol as eluent. (Yield 4.15 g, 75%).
1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 6.8 Hz, 4H), 1.06–1.61 (m, 33H), 1.61–2.01 (m, 11H), 3.33 (q, J = 7.3 Hz, 7H), 3.94 (s, 2H), 6.21 (s, 1H), 7.22 (d, J = 8.7 Hz, 1H), 7.28 (s, 3H), 7.35 (d, J = 7.6 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.85 (dd, J = 19.3 Hz, 8.4, 2H), 8.12 (d, J = 8.5 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 8.91, 14.12, 15.83, 22.68, 26.80, 29.33, 29.51, 29.60, 31.90, 39.20, 46.83, 69.71, 76.72, 77.04, 77.24, 77.35, 116.92, 118.39, 172.74, 179.07, 181.96, 192.83. MS (FTMS+) M+ calculated = 623, M+ observed = 623. m/z accurate mass (FTMS+), M+ calculated = 623.4320, M+ observed = 623.4317. FT-IR (KBr) ν/cm−1 3473–3010 (s), 2960 (s), 2230 (s), 2210 (s), 1740 (s), 1600 (s). UV-Visible λmax 476 nm (57700 M−1 cm−1) in ethanol.
Preparation of 3-ethoxy-(2-carboxyethyl)-[1,1,2-trimethyl-1H-benzo[e]indolium iodide-2-ylidene)methyl]-3-cyclobutene-1,2-dione (10)
(3 g, 70%). 1H NMR (400 MHz, THF) δ 1.42 (t, J = 7.3 Hz, 8H), 1.47–1.89 (m, 38H), 1.94 (s, 13H), 2.08 (s, 1H), 2.76 (t, J = 7.3 Hz, 5H), 3.16 (q, J = 7.3 Hz, 6H), 3.30 (s, 1H), 3.62 (s, 23H), 4.32 (t, J = 7.3 Hz, 5H), 4.90 (q, J = 7.1 Hz, 5H), 5.53 (s, 2H), 7.35 (t, J = 7.5 Hz, 3H), 7.46–7.58 (m, 5H), 7.89 (dd, J = 8.3 Hz, 5.7, 5H), 8.22 (d, J = 8.5 Hz, 2H). 13C NMR (101 MHz, THF) δ 5.83, 13.37, 24.18, 28.53, 36.63, 43.61, 47.56, 67.60, 79.00, 108.23, 120.23, 121.28, 124.79, 126.79, 127.34, 127.59, 129.09, 129.82, 138.01, 166.88, 169.68, 171.21, 185.32, 186.53, 189.90. MS (FTMS+) [M + H]+ calculated = 406, [M + H]+ observed = 406, m/z accurate mass (FTMS+), reference compound: NH4OAc, [M + H]+ calculated = 406.1649, [M + H]+ observed = 406.1647. FT-IR (KBr) ν/cm−1 3543–3060 (s), 2978 (w), 1738 (s), 1707 (s). UV-Visible λmax 440 nm (116000 M−1 cm−1) in ethanol.
Device manufacture and testing
Photo-electrodes (0.5 × 2 cm = 1.0 cm−2) were prepared by doctor blading two layers of TiO2 colloid (DSL18NRT, Dyesol) followed by one layer of scattering TiO2 colloid (WER4-O, Dyesol) onto fluorine tin oxide-coated glass (TEC15, 15 Ω □−1). Each TiO2 layer was calcined at 723 K for 30 min, then immersed in TiCl4:THF2 in deionised water (50 mM) at 343 K for 30 min, rinsed with de-ionised water, air-dried for 10 min and re-sintered at 723 K for 30 min. Counter electrodes were prepared by drilling two holes ca. 2 cm apart through TEC8™ glass (NSG), washing with iPrOH and drying in air before depositing Pt colloid (Pt-1, Dyesol) and sintering at 400 °C for 30 min. The two electrodes were sealed together using a Surlyn™ (DuPont) gasket at 120 °C. Contacts were applied onto both electrodes using conductive silver paint (Agar).
Current–voltage characteristics were measured using an ABET Solar Simmulator with Xe arc lamp and a Keithley 2400 at 100 mW cm−2 or 1 Sun between 0 and 1 V. Spectral response measurements were made on a QEX10 Quantum Efficiency Measurement System in DC mode at resolution of 10 nm. Lamps were calibrated to 1 Sun (100 mW cm−2) using a certified (Oriel 91150V) monocrystalline silicon reference cell traceable to the National Renewable Energy Laboratory (NREL).
