 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zn, Ga, and Ca substituted transition-metal-free oxides with K2NiF4 structures
Ning
Ye
*a,
Alexandra
Hasbani
b,
Jun
Jiang
c and
Joshua L.
Hertz
ac
aDepartment of Mechanical Engineering, University of Delaware, 126 Spencer Labs, Newark, DE 19716, USA. E-mail: ningye@udel.edu
bDowningtown S.T.E.M. Academy, 335 Manor Ave, Downingtown, PA 19335, USA
cDepartment of Materials Science and Engineering, University of Delaware, 201 DuPont Hall, Newark, DE 19716, USA
First published on 5th March 2014
Abstract
Recently, an oxide with K2NiF4 structure but free of transition metals was developed with the intent of creating a new solid electrolyte material. Here, the original composition, La1.6+xSr0.4−xAl0.4Mg0.6O4+x/2, was modified by replacing Sr, Al, or Mg, with the isovalent substitutes Ca, Ga, or Zn, respectively. The phase stability of these compounds was investigated to determine, specifically, the maximum concentration of oxygen defects allowed by the crystal structure. It was found that a greater concentration of oxygen vacancy defects than interstitials could be created in all of the compositions. In addition, the influence of the cation substitutions on the lattice parameters and the electrical conduction behavior was analyzed. At 650 °C in air, the highest conductivity achieved for B-site substituted compositions was 2.63 × 10−4 S cm−1 in La1.5Sr0.5Al0.4Zn0.6O3.9, much higher than 5.82 × 10−7 S cm−1 in La1.65Sr0.35Al0.4Mg0.6O4.025, while the highest value for A-site substituted compositions was only 7.55 × 10−7 S cm−1 in La1.65Ca0.35Al0.4Mg0.6O4.025. The electrochemical results indicated that A-site substituted compositions, although with low conductivity, were possible oxygen ion conductors, while B-site substituted compositions exhibited either mixed ionic and hole conductivity or pure hole conductivity.
1. Introduction
Solid oxide fuel cells (SOFCs) are a promising alternative power generation source with an electrolyte that is a ceramic conductor of oxygen ions. Compared to proton exchange membrane fuel cells (PEMFCs) and alkaline fuel cells (AFCs), which are typically limited in the choice of operational fuel, SOFCs can make use of a wide variety of fuels including hydrogen, hydrocarbons, alcohols, and other alternatives.1,2 In order to achieve sufficiently high electrolyte conductivity, SOFCs are usually operated at temperatures of 800 °C or above, which causes a number of issues including delicate gas sealing,3 material degradation,4,5 and slow start-up and shut-down. Therefore, new electrolytes with larger oxygen ion conductivity are required to reduce the operating temperature. Materials with the cubic fluorite crystal structure, such as doped zirconia and ceria, are the conventional electrolyte materials for SOFCs.6–9 More recently, (La, Sr)(Ga, Mg)O3−δ (LSGM) with a perovskite structure has been developed as a solid electrolyte with high ionic conductivity,10–13 and materials with more exotic crystal structures are being actively explored too.14–17K2NiF4-type materials, which consist of alternatively stacked perovskite (P-layer) and rocksalt (R-layer) layers, have attracted attention due to their high oxygen ion conductivity based on an interstitial mechanism.18–20 Most of the known K2NiF4-type materials have transition-metal elements on the B-site and consequently are mixed ionic electronic conductors (MIEC).20–23 This prevents their application as solid electrolytes. In our previous work,24 transition-metal-free K2NiF4 structure materials with the composition La1.6Sr0.4Al0.4Mg0.6O4 (LSAM) were synthesized and shown to have significantly reduced the electron conductivity.
Ionic defects were created in La1.6+xSr0.4−xAl0.4Mg0.6O4+x/2 compositions by adjusting the La/Sr ratio on the A-site. The stability of the crystal structure to significant defect concentrations can be compared to that of LaSrAlO4,25 where second phases are found for any composition with La/Sr ≠ 1. Based on this result, we proposed that anion defects are stabilized in the K2NiF4 structure by Coulombic attraction between the defect and the layer of the crystal structure in which the defect resides: vacancies in the positively charged P-layer and interstitials in the negatively charged R-layer. Thus, a high charge separation between the P-layer with the nominal composition ABO3 and R-layer with the nominal composition AO is desired. In LaSrAlO4, the P-layer consists of (La0.5Sr0.5AlO3)−0.5 while the R-layer consists of (La0.5Sr0.5O)+0.5. In comparison, the prototypical composition La2NiO4, which supports large anion defect concentrations, consists of a (LaNiO3)−1 P-layer and a (LaO)+1 R-layer (neglecting partial oxidation of the Ni). La1.6Sr0.4Al0.4Mg0.6O4 was found in our previous work to provide maximal charge separation in the La2−xSrxAlxMg1−xO4 composition space while maintaining the K2NiF4 crystal structure, with nominal P-layer and R-layer charges of −0.8 and +0.8, respectively.
