A high conductivity oxide–sulfide composite lithium superionic conductor†
Ezhiylmurugan
Rangasamy
a,
Gayatri
Sahu
a,
Jong Kahk
Keum
a,
Adam J.
Rondinone
a,
Nancy J.
Dudney
b and
Chengdu
Liang
*a
aCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA. Fax: +1-865-574-1753; E-mail: liangcn@ornl.gov
bMaterials Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
First published on 14th January 2014
Abstract
Best of both worlds: a composite electrolyte of LLZO and LPS successfully combines low grain boundary resistance, room temperature processability and low interfacial resistance of LPS with the excellent electrochemical stability and ionic conductivity of LLZO. The composite electrolyte improves the ionic conductivity of parent electrolytes and augments exceptional compatibility with metallic lithium, thereby making the electrolyte attractive for practical solid-state batteries.
Introduction
Since its introduction in 1991,1 modern Li-ion battery technology has found a wide range of applications from portable electronics to transportation systems. Due to the increasing demand on advanced energy storage devices, high energy density technologies such as Li–S and Li–O2 are now being extensively researched.2 Li–S cells offer a 6-fold increase in specific energy over conventional Li-ion systems.2 Their implementation is currently limited by the dissolution and migration of polysulfides in conventional liquid electrolytes.3,4 The polysulfide shuttle phenomenon leads to columbic inefficiencies and rapid electrode degradation. Additionally, dendritic growth results from the cycling of the metallic lithium anode, leading to internal cell shorting. This raises safety concerns due to the flammability of organic solvents. Solid state Li-ion conductors offer a solution to these issues by providing a typically impermeable membrane that prevents the penetration of lithium dendrites and the migration of polysulfides.5,6 Additionally, they offer improved electrochemical, mechanical and thermal stability.7,8 Ionic conductivity of solid electrolytes can be as high as liquid electrolytes. For example, the sulfide based Li10GeP2S12 has an unprecedented ionic conductivity of 1.2 × 10−2 S cm−1, which is comparable to that of 1 M LiPF6 in the carbonate solvents.9 Solid electrolytes are single ion conductors; the lithium ion transference number is hence 1. With comparable ionic conductivities, the effective lithium ion conductivity of solid electrolytes is much higher than that of liquid electrolytes. However, a majority of the solid state conductors suffer from poor ionic conductivity (10−6 to 10−8 S cm−1), while the better ionic conductors are not stable with metallic Li anodes.10The practical use of solid electrolytes in batteries is far beyond the investigation of ionic conductivity. Chemical compatibility of solid electrolytes with electrode materials, interfacial resistance, and processability of the solid electrolytes restrict the application of solid electrolytes in batteries. Thus, it is very challenging to have a single electrolyte that meets all the above requirements for practical use in batteries. The garnet structured Li7La3Zr2O12 (LLZO)11–14 and the nanoporous β-Li3PS4 (LPS)15 are two promising electrolytes with their own merits and limitations for application in batteries. The LLZO combines good ionic conductivity (>10−4 S cm−1) with excellent electrochemical stability and mechanical properties.12,14,16,17 Despite the advantages, LLZO requires aliovalent substitution to stabilize the higher conducting cubic phase11–13,18–20 and temperatures in excess of 1000 °C to achieve high relative densities (>95%) via sintering. LLZO processed under ambient conditions via cold pressing does not possess the excellent ionic conductivity of hot pressed membranes as a result of the high resistance from non-sintered grain boundaries and porosity. It is also limited by high interfacial resistances with the electrode materials.21 The nanoporous LPS is a superionic conductor offering good electrochemical stability and favorable ionic conductivity.15 Due to the negligible grain boundary resistance for sulfide electrolytes, LPS exhibits excellent conductivity even under cold pressed conditions.22 These sulfides can be dry-pressed to high relative densities under ambient conditions, while the LPS exhibits minimal interfacial resistance in a non-blocking electrode configuration.5,15 Thus, LPS combines good electrochemical properties, a facile synthesis procedure, and an easy membrane fabrication. However, the room-temperature conductivity of 1.6 × 10−4 S cm−1 leaves scope for improvement. From the aforementioned properties, it can be observed that LLZO and LPS are complementary to each other. Is it possible to form a composite of LLZO and LPS to achieve a whole greater than the sum of its parts? Herein, we report a composite superionic conductor utilizing an oxide–sulfide system that enhances the properties of its parent electrolytes: (1) excellent processability through cold pressing; (2) enhanced ionic conductivity; and (3) high chemical compatibility and low interfacial resistance with metallic lithium anode.