Conclusions
This study shows that the position of dye anchoring points in half-squarylium dyes influences hydrophobicity and contact angle of dyes sorbed to TiO2 surfaces. These phenomena are ascribed to the orientation of both individual dyes on the surface and the way in which sorbed dyes interact with each other such that hydrophobic surfaces consist of dye monolayers with hydrophobic chains perpendicularly arranged to the surface. Cyclic voltammetry measurements suggest the LUMO levels of all the dyes tested should be correctly positioned to enable electron injection from the dye excited state. However, the position of the linker group on the chromophore does affect the dye performance in DSC devices where improved Jsc and Voc are observed for dyes where DFT calculations suggest the LUMO is located near to the squaraine linker moiety. Finally, we believe this is the first report of the ultra-fast co-sensitization of a dye precursor (Hf-SQ) with the associated full dye (squaraine) which is important in the context of extending spectral response whilst minimizing dye costs. Thus, co-sensitizing with two dyes from the same synthetic pathway reduces synthetic costs and results in similar device performance to panchromatic squaraines reported in the literature, which require much more complex and expensive synthetic procedures.Acknowledgements
We gratefully acknowledge ERDF-WAG LCRI funding for SPARC (AC), EPSRC/TSB funding for SPECIFIC (EP/I019278/1) for MLD and AC, and the EPSRC UK National Mass Spectrometry Facility at Swansea University. We thank EPSRC for funding the National Crystallography Service (Southampton and Newcastle), and Diamond Light Source for access to synchrotron facilities.Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CAS.
- A. Hagfeldt, G. Boschloo, L. C. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835 CrossRef CAS PubMed.
- Y. Cao, Y. Bai, Q. Yu, Y. Cheng, S. Liu, D. Shi, F. Gao and P. Wang, J. Phys. Chem. C, 2009, 113, 6290 CAS.
- H. Im, S. Kim, C. Park, S.-H. Jang, C.-J. Kim, K. Kim, N.-G. Park and C. Kim, Chem. Commun., 2010, 46, 1335 RSC.
- K. Hara, Z. S. Wang, T. Sato, A. Furube, R. Katoh, H. Sugihara, Y. D. Oh, C. Kasada, A. Shinpo and S. Suga, J. Phys. Chem. B, 2005, 109, 15476 CrossRef CAS PubMed.
- S. Ito, S. M. Zakeeruddin, R. Humphry-Baker, P. Liska, R. Charvet, P. Comte, M. K. Nazeeruddin, P. Pechy, M. Takata, H. Miura, S. Uchida and M. Grätzel, Adv. Mater., 2006, 18, 1202 CrossRef CAS.
- M. K. Nazeeruddin, P. Péchy and M. Grätzel, Chem. Commun., 1997, 1705 RSC.
- S. Pack, H. Choi, C. Kim, N. Cho, S. So, K. Song, M. K. Nazeeruddin and J. Ko, Chem. Commun., 2011, 47, 2874 RSC.
- J. H. Delcamp, Y. Shi, J.-H. Yum, T. Sajoto, E. Dell'Orto, S. Barlow, M. K. Nazeeruddin, S. R. Marder and M. Grätzel, Chem. – Eur. J., 2013, 19, 1819 CrossRef CAS PubMed.
- Y. Shi, R. B. M. Hill, J.-H. Yum, A. Dualeh, S. Barlow, M. Grätzel, S. R. Marder and M. K. Nazeeruddin, Angew. Chem., 2011, 123, 6749 CrossRef.
- T. Maeda, S. Arikawa, H. Nakao, S. Yagi and H. Nakazumi, New J. Chem., 2013, 37(71), 701–708 RSC.
- M. Guo, P. Diao, Y.-J. Ren, F. Meng, H. Tian and S.-M. Cai, Sol. Energy Mater. Sol. Cells, 2005, 88, 23 CrossRef CAS PubMed.
- Y. Chen, Z. Zeng, C. Li, W. Wang, X. Wang and B. Zhang, New J. Chem., 2005, 29, 773 RSC.
- D. Kuang, P. Walter, F. Nüesch, S. Kim, J. Ko, P. Comte, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, Langmuir, 2007, 23, 10906 CrossRef CAS PubMed.
- J.-J. Cid, J.-H. Yum, S.-R. Jang, M. K. Nazeeruddin, E. M. Nez-Ferrero, E. Palomares, J. Ko, M. Grätzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358 CrossRef CAS PubMed.
- P. J. Holliman, M. L. Davies, A. Connell, B. Vaca Velasco and T. M. Watson, Chem. Commun., 2010, 7256 RSC.
- P. J. Holliman, M. Mohsen, A. Connell, M. L. Davies, K. Al-Salihi, M. B. Pitak, G. J. Tizzard, S. J. Coles, R. W. Harrington, W. Clegg, C. Serpa, O. H. Fontes, C. Charbonneau and M. J. Carnie, J. Mater. Chem., 2012, 22(26), 13318 RSC.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed.
- J.-H. Yum, P. Walter, S. Huber, D. Rentsch, T. Geiger, F. Nüesch, F. De Angelis, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2007, 129, 10320 CrossRef CAS PubMed.
- T. Geiger, S. Kuster, J.-H. Yum, S.-J. Moon, M. K. Nazeeruddin, M. Grätzel and F. Nüesch, Adv. Funct. Mater., 2009, 19, 2720 CrossRef CAS.
- H. Choi, J.-J. Kim, K. Song, J. Ko, M. K. Nazeeruddin and M. Grätzel, J. Mater. Chem., 2010, 20, 3280 RSC.