In this paper, Ca, Ga, or Zn substitute for Sr, Al, or Mg, respectively, in LSAM and the effects of these substitutions on the phase stability and conductivity are studied. Ca substitution is chosen because its smaller ionic radius is known to increase the concentration of oxygen interstitials in La2NiO4.26 Ga is selected to replace Al based on the previous success of high oxygen ion conductivity in the related perovskite LSGM. Zn is examined in the interest of increasing the cell volume to promote ion mobility.27,28 Of specific interest is whether these substitutions are able to increase oxygen defect mobility and/or increase available defect concentrations by allowing increased P-layer/R-layer charge separation with nominal compositions beyond (A3+1.6A2+0.4)(B3+0.4B2+0.6)O4.
2. Experiments
2.1 Synthesis and structural characterization
The samples were all prepared by solid state reactions. Reagents of La2O3 (99.999%, Inframat Advanced Materials), SrCO3 (99.5+%, Inframat Advanced Materials), Mg(OH)2 (98.4%, Alfa Aesar), Al2O3 (99.5% metal basis, Alfa Aesar), Ga2O3 (99.99% metal basis, Alfa Aesar), ZnO (99.9% metal basis, Alfa Aesar) and CaCO3 (99.5% metal basis, Alfa Aesar) were used as the starting raw materials. These reagents were weighed stoichiometrically and then ball milled using spherical YSZ milling media for 20 hours in deionized water. The grinding media were removed from the mixed reagents and then rinsed with DI water for 5 min in an ultrasonic bath 5 times, after which the rinse water was visually clean. All of the rinsed water was collected and heated with the milled dispersion on a hotplate to reduce the loss of chemicals. Finally, the dried precursors were milled in an agate mortar and calcined at 1350 °C for 5 hours with equal heating and cooling rates of 100 °C h−1. The as-synthesized materials were structurally characterized in conventional θ–2θ geometry on a PANalytical X'Pert X-ray diffractometer (XRD) using Cu-Kα radiation. The lattice parameters were determined from XRD patterns using Jade software (ver. 5.0, Materials Data, Inc.). Calcined powders were uniaxially pressed without a binder at a pressure of 300 MPa and then sintered at 1450 °C for 10 h with an equal heating and cooling rate of 100 °C h−1 to obtain pellets with density >95%. The microstructures of the sintered pellets were characterized by scanning electron microscopy (SEM, JOEL JSM-7400F) with a 3 keV accelerating voltage. Imaged surfaces were polished to 800-grit, thermally etched in air at 1150 °C for 6 h, and then coated with roughly 10 nm of gold–palladium (60% Au, 40% Pd).2.2 Conductivity measurement
The sintered pellets were polished by hand into a rectangular shape with dimensions 12 mm × 5 mm × 1 mm. Four silver paint electrodes were applied along the length of the sample for four-probe DC conductivity measurements. Electrical measurements were collected using a Keithley 2601A Sourcemeter with linear sweep voltages between −1 V and +1 V. This relatively wide voltage range was used to reduce the noise in the current signal despite high sample resistances. The linearity of the current–voltage relationship over the entire range suggested that no oxidization or reduction of the materials under test occurred. The temperature was controlled between 300 °C and 650 °C in a tube furnace and directly measured with a thermocouple in close proximity to the sample. Flowing dry air and commercial Ar–O2 mixtures were used to change the oxygen partial pressure.3. Results and discussions
Starting from a composition of La1.6Sr0.4Al0.4Mg0.6O4, efforts were made to increase the P-layer/R-layer charge separation when substituting Ga, Zn, or Ca. As seen in Fig. 1, the direct substitution for Al, Mg, and Ca maintained the K2NiF4 crystal structures; however, for all three substitutions, the charge separation could not increase from −0.8/+0.8 to −0.85/+0.85 by changing the nominal composition from (A3+1.6A2+0.4)(B3+0.4B2+0.6)O4 to (A3+1.7A2+0.3)(B3+0.3B2+0.7)O4. Increasing the charge separation in all cases led to the appearance of second phase La2O3. | ||
Fig. 1 XRD patterns of Ga, Zn, and Ca substituted samples. Peaks marked by  indicate the secondary phase of La2O3. indicate the secondary phase of La2O3. | ||
By comparing the XRD results of the samples with the nominal composition (A3+1.6A2+0.4)(B3+0.4B2+0.6)O4 in Fig. 1, the peak locations of the Ca substituted sample are clearly shifted to a higher angle compared to the corresponding peaks of the Ga or Zn containing samples, indicating reduced lattice parameters. Lattice parameters for these and other compositions as determined from the XRD patterns are given in Table 1. The table shows that both a and c lattice parameters of LSAM are increased by Ga and Zn substitution, while they are both reduced by Ca substitution. These results are intuitive, since the ionic radii of Ga and Zn are larger than those of Al and Mg, but the ionic radius of Ca is smaller than that of Sr.