Experimental methods
Synthesis of LPS, LLZO and their composites
Li3PS4 was synthesized through a solution based procedure reported earlier by this group.15 The synthesized Li3PS4 was heat treated at 140 °C for 1 hour to obtain the nano-crystalline β-Li3PS4 (LPS). LLZO was synthesized utilizing the following precursors: Li2CO3 – Acros International 99.999% pure, La2O3 – Acros International 99.9% pure, ZrO2 – Inframat Advanced Materials 99.9% pure 30–60 nm and Al2O3 – Sigma Aldrich <50 nm. The precursors were mixed (8000M Spex Mixer Mill) in the molar ratio 3.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:2:0.12 in a 30 ml high density poly-ethylene (HDPE – VWR Scientific) vial for 1 hour. Agate balls (5 mm diameter) were used as the milling media. The milled powders were collected and cold pressed in a 1′′ steel die at 40 MPa. The precursor mixtures were cold pressed into pellets followed by calcination at 1000 °C for 8 hours with a heating rate of 250 °C h−1. A MgO crucible was used as the calcination medium. The fired LLZO was ground to fine powder by a mortar and pestle. Since LLZO was prepared in air, possible moisture uptake was removed by heating the LLZO powder in a vacuum at 160 °C for 2 hours prior to the preparation of the composite. Tetragonal LLZO was synthesized through the aforementioned procedure without Al2O3. For the preparation of the composite electrolyte, the LLZO and LPS precursors were mixed (Mixer Mill) in the requisite ratios in a HDPE vial with 2 mm dia ZrO2 milling media (1:50 mass ratio) for 3 minutes.
:1.5:2:0.12 in a 30 ml high density poly-ethylene (HDPE – VWR Scientific) vial for 1 hour. Agate balls (5 mm diameter) were used as the milling media. The milled powders were collected and cold pressed in a 1′′ steel die at 40 MPa. The precursor mixtures were cold pressed into pellets followed by calcination at 1000 °C for 8 hours with a heating rate of 250 °C h−1. A MgO crucible was used as the calcination medium. The fired LLZO was ground to fine powder by a mortar and pestle. Since LLZO was prepared in air, possible moisture uptake was removed by heating the LLZO powder in a vacuum at 160 °C for 2 hours prior to the preparation of the composite. Tetragonal LLZO was synthesized through the aforementioned procedure without Al2O3. For the preparation of the composite electrolyte, the LLZO and LPS precursors were mixed (Mixer Mill) in the requisite ratios in a HDPE vial with 2 mm dia ZrO2 milling media (1:50 mass ratio) for 3 minutes.
Structural and electrochemical characterization
Crystallographic phase identification was conducted by using a PANalytical X'Pert Pro Powder Diffractometer with Cu Kα radiation. XRD samples were prepared in a glove box with an Ar atmosphere. Kapton® films were used to seal the quartz slide to exclude air contact. Rietveld refinement and structural analyses were conducted by the software of HighScore Plus, which is developed by PANalytical. The powder samples were cold pressed into pellets (0.5′′ dia) in a steel die at 600 MPa inside an Ar atmosphere. Carbon coated Al-foils were used as blocking electrodes. Relative densities for the respective pellets were calculated from their gravimetric and geometric measurements. Electrochemical Impedance Spectroscopy (EIS) measurements were conducted at a 100 mV amplitude in the frequency range of 1 MHz to 1 Hz using a Solartron 1260 Frequency Response Analyzer. For the preparation of the Li/LLZO–LPS/Li symmetric cell, the powders were cold pressed along with Li foils at 300 MPa in 0.5′′ die. The cycling measurements were conducted at a current density of 0.1 mA cm−2 by using a MACCOR 4000 battery tester. Arrhenius measurements were conducted between 25 °C and 100 °C with the temperature controlled by an environmental chamber. Each temperature point was equilibrated for 60 min before the impedance measurement. The cyclic voltammetry measurements were conducted between −0.2 V and 5 V vs. Li/Li+ using a scan rate of 10 mV s−1. The cell was fabricated using a Pt working electrode and a Li counter electrode cold pressed with the composite powders at 300 MPa in a 0.5′′ die. The Li counter electrode was employed as the quasi-reference electrode.Results and discussion
LPS imparts excellent processability to the composite
LLZO is a hard oxide crystal, which is impossible to densify at room temperature through hydraulic pressing.23 Thus, the high ionic conductivity of LLZO cannot be achieved without sintering or hot pressing at a temperature in excess of 1000 °C, which restricts its application in batteries. As a stark contrast to the hard oxides, the sulfide based solid electrolytes are relatively soft and dense materials with high ionic conductivity being achievable through cold-pressing.15,22 A mixture of the LLZO and LPS precursors was subjected to a simple ball-milling procedure.The mechanical mixing of these two materials results in a coating of LPS over LLZO by taking advantage of the soft and sticky nature of the sulfides. Thus by employing a simple dry milling procedure, a core–shell structure is obtained (see Fig. 1a). The Li distribution data (see Fig. 1b) from Electron Energy Loss Spectroscopy (EELS) image confirm a Li-rich shell of LPS (arising from the higher molar concentration of Li in LPS vs. LLZO). La and Zr distribution data provided information complementary to the Li data confirming a LLZO core (see Fig. S1†). XRD analysis of the synthesized powder and the prepared composites (Fig. 1e) clearly indicate that there are no chemical/crystallographic changes during the milling procedure. This observation rules out the possibility of a solid-state reaction between the LLZO and the LPS. Further, Rietveld analysis of the parent electrolytes estimated the samples to be near phase purity (LLZO – 98.1%, Li2Zr2O7 1%, LiAlO3 0.8% and LPS – 99.2%, Li2S – 0.8%), while the phase compositions of the composite electrolyte were estimated at 81.8% LPS and 18.2% LLZO for an 80:20 mixture. The analysis also confirmed the absence of any contamination from the milling media (ZrO2). The stress, strain and particle size properties of the parent electrolytes were derived to be unchanged in the composite electrolyte thus confirming the absence of structural or chemical changes during the composite preparation.