- A. L. Tatarets, I. A. Fedyunyaeva, E. Terpetschnig and L. D. Patsenker, Dyes Pigm., 2005, 64, 125 CrossRef CAS PubMed.
- L. Beverina and P. Salice, Eur. J. Org. Chem., 2010, 1207 CrossRef CAS.
- M. Matsui, T. Shibata, M. Fukushima, Y. Kubota and K. Funabiki, Tetrahedron, 2012, 68, 9936 CrossRef CAS PubMed.
- M. Matsui, H. Mase, J.-Y. Jin, K. Funabiki, T. Yoshida and H. Minoura, Dyes Pigm., 2006, 70, 48 CrossRef CAS PubMed.
- G. Cicero, G. Musso, A. Lambertini, B. Camino, S. Bianco, D. Pugilese, F. Risplendi, A. Sacco, N. Shahzad, A. M. Ferrari, B. Ballarin, C. Barolo, E. Tresso and G. Caputo, Phys. Chem. Chem. Phys., 2013, 15, 7198 RSC.
- G. Cicero, G. Musso, A. Lambertini, B. Camino, S. Bianco, D. Pugilese, F. Risplendi, A. Sacco, N. Shahzad, A. M. Ferrari, B. Ballarin, C. Barolo, E. Tresso and G. Caputo, Phys. Chem. Chem. Phys., 2013, 15, 7198 RSC.
- L. Alibabaei, J.-H. Kim, M. Wang, N. Pootrakulchote, J. Teuscher, D. Di Censo, R. Humphry-Baker, J.-E. Moser, Y.-J. Yu, K.-Y. Kay, S. M. Zakeeruddin and M. Grätzel, Energy Environ. Sci., 2010, 3(11), 1757 CAS.
- E. Terpetschnig and J. R. Lakowicz, Dyes Pigm., 1993, 21(3), 227 CrossRef CAS.
- L. J. S. Brown and P. A. Siesel, Tetrahedron, 1989, 45(15), 4845 CrossRef.
- T. Maeda, S. Mineta, H. Fujiwara, H. Nakao, S. Yagi and H. Nakazumi, J. Mater. Chem. A, 2013, 1, 1303 CAS.
- A. Burke, L. Schmidt-Mende, S. Ito and M. Grätzel, Chem. Commun., 2007, 234 RSC.
- S. Kuster, F. Sauvage, Md. K. Nazeeruddin, M. Grätzel, F. A. Nüesch and T. Geiger, Dyes Pigm., 2010, 87, 30–38 CrossRef CAS PubMed.
- M. Wang, S.-J. Moon, D. Zhou, F. Le Formal, N.-L. Cevey-Ha, R. Humphry-Baker, C. Grätzel, P. Wang, S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2010, 20, 1821 CrossRef CAS.
- G. C. Vougioukalakis, T. Stergiopoulos, A. G. Kontos, E. K. Pefkianakis, K. Papadopoulos and P. Falaras, Dalton Trans., 2013, 42, 6582 RSC.
- J.-H. Yum, S. J. Moon, R. Humphry-Baker, P. Walter, T. Geiger, F. Nüesch, M. Grätzel and M. D. K. Nazeeruddin, Nanotechnology, 2008, 19, 424005 CrossRef CAS PubMed.
- L. Jing, W. WenJun, Y. JiaBao, T. Jin, L. YiTao and H. JianLi, Sci. China: Chem., 2011, 54, 699 Search PubMed.
- M. G. Iglesias, J. J. Cid, J.-H. Yum, A. Forneli, P. Vazquez, M. K. Nazeeruddin, E. Palomares, M. Grätzel and T. Torres, Energy Environ. Sci., 2011, 4, 189 Search PubMed.
- X. Wang, J. Xu, M. Li, D. Fang, B. Chen, L. Wang and W. Xu, RSC Adv., 2013, 2, 5227 RSC.
- A. L. Puyad, G. K. Chaitanya, A. Thomas, M. Paramasivam and K. Bhanuprakash, J. Phys. Org. Chem., 2013, 26, 37 CrossRef CAS.
- P. J. Holliman, K. J. Al-Salihi, A. Connell, M. L. Davies, E. W. Jones and D. A. Worsley, RSC Adv., 2014, 4(5), 2515–2522 RSC.
- R. Hooft, Collect: Data Collection Software, B. V. Nonius, Delft: Netherlands, 1998 Search PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- G. M. Sheldrick, SADABS, Version 2.10, Bruker AXS Inc., Madison, Wisconsin, USA, 2003 Search PubMed.
- CrystalClear-SM Expert 3.1 b26 (Rigaku, 20112).
- SHELXL97/SHELX2013, G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. J. M. Duisenberg, L. M. J. Kroon-Batenburg and A. M. M. Schreurs, An intensity evaluation method: EVAL-14, J. Appl. Crystallog, 2003, 36, 220–229 CrossRef CAS.
- PLATON/SQUEEZE; (a) A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, C34 Search PubMed; (b) P. v. d. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990,(46), 194–201 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 977417–977428. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ta15278b |
| This journal is © The Royal Society of Chemistry 2014 |