| Nominal composition | Cation radius ratioa | Phase | K2NiF4 phase lattice parameter (Å) | ||
|---|---|---|---|---|---|
| r A(IX)/rB(VI) | a | c | |||
| a The ionic radii are from R. D. Shannon.29r(La3+,IX) = 1.216 Å; r(Ni2+,VI) = 0.69 Å; r(Sr2+,IX) = 1.31 Å; r(Al3+,VI) = 0.535 Å; r(Mg2+,VI) = 0.72 Å; r(Ca2+,IX) = 1.18 Å; r(Ga3+,VI) = 0.62 Å; r(Zn2+,VI) = 0.74 Å. b Lattice parameters of La2NiO4 are from JCPDS card 34-0314. | |||||
| La2NiO4b | 1.76 | K2NiF4 | 3.861 | 12.68 | |
| La1.6Sr0.4Al0.4Mg0.6O4 (ref. 24) | 1.91 | K2NiF4 | 3.829 | 12.646 | |
| Ca substitution | La1.7Ca0.3Al0.4Mg0.6O4.05 | 1.87 | K2NiF4, La2O3 | 3.818 | 12.490 |
| La1.65Ca0.35Al0.4Mg0.6O4.025 | 1.87 | K2NiF4 | 3.820 | 12.548 | |
| La1.6Ca0.4Al0.4Mg0.6O4 | 1.87 | K2NiF4 | 3.804 | 12.530 | |
| La1.5Ca0.5Al0.4Mg0.6O3.95 | 1.87 | K2NiF4 | 3.803 | 12.512 | |
| La1.4Ca0.6Al0.4Mg0.6O3.9 | 1.87 | K2NiF4 | 3.800 | 12.507 | |
| La1.3Ca0.7Al0.4Mg0.6O3.85 | 1.86 | K2NiF4, CaO | 3.798 | 12.514 | |
| La1.7Ca0.3Al0.3Mg0.7O4 | 1.82 | K2NiF4, La2O3 | 3.819 | 12.538 | |
| La1.5Ca0.5Al0.5Mg0.5O4 | 1.92 | K2NiF4, Al2Ca3O6 | 3.792 | 12.508 | |
| Ga substitution | La1.7Sr0.3Ga0.4Mg0.6O4.05 | 1.81 | K2NiF4, La4Ga2O9 | 3.866 | 12.677 |
| La1.6Sr0.4Ga0.4Mg0.6O4 | 1.82 | K2NiF4 | 3.861 | 12.681 | |
| La1.5Sr0.5Ga0.4Mg0.6O3.95 | 1.82 | K2NiF4 | 3.862 | 12.689 | |
| La1.4Sr0.6Ga0.4Mg0.6O3.9 | 1.83 | K2NiF4, LaSr2GaO5 | 3.862 | 12.684 | |
| La1.7Sr0.3Ga0.3Mg0.7O4 | 1.78 | K2NiF4, La2O3 | 3.866 | 12.670 | |
| Zn substitution | La1.7Sr0.3Al0.4Zn0.6O4.05 | 1.87 | K2NiF4, La2O3, LaAlO3 | 3.832 | 12.723 |
| La1.65Sr0.35Al0.4Zn0.6O4.025 | 1.87 | K2NiF4 | 3.836 | 12.741 | |
| La1.6Sr0.4Al0.4Zn0.6O4 | 1.88 | K2NiF4 | 3.831 | 12.762 | |
| La1.55Sr0.45Al0.4Zn0.6O3.975 | 1.88 | K2NiF4 | 3.835 | 12.757 | |
| La1.5Sr0.5Al0.4Zn0.6O3.95 | 1.88 | K2NiF4 | 3.827 | 12.785 | |
| La1.4Sr0.6Al0.4Zn0.6O3.9 | 1.89 | K2NiF4 | 3.815 | 12.832 | |
| La1.3Sr0.7Al0.4Zn0.6O3.85 | 1.90 | K2NiF4, La2O3 | 3.807 | 12.909 | |
| La1.7Sr0.3Al0.3Zn0.7O4 | 1.81 | K2NiF4, La2O3 | 3.844 | 12.759 | |
| La1.5Sr0.5Al0.5Zn0.5O4 | 1.94 | K2NiF4, La2O3 | 3.821 | 12.743 | |
As shown in Fig. 2, the microstructures of the sintered pellets reveal that dense ceramic structures with little porosity are obtained for all three stoichiometric samples. This result is consistent with the >95% pellet densities determined by Archimedes' method. The grain sizes for the sintered pellets are between 1 μm and 8 μm. No intergranular phases are observed in the micrographs, though they may be present at quantities/thicknesses smaller than could be observed presently.