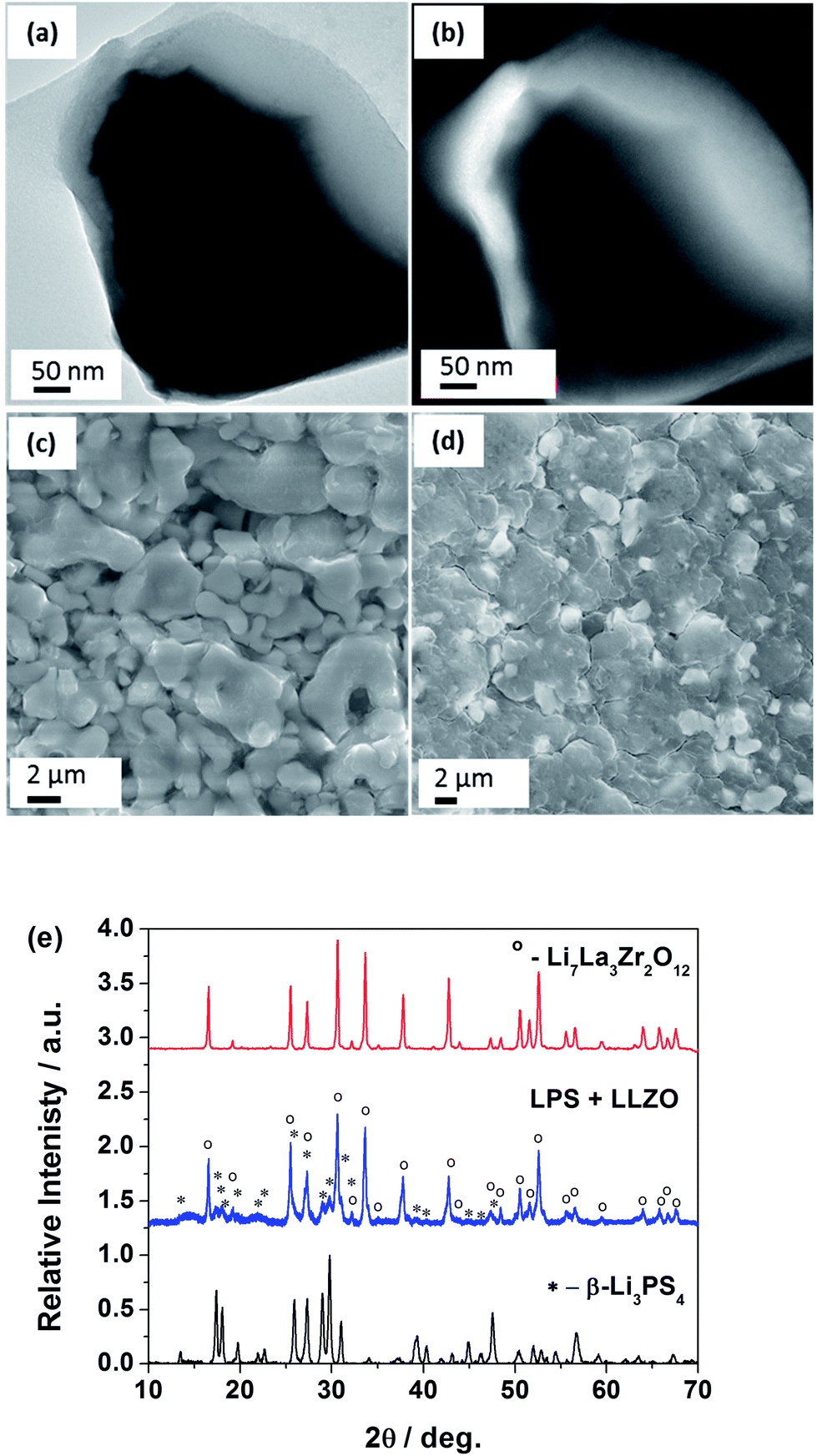 | ||
| Fig. 1 (a) A TEM image of the LLZO–LPS (nanocrystalline) composite electrolyte clearly illustrates the core–shell structure; (b) an EELS map shows a higher Li concentration across the LLZO–LPS interface (the bright part has a high concentration of Li); (c) an SEM image of the cold-pressed LLZO shows evident open porosity; (d) an SEM image of the cold-pressed composite electrolyte illustrates processability to a dense membrane; (e) XRD spectra confirm that no chemical reaction between the LLZO and the LPS. All peaks in the composite were identified as from its parent electrolytes. | ||
LLZO under the cold-press condition yields a pellet with significant amount of open porosity (39% calculated) (Fig. 1c). As a result, this pellet suffers from lower ionic conductivity and a lack of structural stability. The mechanical processing of LLZO along with the soft and sticky LPS results in a LPS shell over the hard LLZO core (Fig. 1a). This sticky shell significantly improves in the particle–particle adhesion and hence the material processability of LLZO. The addition of LPS, as little as 10% wt, aids in improving the structural stability of the fabricated membrane. This improvement is seen in the difference between the blank LLZO membranes (Fig. 1c) and the 70:30 (LPS:LLZO) membranes (Fig. 1d). Membranes with the addition of LPS result in a significant reduction in the porosity at higher weight fractions of LPS (>50%). The 70:30 membranes are extremely dense membranes with no observed open porosity which is in contrast to the blank LLZO. Thus the composite electrolyte vastly improves the room temperature processability of LLZO.
LPS–LLZO composite improves the ionic conductivity of its parent components
The maximum Li-ion conductivity of 5.36 × 10−4 S cm−1 is measured at the 70:30 (LPS:LLZO) composition, while the samples with ≤40 wt% LLZO exhibit ionic conductivity greater than the parent LPS electrolyte (Fig. 2a). The maximum conductivity of the composite is greater than the individual conductivities of LPS (1.6 × 10−4 S cm−1) and LLZO (4.0 × 10−4 S cm−1).12,15 Activation energies calculated from the Arrhenius measurements (see Fig. 2a, line b) reveal a contrasting behavior to the conductivity. Activation energy starts increasing for samples beyond 20 wt% of LLZO. The activation energies for the higher conducting samples (>40% LPS) range between 0.349 eV and 0.397 eV, falling within the range of the reported values for LPS (0.356 eV (ref. 15)) and the sintered Al-substituted cubic LLZO 0.26–0.34 eV.11–13,17 The increase in the activation energy is a direct result of the inclusion of higher weight fractions of non-sintered LLZO. The absence of well sintered and formed grain boundaries increases their contribution to resistance. Since grain boundaries have higher activation energy than the bulk, the activation energy increases with increasing LLZO weight fractions beyond the optimum concentration. Hence, increasing weight fractions of LLZO show an increased resistance arising from cold pressing and sample porosity. The sample porosity is evident under SEM where the images (see Fig. 1c and d) clearly indicate the composite membranes with minimal porosity while the LLZO sample contains considerable porosity (Fig. 1a). The blank LPS sample is the least porous of all samples (Table S1†). Thus, further reduction in porosity of the composite samples can significantly enhance the measured conductivities for the composite electrolyte.