 | ||
| Fig. 2 Representative SEM micrographs of the polished and thermally etched surfaces of sintered pellets with compositions (a) La1.6Ca0.4Al0.4Mg0.6O4, (b) La1.6Sr0.4Ga0.4Mg0.6O4, and (c) La1.6Sr0.4Al0.4Zn0.6O4. | ||
Fig. 3 shows the results of adjusting the A-site ratio in order to create ionic defects. Increasing the ratio of A3+/A2+ (x > 0) in (A3+1.6+xA2+0.4−x)(B3+0.4B2+0.6)O4+x/2 ideally creates oxygen interstitials, while decreasing the ratio (x < 0) creates oxygen vacancies. The XRD results in Fig. 3 indicate that, for all samples studied, a higher concentration of oxygen vacancies could be created relative to oxygen interstitials before the appearance of secondary phases. In Ga containing samples, x could be −0.1 without second phases, whereas La4Ga2O9 peaks appear when x = +0.1. For both Zn and Ca substitutions, the K2NiF4 structure was stable when −0.2 ≤ x ≤ +0.05. In the Zn containing specimen, increasing amounts of oxygen vacancies lead to reduction in the a lattice parameter and increase in the c parameter. This leads to increased peak splitting between the (133) and (008) peaks. Thus, the peak at 2θ = 57.41° marked with ○ for the x = −0.2 sample in Fig. 3(b) is the (008) peak of the K2NiF4 structure. Since the ionic radii of the A-site cations are r(Sr2+,IX) = 1.31 Å > r(La3+,IX) = 1.216 Å > r(Ca2+,IX) = 1.18 Å, reducing the La/Sr ratio in Zn containing samples caused a significant increase of the lattice parameter c, while reducing the La/Ca ratio in Ca containing samples led to a significant decrease of the lattice parameter c. In general, reducing the La/Sr or La/Ca ratio caused a slight decrease in the lattice parameter a.
 | ||
| Fig. 3 (a) XRD patterns of La1.6+xSr0.4−xGa0.4Mg0.6O4+x/2; (b) XRD patterns of La1.6+xSr0.4−xAl0.4Zn0.6O4+x/2; (c) XRD patterns of La1.6+xCa0.4−xAl0.4Mg0.6O4+x/2. Peaks marked by indicate La2O3, ▲ indicate La4Ga2O9, ● indicate LaSr2GaO5, ■ indicate LaAlO3, and ♦ indicate CaO. | ||
Table 1 gives the calculated lattice parameters based on XRD alongside the ratio of the ionic radii of the A-site cation to that of the B-site cation. Since multiple cations are present on each site, weighted average ionic radii are used, taking the radii in a 9-fold coordination for the A-site and a 6-fold coordination for the B-site. Ganguli suggested that the ratio of ionic radii, rA(IX)/rB(VI), should be between about 1.7 and 2.4 in order to retain the K2NiF4 phase.30 As indicated by the results in Table 1, all compositions in this study, even those with multiple phases present, satisfy the rule.
The substitution of Ca on the A-site caused relatively little change in the total electrical conductivity. As shown in Fig. 4, increasing the La/Ca ratio in order to create oxygen interstitials caused an increase in both the high temperature conductivity and activation energy of conduction. The electrical conductivity of La1.6+xCa0.4−xAl0.4Mg0.6O4 is very similar to the previously reported values for La1.6+xSr0.4−xAl0.4Mg0.6O4.24 The inset in Fig. 4 plots the conductivity at 650 °C vs. the oxygen stoichiometry and suggests a moderate amount of interstitial conductivity in both the superstoichiometric and stoichiometric compositions. When the La/Ca ratio is reduced such that vacancies are expected on the oxygen sublattice, the conductivity values are within the experimental error of that of the previously reported La1.6Sr0.4Al0.4Mg0.6O4 sample. These results indicate that the vacancies have low mobility. The similarity of the conductivity values for these samples, despite the increasing vacancy concentration, suggests that the charge carrier concentration does not change with the composition. The carrier concentration is thus determined by either intrinsic defects or impurities acting as dopants.