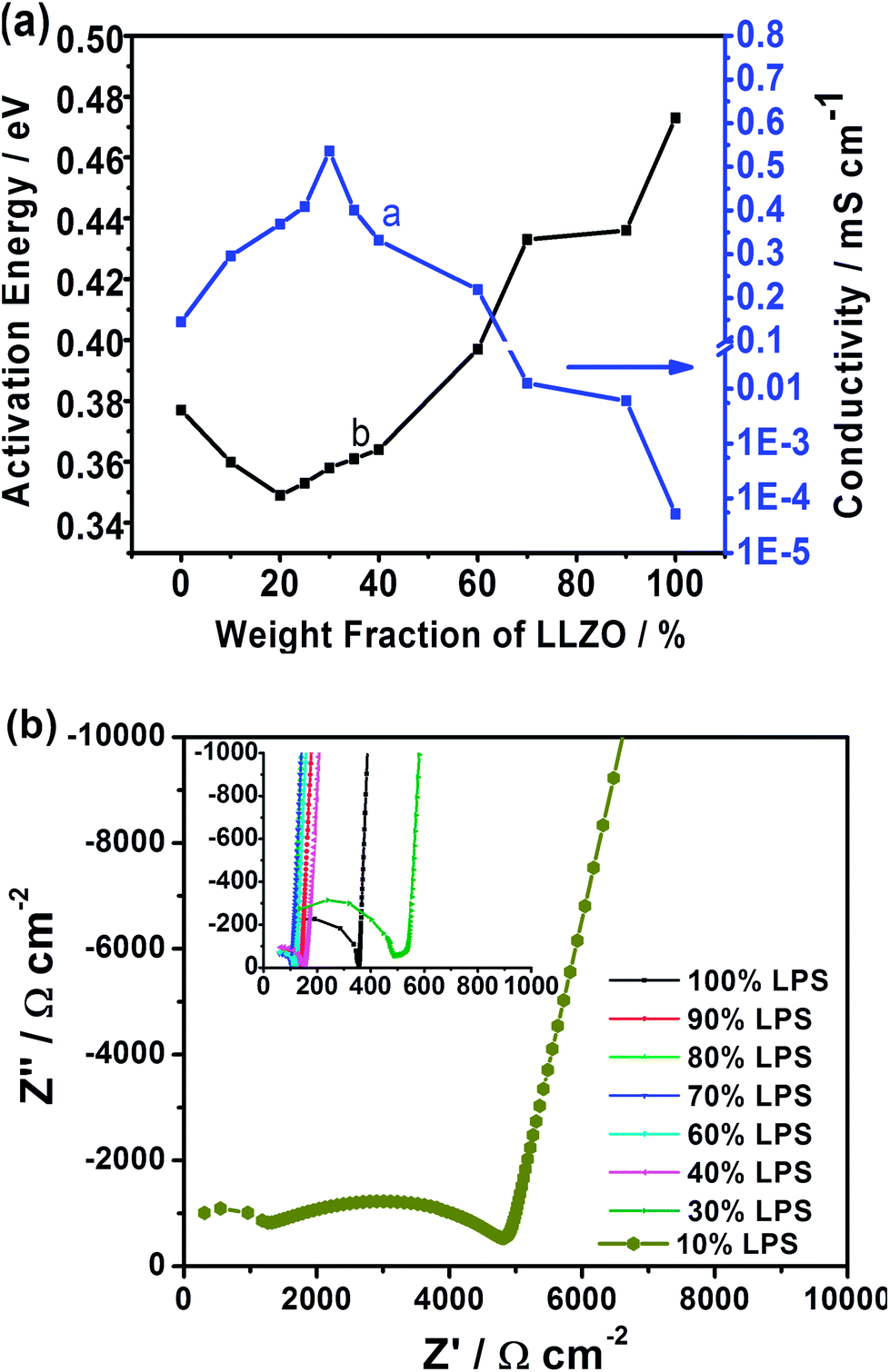 | ||
| Fig. 2 (a) Plot of ionic conductivity (right y-axis) and activation energy (left y-axis) as a function of the weight fractions of LLZO in the composite; (b) room temperature Nyquist data for the 10:90 LPS:LLZO mixture resolving the contribution from the space charge layer in comparison (inset plot) with the remaining compositions exhibiting a single component behavior. | ||
As mentioned in the earlier text, the Rietveld refinement shows the absence of secondary phase formation in the composite. Therefore, an interesting question is that why the composite electrolyte has an ionic conductivity higher than the parent compounds, which are superionic conductors. What then is the contributing factor to the enhancement in ionic conductivity of the composite? Apparently, the enhancement is from the interface between LLZO and LPS, since it is the only addition to the bulk LLZO and LPS within the composite system. A detailed examination of the room temperature Nyquist plots (see Fig. 2b) reveals a subtle change in behavior at the 10:90 composition of LPS:LLZO. Unlike the other compositions, this mixture exhibits two semi-circular regions indicating a higher conducting and a lower conducting component. Since both the LPS and LLZO typically present a single component,11,12,15,18,19,24 the dual component behavior is induced by the interfacial layer. At the aforementioned composition, the bulk matrix is still occupied by the porous non-dense LLZO that results in a low conductivity. However, the presence of 10 wt% LPS is significant enough to improve the ionic conductivity by two orders of magnitude over the blank LLZO (6.03 × 10−6 S cm−1vs. 5.18 × 10−8 S cm−1). The addition of this minor fraction of LPS results in an interfacial layer significant enough to improve the total conductivity, but still limited by the sample porosity and grain boundary resistance. Thus the resulting Nyquist data are able to resolve the contributions from the interfacial layer. At the higher conducting compositions, the data are limited by the frequency limitations of the impedance analyzer from resolving these interfacial contributions.
The interface between the LLZO and the LPS leads to the formation of a layer of ionic and electronic point defects, termed as the space-charge layer.25 A space-charge layer can significantly enhance the ionic conductivity of solid state electrolyte mixtures depending on its nature.25–33 Higher ionic conductivities have been achieved in a Li4GeS4–Li3PS4 composite system utilizing the space charge effect.34 The space charge effect is also observed at an interface between the sulfide electrolyte and oxide cathodes due to the vast difference in chemical potentials across the interface.35 Most likely the space-charge layer of the LLZO–LPS composite is akin to that of the above systems with a redistribution of the vacancies and interstitial sites across the interface. This interface now contributes to an enhancement in ionic conductivity as observed with the room temperature Nyquist plots (Fig. 2b). An enhanced ionic conductivity is explained by the space charge layer effect. In addition, the phase of LLZO is important to the overall conductivity of the composite electrolyte. A control sample was prepared by using the lower conducting tetragonal phase of LLZO.13,36,37 The resulting composite electrolyte of the tetragonal LLZO with LPS has a 5-fold drop in ionic conductivity (5.21 × 10−5 S cm−1 as against 2.6 × 10−4 S cm−1 observed with cubic LLZO for the 50:50 composition), thus confirming that a high conduction phase of LLZO favors the overall conductivity of the composite. A set of control experiments conducted on various ionic conductors revealed that this enhancement is unique to the LPS–LLZO system for the two parent systems. A different combination always resulted in significantly lower ionic conductivity (Fig. S2†) in comparison with the parent components.
Composite electrolyte exhibits excellent electrochemical stability and cyclability
The electrochemical stability of the composite was investigated using cyclic voltammetry (Fig. 3a). The composite is stable up to a potential of 5 V vs. Li/Li+ as observed for its parent electrolytes.15,16 Additionally, the anodic current is present only below 0 V vs. Li/Li+ (arising from electrodeposition of Li) thus confirming the stability of the composite electrolyte with metallic Li. A symmetric cell was fabricated with Li/LPS–LLZO/Li setup to demonstrate excellent stability and cyclability of the composite electrolyte with metallic Li. A minimal polarization of 32.9 mV is observed under a current density of 0.1 mA cm−2 under ambient conditions (25 °C). The direct-current (DC) conductivity of the full cell was calculated to be 3.36 × 10−4 S cm−1 in comparison with the ionic conductivity of the electrolyte measured at 5.36 × 10−4 S cm−1 using EIS. It must be noted that the total DC conductivity includes interfacial resistances from two Li/LLZO–LPS interfaces. The resistance at the interface is thus at the same order of the composite electrolyte, clearly indicating that the lithium ion transport in the symmetric cell is not kinetically limited at the interface. The composite electrolyte exhibited a lower polarization than that was observed for the parent LPS electrolyte.15 The low interfacial resistance is in stark contrast to the high interfacial resistances for the LLZO system.21 Once