 | ||
| Fig. 4 Arrhenius plot of total electrical conductivity measured in air for (A) La1.6Sr0.4Al0.4Mg0.6O4;24 and the Ca substituted samples (B) La1.65Ca0.35Al0.4Mg0.6O4.025; (C) La1.6Ca0.4Al0.4Mg0.6O4; (D) La1.5Ca0.5Al0.4Mg0.6O3.95; (E) La1.4Ca0.6Al0.4Mg0.6O3.9. The inset figure displays the conductivity at 650 °C vs. the oxygen stoichiometry. | ||
Fig. 5 shows the conductivity of the La1.6+xCa0.4−xAl0.4Mg0.6O4+x/2 samples at 550 °C over a range of oxygen partial pressures. The conductivities of all compositions are essentially independent of the oxygen partial pressure, with the slopes of the best-fit lines ≤0.007. This result gives further indication that the dominant charge carriers are ionic,31–34 since electronic carriers are expected to have concentrations that are determined by oxidation reactions and are thus dependent upon the oxygen partial pressure.34–37 Nevertheless, the small change in conductivity despite x varying between +0.05 and −0.2 implies a low mobility of the created defects. A similar phenomenon was observed in La1.6+xSr0.4−xAl0.4Mg0.6O4+x/2,24 which has the same B-site composition. The reason that defect mobilities are low in K2NiF4 materials that have only period 3 elements in the B-site remains an open question, but is perhaps due to the small cell volume27,28 and/or low polarizability38 related to their small ionic radii.
 | ||
| Fig. 5 Total electrical conductivity at 550 °C of La1.6+xCa0.4−xAl0.4Mg0.6O4+x/2vs. oxygen partial pressure for compositions with x = −0.2, −0.1, 0, and +0.05. The near-zero slopes of the best fit lines are indicated next to each line. | ||
Fig. 6 shows that B-site substitutions caused a much greater change in conductivity than A-site substitutions. For the oxygen stoichiometric compositions, Ga substitution yielded roughly one order of magnitude increase in the electrical conductivity relative to the original LSAM composition, and Zn substitution caused a further increase by a little over one order of magnitude. Unlike the LSAM or the Ca-substituted compositions, decreasing the La/Sr ratio from that of the stoichiometric composition in the Ga- and Zn-substituted samples increased the conductivity. In the Zn-containing compositions, the conductivity improves from x = 0 to x = −0.1, however, further increases in the defect concentration at x = −0.2 yields lower conductivity. The reason for this may be an impurity phase not perceptible by XRD or SEM. A strong possibility is a Sr-rich phase, which is often observed to form a poorly-conducting layer at grain boundaries in perovskite oxide ion conductors.39,40 An increase in activation energy from 1.15 eV for x = −0.1 to 1.30 eV for x = −0.2 could be seen as evidence for this hypothesis, but confirmation is still needed. Increasing the La/Sr ratio from that of the stoichiometric composition in the Zn-substituted samples (which was not possible in the Ga-substituted samples) reduced the conductivity significantly.
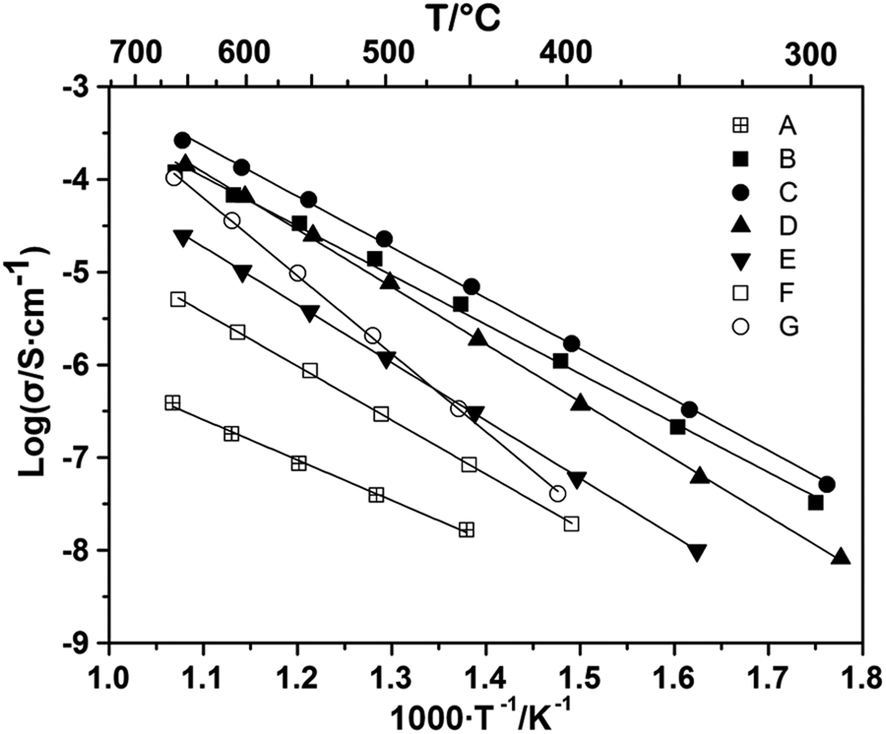 | ||
| Fig. 6 Arrhenius plot of the total electrical conductivity measured in air for (A) La1.6Sr0.4Al0.4Mg0.6O4;24 and the B-site substituted samples (B) La1.6Sr0.4Al0.4Zn0.6O4; (C) La1.5Sr0.5Al0.4Zn0.6O3.95; (D) La1.4Sr0.6Al0.4Zn0.6O3.9; (E) La1.65Sr0.35Al0.4Zn0.6O4.025; (F) La1.6Sr0.4Ga0.4Mg0.6O4; (G) La1.5Sr0.5Ga0.4Mg0.6O3.95. In all cases, square symbols indicate compositions with stoichiometric oxygen content. | ||
Fig. 7 shows the total electrical conductivity at 550 °C of La1.6+xSr0.4−xGa0.4Mg0.6O4+x/2 measured in various oxygen partial pressures. The electrical conductivity of stoichiometric La1.6Sr0.4Ga0.4Mg0.6O4 has near-zero dependence on the oxygen partial pressure, indicating that the conduction mechanism is via intrinsic defects at a concentration larger than that created by redox reactions. Studies of the similar compositions LaSrGa1−xMgxO4−x/2 (for which a low value of 0.03 was reported as the maximum possible x, consistent with our theory of defects being stabilized by charge separation between the P-layer and R-layer), found evidence of oxygen ion conduction,41 but ionic conduction cannot be definitively determined here. Oxygen substoichiometric La1.5Sr0.5Ga0.4Mg0.6O3.95 exhibited a more clear oxygen partial pressure dependence of conductivity. The total conductivity can be represented by:34–37