again, the composite electrolyte shows an interfacial property superior to its parent compounds. Excellent cycling performance was achieved in the symmetric cell. Representative cycles are shown in Fig. 3b. The commonly observed voltage deviations and cell shorting in some less conductive solid electrolytes or defective membranes have not been observed in the Li/LLZO–LPS/Li symmetric cell, even after a few hundred cycles. All cycles have a characteristic flat voltage profile, which indicates an exceptional stability for symmetric cycling. These observations provide additional evidence to the high compatibility of the composite electrolyte with metallic lithium and an astonishing processability of the material through cold-pressing.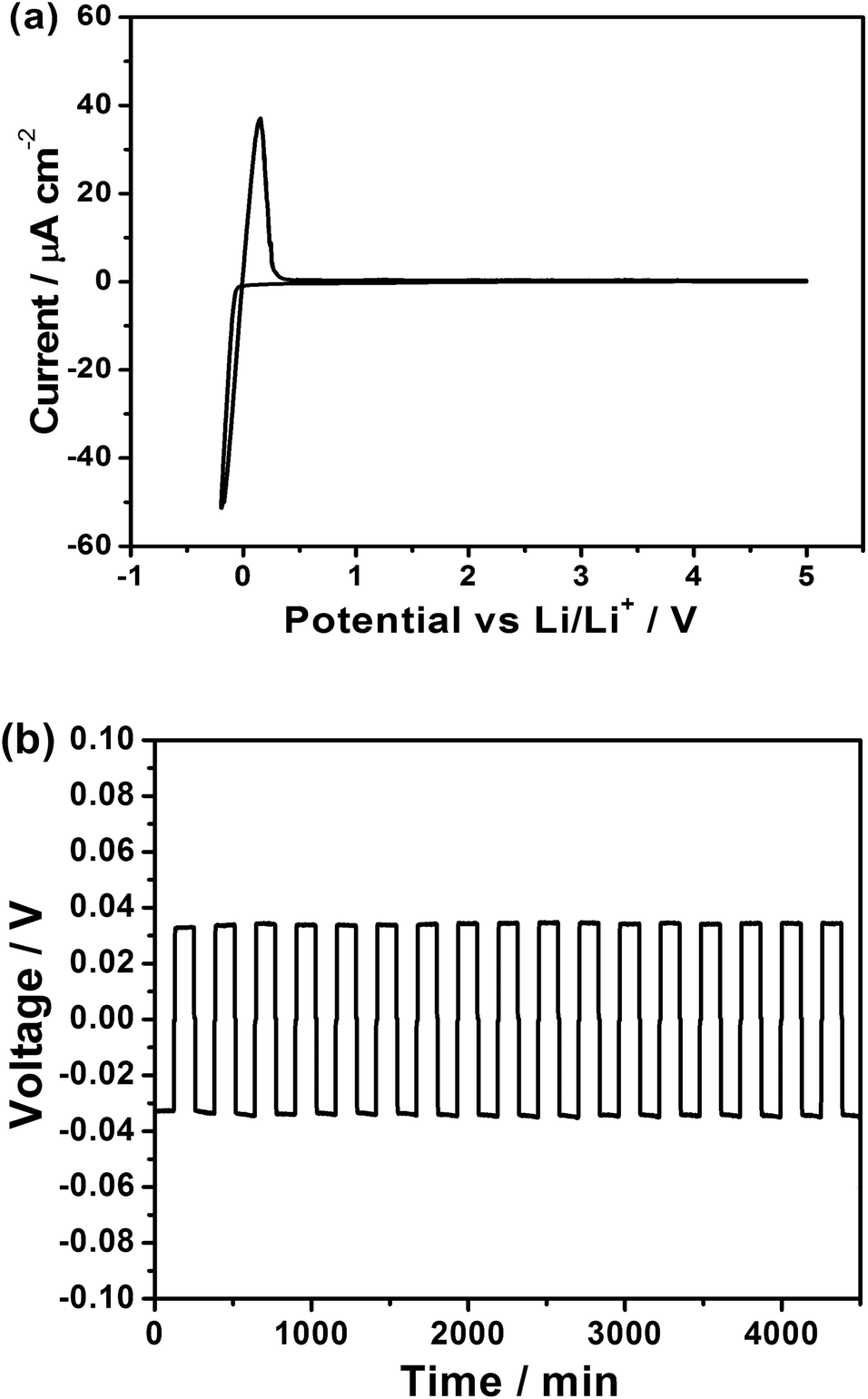 | ||
| Fig. 3 (a) Cyclic voltammetry measurement of a Pt/LLZO–LPs/Li cell demonstrates a wide electrochemical window of 5 V versus metallic lithium; (b) representative cycling data of a Li/LPS–LLZO/Li symmetric cell at a current density of 0.1 mA cm−2 and room temperature. | ||
Conclusions
To summarize, the “hard” oxide LLZO and “soft” sulfide LPS form an excellent composite ceramic superionic conductor through a facile mechanical mixing method. This composite electrolyte successfully combines and enhances the properties of its parent electrolytes. The soft sulfide aids in overcoming the processability barrier of the hard oxide electrolytes. Dense electrolyte membranes can thus be prepared through cold-pressing. The improved processability brings the solid electrolytes a step closer to the use of ceramic solid electrolytes in practical batteries. The composite electrolyte has a higher Li-ion conductivity than that of its parent electrolytes. The enhanced ionic conductivity is believed as an effect of the space charge layer formed at the interfaces of LLZO and LPS particles. The composite electrolyte has an excellent electrochemical stability and also succeeds in achieving low interfacial resistance with the metallic Li anode. The concept of compositing oxides with sulfides to achieve improved mechanical properties, ionic conductivity, and electrochemical properties of existing solid electrolytes opens an avenue for the discovery of new materials that are enablers for future all-solid state batteries. Large scale energy storage needs batteries with high energy and inherent safety. All-solid state batteries meet these needs.Acknowledgements
This work was sponsored by the Division of Materials sciences and Engineering, Office of Basic Energy Sciences U.S. Department of Energy (DOE). The synthesis and characterization of materials were conducted at the Center for Nanophase Materials Sciences, which is sponsored at Oak Ridge National Laboratory by the Division of Scientific User Facilities, U.S. DOE. Potential use of this solid electrolyte in lithium–sulfur batteries was supported by the U.S. Department of Energy (DOE)/Energy Efficiency and Renewable Energy (EERE) through the Office of Vehicle Technologies.Notes and references
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed
.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed
- X. L. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821–9826 RSC
- C. D. Liang, N. J. Dudney and J. Y. Howe, Chem. Mater., 2009, 21, 4724–4730 CrossRef CAS