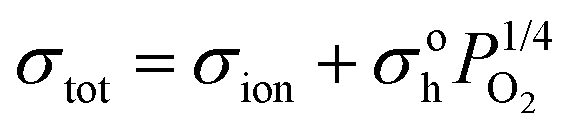 | (1) |
 | (2) |
 is a vacant oxygen site, OXO is an occupied oxygen site, and h˙ is a hole. With the concentration of oxygen vacancies held approximately constant by the stoichiometry, the mass action law of reaction (2) provides a 1/4 power law dependence of the hole concentration.34–37,42 The oxygen partial pressure dependence of conductivity measured for La1.5Sr0.5Ga0.4Mg0.6O3.95 is about 0.102. The deviation from the classic 1/4 power dependence is very common in other mixed conducting materials.43,44 Since the conductivity increases with oxygen substoichiometry, the predominant charge carrier in La1.5Sr0.5Ga0.4Mg0.6O3.95 seems to be mixed between vacancies and holes.
is a vacant oxygen site, OXO is an occupied oxygen site, and h˙ is a hole. With the concentration of oxygen vacancies held approximately constant by the stoichiometry, the mass action law of reaction (2) provides a 1/4 power law dependence of the hole concentration.34–37,42 The oxygen partial pressure dependence of conductivity measured for La1.5Sr0.5Ga0.4Mg0.6O3.95 is about 0.102. The deviation from the classic 1/4 power dependence is very common in other mixed conducting materials.43,44 Since the conductivity increases with oxygen substoichiometry, the predominant charge carrier in La1.5Sr0.5Ga0.4Mg0.6O3.95 seems to be mixed between vacancies and holes.
 | ||
| Fig. 7 Total electrical conductivity at 550 °C of La1.6+xSr0.4−xGa0.4Mg0.6O4+x/2vs. oxygen partial pressure for compositions with x = −0.1 and 0. The slopes of the best-fit lines are indicated. | ||
Fig. 8 shows the total electrical conductivity at 550 °C of the La1.6+xSr0.4−xAl0.4Zn0.6O4+x/2 series samples over a range of oxygen partial pressures. When x is between 0 and −0.2, the conductivity has a roughly 1/4 power law dependence over the entire range of oxygen partial pressures. This result indicates that the main charge carriers are holes, as described previously. For the x = +0.05 composition, where interstitial defects are expected, the slope of the oxygen partial pressure dependence appears to be 1/6. This result suggests that the defect concentration is no longer fixed by the stoichiometry, but rather the charge carrier concentrations are determined by the oxidation reaction.42
 | ||
| Fig. 8 Total electrical conductivity at 550 °C of La1.6+xSr0.4−xAl0.4Zn0.6O4+x/2vs. oxygen partial pressure for compositions with x = −0.2, −0.1, 0, and +0.05. The slopes of the best fit lines are indicated. | ||
In this work, A-site and B-site substitutions caused contrasting effects on the conductivity. While all of the tested compositional changes caused increases in the conductivity of stoichiometric samples, the A-site substitution of Ca caused the conductivity to increase with increasing interstitial defects and decrease with vacancy defects. In contrast, both B-site substitutions caused the conductivity to increase with the presence of vacancies and decrease with interstitial defects—if the crystal structure even allowed them. This result further supports the notion that positively charged vacancies prefer to inhabit a negatively charged perovskite layer with the composition ABO3 and negatively charged interstitials prefer to inhabit a positively charged rocksalt layer with the composition AO. Still, the presence of mixed conduction in the B-site substituted samples indicates that getting sufficient oxide ion mobility without coincident electron/hole conductivity in the K2NiF4 structure will be difficult.