- Z. Lin, Z. Liu, N. J. Dudney and C. Liang, ACS Nano, 2013, 7, 2829–2833 CrossRef CAS PubMed
- Z. Lin, Z. Liu, W. Fu, N. J. Dudney and C. Liang, Angew. Chem., Int. Ed., 2013, 52, 7460–7463 CrossRef CAS PubMed
- P. Knauth, Solid State Ionics, 2009, 180, 911–916 CrossRef CAS PubMed
- Y. H. Cho, J. Wolfenstine, E. Rangasamy, H. Kim, H. Choe and J. Sakamoto, J. Mater. Sci., 2012, 47, 5970–5977 CrossRef CAS PubMed
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed
- Y. F. Mo, S. P. Ong and G. Ceder, Chem. Mater., 2012, 24, 15–17 CrossRef CAS
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed
- E. Rangasamy, J. Wolfenstine and J. Sakamoto, Solid State Ionics, 2012, 206, 28–32 CrossRef CAS PubMed
- C. A. Geiger, E. Alekseev, B. Lazic, M. Fisch, T. Armbruster, R. Langner, M. Fechtelkord, N. Kim, T. Pettke and W. Weppner, Inorg. Chem., 2011, 50, 1089–1097 CrossRef CAS PubMed
- J. E. Ni, E. D. Case, J. S. Sakamoto, E. Rangasamy and J. B. Wolfenstine, J. Mater. Sci., 2012, 47, 7978–7985 CrossRef CAS
- Z. Liu, W. Fu, E. A. Payzant, X. Yu, Z. Wu, N. J. Dudney, J. Kiggans, K. Hong, A. J. Rondinone and C. Liang, J. Am. Chem. Soc., 2013, 135, 975–978 CrossRef CAS PubMed
- S. Ohta, T. Kobayashi and T. Asaoka, J. Power Sources, 2011, 196, 3342–3345 CrossRef CAS PubMed
- J. Sakamoto, E. Rangasamy, H. Kim, Y. Kim and J. Wolfenstine, Nanotechnology, 2013, 24, 424005 CrossRef PubMed
- J. L. Allen, J. Wolfenstine, E. Rangasamy and J. Sakamoto, J. Power Sources, 2012, 206, 315–319 CrossRef CAS PubMed
- E. Rangasamy, J. Wolfenstine, J. Allen and J. Sakamoto, J. Power Sources, 2013, 230, 261–266 CrossRef CAS PubMed
- J. Wolfenstine, J. Ratchford, E. Rangasamy, J. Sakamoto and J. L. Allen, Mater. Chem. Phys., 2012, 134, 571–575 CrossRef CAS PubMed
- Y. Jin and P. McGinn, J. Power Sources, 2011, 196, 8683–8687 CrossRef CAS PubMed
- K. Takada, Acta Mater., 2013, 61, 759–770 CrossRef CAS PubMed
- J. Akedo, J. Am. Ceram. Soc., 2006, 89, 1834–1839 CrossRef CAS
- W. E. Tenhaeff, E. Rangasamy, Y. Wang, A. P. Sokolov, J. Wolfenstine, J. Sakamoto and N. J. Dudney, ChemElectroChem, 2013 DOI:10.1002/celc.201300022
- J. Maier, Prog. Solid State Chem., 1995, 23, 171–263 CrossRef CAS
- C. C. Liang, J. Electrochem. Soc., 1973, 120, 1289–1292 CrossRef CAS PubMed
- N. J. Dudney, Annu. Rev. Mater. Sci., 1989, 19, 103–120 CrossRef CAS
- N. J. Dudney, Solid State Ionics, 1988, 28, 1065–1072 CrossRef
- N. J. Dudney, J. Am. Ceram. Soc., 1985, 68, 538–545 CrossRef CAS
- J. Maier, Nat. Mater., 2005, 4, 805–815 CrossRef CAS
- J. Maier, Solid State Ionics, 1987, 23, 59–67 CrossRef CAS
- J. Maier, J. Phys. Chem. Solids, 1985, 46, 309–320 CrossRef CAS
- J. Maier, Ber. Bunsen-Ges, 1985, 89, 355–362 CrossRef CAS
- R. Kanno and M. Maruyama, J. Electrochem. Soc., 2001, 148, A742–A746 CrossRef CAS PubMed
- N. Ohta, K. Takada, L. Q. Zhang, R. Z. Ma, M. Osada and T. Sasaki, Adv. Mater., 2006, 18, 2226–2229 CrossRef CAS
- J. Awaka, N. Kijima, H. Hayakawa and J. Akimoto, J. Solid State Chem., 2009, 182, 2046–2052 CrossRef CAS PubMed
- J. Wolfenstine, E. Rangasamy, J. L. Allen and J. Sakamoto, J. Power Sources, 2012, 208, 193–196 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ta15223e |
| This journal is © The Royal Society of Chemistry 2014 |