In the transition-metal-free K2NiF4 oxides, the electronic conductivity could be greatly suppressed compared to prototype La2NiO4 materials. Large amounts of oxygen defects could also be created. Nonetheless, probably due to the presence of alkaline earth metal/post-transition metals in the B-site, the as-created oxygen defects had low mobility. Therefore, although oxygen ion conductors were achieved with A-site substitution of Ca, the ionic conductivity remained extremely low. In future work, a new strategy to select B-site atoms that increase the mobility of oxygen defects without reintroducing electronic conductivity would be key to the success of designing K2NiF4 electrolyte materials.
4. Conclusions
La1.6+xSr0.4−xGa0.4Mg0.6O4+x/2 (−0.1 ≤ x ≤ 0), La1.6+xSr0.4−xAl0.4Zn0.6O4+x/2 (−0.2 ≤ x ≤ +0.05) and La1.6+xCa0.4−xAl0.4Mg0.6O4+x/2 (−0.2 ≤ x ≤ +0.05) with K2NiF4 structures were synthesized without impurity phases. Compared to the electrical conductivity of LSAM, the electrical conductivity was relatively constant when replacing Sr with Ca, but increased between 1 and 2.5 orders of magnitude when substituting Al or Mg with Ga or Zn. Electrochemical analysis revealed that La1.6+xCa0.4−xAl0.4Mg0.6O4+x/2 appeared to support the electrolytic conduction of interstitial oxygen ions, though with low conductivity. Conversely, the main carriers in La1.6+xSr0.4−xAl0.4Zn0.6O4+x/2 seemed to be holes. In La1.6+xSr0.4−xGa0.4Mg0.6O4+x/2, x = 0 appeared to be predominantly an ionic conductor while x = −0.1 was a mixed conductor of holes and oxygen vacancies. The results indicate that electrolytic conduction is possible in transition-metal-free K2NiF4 oxides, but high ionic mobility is yet to be achieved.References
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- E. D. Wachsman, Science, 2011, 334, 935–939 CrossRef CAS PubMed.
- R. N. Singh, J. Mater. Eng. Perform., 2006, 15, 422–426 CrossRef CAS.
- N. Sakai, T. Horita, K. Yamaji, Y. Xiong, H. Kishimoto, M. Brito and H. Yokokawa, Solid State Ionics, 2006, 177, 1933–1939 CrossRef CAS PubMed.
- Z. Yang, M. S. Walker, P. Singh and J. W. Stevenson, Electrochem. Solid-State Lett., 2003, 6, B35–B37 CrossRef CAS PubMed.
- H. L. Tuller, Solid State Ionics, 2000, 131, 143–157 CrossRef CAS.
- C. S. Tedmon, H. S. Spacil and S. P. Mitoff, J. Electrochem. Soc., 1969, 116, 1170–1175 CrossRef PubMed.
- J. L. Hertz and H. L. Tuller, J. Electrochem. Soc., 2007, 154, B413–B418 CrossRef CAS PubMed.
- H. L. Tuller and A. S. Nowick, J. Electrochem. Soc., 1975, 122, 255–259 CrossRef CAS PubMed.
- T. Ishihara, H. Matsuda and Y. Takita, J. Am. Chem. Soc., 1994, 116, 3801–3803 CrossRef CAS.
- S. Tao, F. W. Poulsen, G. Meng and O. T. Sørensen, J. Mater. Chem., 2000, 10, 1829–1833 RSC.
- C. N. Munnings, S. J. Skinner, G. Amow, P. S. Whitfield and I. J. Davidson, J. Fuel Cell Sci. Technol., 2005, 2, 34–37 CrossRef CAS.
- M. Li, Y. Zhang, M. An, Z. Lü, X. Huang, J. Xiao, B. Wei, X. Zhu and W. Su, J. Power Sources, 2012, 218, 233–236 CrossRef CAS PubMed.
- X. Kuang, M. A. Green, H. Niu, P. Zajdel, C. Dickinson, J. B. Claridge, L. Jantsky and M. J. Rosseinsky, Nat. Mater., 2008, 7, 498–504 CrossRef CAS PubMed.
- M.-R. Li, X. Kuang, S. Y. Chong, Z. Xu, C. I. Thomas, H. Niu, J. B. Claridge and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2010, 49, 2362–2366 CrossRef CAS PubMed.
- C. I. Thomas, X. Kuang, Z. Deng, H. Niu, J. B. Claridge and M. J. Rosseinsky, Chem. Mater., 2010, 22, 2510–2516 CrossRef CAS.
- P. R. Slater, J. E. Sansom and J. R. Tolchard, Chem. Rec., 2004, 4, 373–384 CrossRef CAS PubMed.
- H. S. Kim and H. I. Yoo, Phys. Chem. Chem. Phys., 2010, 12, 4704–4713 RSC.
- A. L. Shaula, E. N. Naumovich, A. P. Viskup, V. V. Pankov, A. V. Kovalevsky and V. V. Kharton, Solid State Ionics, 2009, 180, 812–816 CrossRef CAS PubMed.
- E. V. Tsipis, E. N. Naumovich, A. L. Shaula, M. V. Patrakeev, J. C. Waerenborgh and V. V. Kharton, Solid State Ionics, 2008, 179, 57–60 CrossRef CAS PubMed.
- S. Abou-Warda, W. Pietzuch, G. Berghöfer, U. Kesper, W. Massa and D. Reinen, J. Solid State Chem., 1998, 138, 18–31 CrossRef CAS.
- S. J. Skinner and J. A. Kilner, Ionics, 1999, 5, 171–174 CrossRef CAS.
- J. B. Goodenough and S. Ramasesha, Mater. Res. Bull., 1982, 17, 383–390 CrossRef CAS.
- N. Ye and J. L. Hertz, Acta Mater., 2014, 63, 123–129 CrossRef CAS PubMed.
- E. S. Raj, S. J. Skinner and J. A. Kilner, Solid State Sci., 2004, 6, 825–829 CrossRef CAS PubMed.
- H. S. Kim and H. I. Yoo, Solid State Ionics, 2013, 232, 129–137 CrossRef CAS PubMed.
- J. Liu, S. Hull, I. Ahmed and S. J. Skinner, Nucl. Instrum. Methods Phys. Res., Sect. B, 2011, 269, 539–543 CrossRef CAS PubMed.
- T. Ishihara, H. Matsuda, Y. Mizuhara and Y. Takita, Solid State Ionics, 1994, 70, 234–238 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- D. Ganguli, J. Solid State Chem., 1979, 30, 353–356 CrossRef CAS.
- A. Tarancón, T. Norby, G. Dezanneau, A. Morata, F. Peiró and J. Morante, Electrochem. Solid-State Lett., 2004, 7, A373–A375 CrossRef PubMed.
- T. Ishihara, T. Akbay, H. Furutani and Y. Takita, Solid State Ionics, 1998, 113, 585–591 CrossRef.
- K. Huang and J. B. Goodenough, J. Alloys Compd., 2000, 303, 454–464 CrossRef.
- D. Lybye, F. W. Poulsen and M. Mogensen, Solid State Ionics, 2000, 128, 91–103 CrossRef CAS.
- I. Kosacki, V. Petrovsky and H. U. Anderson, J. Electroceram., 2000, 4, 243–249 CrossRef CAS.
- I. Kosacki and H. Tuller, Solid State Ionics, 1995, 80, 223–229 CrossRef CAS.
- J. Y. Park and G. M. Choi, Solid State Ionics, 2002, 154, 535–540 CrossRef.
- S. Hull, Rep. Prog. Phys., 2004, 67, 1233–1314 CrossRef CAS.
- R. Baker, B. Gharbage and F. Marques, J. Eur. Ceram. Soc., 1998, 18, 105–112 CrossRef CAS.
- K. Huang, R. S. Tichy and J. B. Goodenough, J. Am. Ceram. Soc., 1998, 81, 2565–2575 CrossRef CAS.
- N. Kitamura, N. Hamao, S. C. Vogel and Y. Idemoto, Electrochemistry, 2013, 81, 448–453 CrossRef CAS PubMed.
- H. Tuller, Nonstoichiom. Oxides, 1981, 271–335 CAS.
- M. Burriel, M. Casas-Cabanas, J. Zapata, H. Tan, J. Verbeeck, C. Soliís, J. Roqueta, S. J. Skinner, J. A. Kilner, G. Van Tendeloo and J. Santiso, Chem. Mater., 2010, 22, 5512–5520 CrossRef CAS.
- S. Y. Jeon, M. B. Choi, J. H. Hwang, E. D. Wachsman and S. J. Song, J. Electrochem. Soc., 2011, 158, B476–B480 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |