 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High voltage sulphate cathodes Li2M(SO4)2 (M = Fe, Mn, Co): atomic-scale studies of lithium diffusion, surfaces and voltage trends†
John M.
Clark
a,
Christopher
Eames
a,
Marine
Reynaud‡
b,
Gwenaëlle
Rousse
cd,
Jean-Noël
Chotard
b,
Jean-Marie
Tarascon
d and
M. Saiful
Islam
*a
aDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: m.s.islam@bath.ac.uk
bLaboratoire de Réactivité et Chimie des Solides (LRCS), Université de Picardie Jules Verne, CNRS UMR 7314, 33 rue Saint Leu, 80039 Amiens Cedex, France
cIMPMC, CNRS UMR 7590, Université Pierre et Marie Curie UPMC Univ. Paris 06, 4 place Jussieu, 75252 Paris Cedex 05, France
dCollege de France, 11 place Marcelin Berthelot, 75231 Paris Cedex 05, France
First published on 27th March 2014
Abstract
The search for high voltage cathodes for lithium-ion batteries has led to recent interest in the monoclinic Li2Fe(SO4)2 material which has a voltage of 3.83 V vs. lithium, the highest recorded for a fluorine-free iron-based compound. Here we investigate the defect, surface and lithium migration properties of the Li2M(SO4)2 (M = Fe, Mn, Co) materials using combined atomistic modelling and density functional theory (DFT) techniques. All intrinsic defect types including Li/M antisite disorder are found to be of high energy, suggesting insignificant concentrations. Low activation energies are found for lithium migration along the a-axis channels giving rise to long-range 1D diffusion, which are supported by molecular dynamics (MD) simulations. For the crystal morphology a significant surface area is exposed to these 1D diffusion channels, which would allow facile Li insertion and extraction. Using DFT simulations we reproduce the high voltage of the Li2Fe(SO4)2 material in accord with electrochemical data and also examine local structural distortions on lithium extraction.
1. Introduction
The search for alternative cathode materials to replace the layered LiCoO2 system conventionally used within rechargeable lithium batteries has generated considerable research activity.1–7 The Co-based materials pose problems associated with cost, safety and sustainability, particularly for large-scale applications (such as electric vehicles and grid storage). An avenue that has been heavily probed in the quest for new materials involves the combination of abundant Fe and stable phosphate groups (PO43−), with most interest focused on olivine-structured LiFePO4.1–5,8 In recent studies other polyanion framework compounds have been investigated as alternative cathode materials including the following: Li2FeSiO4,9,10 LiFeBO3,11 LiFeSO4F,12–15 Li2FePO4F,16 Li2FeP2O7,17 and LiFeSO4OH.15,18–20More recently, the newly synthesised lithium iron sulphate, with the composition Li2Fe(SO4)2, was found to display a theoretical capacity of 102 mA h g−1 and a high potential of 3.83 V vs. Li/Li+ for the Fe2+/Fe3+ redox couple, which is the highest reported voltage for a fluorine-free iron-based compound.21 Further studies of the structural and transport properties will provide useful insights into this significant iron–sulphate system and may subsequently provide a new platform for related lithium battery electrode research. There is also interest in both the previously reported Li2Co(SO4)2 phase21 and the recently synthesized Li2Mn(SO4)2 phase.22
To fully understand the factors influencing the electrochemical behaviour of the Li2M(SO4)2 (M = Fe, Mn, Co) electrode materials, it is clear that knowledge of the fundamental solid-state properties is needed on the atomic scale. The present study uses well-established atomistic simulation and density functional theory (DFT) methods to investigate key issues related to their defect chemistry, lithium diffusion, surface structures and voltage trends. As shown in our previous studies of other lithium battery cathode materials, atomistic simulation is well suited to treating the extensive lattice relaxation around charged defects, surfaces and migrating ions in polar inorganic solids.23–29 DFT methods30–34 are used to provide insights into the high voltages and the local structural changes observed during electrochemical cycling of these sulphate materials.
2. Methodology
As these atomistic and DFT techniques are described in detail elsewhere,35–37 only a general outline will be given here. For the atomistic simulations, the interatomic interactions consist of a long-range Coulombic term and a short-range component representing electron–electron repulsion and van der Waals interactions. The short-range interactions were modelled using the two-body Buckingham potential.35 An additional three-body term was used for the SO42− units as previously used for sulphates,38–41 silicates27 and phosphates.23–25 The shell model42 was employed to account for polarizability effects. Li–O, Fe–O and O–O interatomic potentials were taken directly from the recent study of the related tavorite LiFeSO4F material,28 whilst the Mn–O and Co–O potentials were refined from those used for the study of LiMnPO4 and LiCoPO4.24 For the more covalent sulphate (SO42−) component, the interatomic potential model successfully formulated to simulate M2SO4 (M = Na, K, Rb and Cs) and XSO4 (X = Sr, Ca, Ba) was used.38–41 Table S1 (ESI†) lists the interatomic potential parameters used in this study.As argued previously, employing these interatomic potential methods are assessed primarily by their ability to reproduce observed crystal properties. Indeed, they are found to work well, even for phosphate and silicate cathodes23–29 where there is undoubtedly a degree of covalency. The lattice relaxation about defects (such as Li vacancies) and migrating ions was treated by the Mott–Littleton scheme incorporated in the GULP code (v4.0),43 which partitions the crystal lattice into two regions, where ions in the inner region immediately surrounding the defect (∼1000 ions) are relaxed explicitly. Hence, the polyanionic framework is not treated as a rigid lattice.
Molecular dynamics (MD) simulations were performed with DL_POLY (v2.2)44 using the same interatomic potentials and a supercell consisting of 8 × 4 × 4 unit cells (3328 atoms) with 10% delithiation in which the lithium vacancies were initially distributed randomly. The systems were first equilibrated for at least 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps (with a time step of 0.5 fs). The main simulation runs of 2 million steps were then performed with an NPT ensemble (at T = 473 K) to give a long simulation time of 1 ns. NPT was used to link up with experimental conditions and slight volume changes. However, it should be noted that the large supercells and long time-scales required are currently not accessible by ab initio methods.
000 steps (with a time step of 0.5 fs). The main simulation runs of 2 million steps were then performed with an NPT ensemble (at T = 473 K) to give a long simulation time of 1 ns. NPT was used to link up with experimental conditions and slight volume changes. However, it should be noted that the large supercells and long time-scales required are currently not accessible by ab initio methods.
The surface structures in this study were simulated with potentials-based methods using the METADISE code.45 Atoms near to the surface (region I) are allowed to relax while bulk atoms (region II), which represent the rest of the crystal, are kept fixed and ensure that all the ions in region I experience the forces associated with the rest of the crystal and that the energies are fully converged. The surface energy, γ, of a crystal face is defined as the excess in energy of a surface simulation over the energy of a bulk system containing the same number of atoms, per unit surface area. The equilibrium morphology of the crystal can be obtained from the surface energies using a Wulff construction in which the equilibrium form of a crystal should possess minimal total surface energy for a given volume. The surface area of a facet is not directly proportional to the surface energy. The shape would be such that the normal vector to the face from a point within the crystal would be proportional to the surface energy of that face.
DFT calculations were performed using the plane wave code VASP (v5.4).46 Since we required optimised lattice parameters, the basis set was converged against the stress which is more sensitive to an under-converged basis set than the forces. A cutoff energy of 750 eV with a k-point mesh density of at least 0.04 Å−1 was needed to adequately converge the stress (6 × 4 × 4 grid). PAW potentials47,48 and the PBE exchange and correlation functional49 were used. An antiferromagnetic ordering of the moments on all transition metal (Fe, Mn and Co) atoms was found to be lower in energy than a ferromagnetic ordering, in line with magnetic measurements (TN < 8 K for Li2MII(SO4)2 with M = Co, Mn, Fe, and TN = 35 K for LiFeIII(SO4)2) and with the magnetic structures deduced at 2 K from neutron powder diffraction.22 DFT + U methodology was used with an effective Hubbard Ueff = U − J = 4.0, 3.9 and 5.7 eV (J = 1.0 eV) for Fe, Mn and Co respectively; these values are in agreement with the U values derived for Fe-, Mn- and Co-based cathodes.50
Previous DFT studies on a variety of oxide electrode materials31,51,52 have shown that such methods are well suited to probing lithium insertion/extraction properties and to predicting precise trends in cell voltages. Using the diffraction refined structural data for the lithiated (Li2M(SO4)2 materials) and delithiated (LiFe(SO4)2) structures we have calculated the open circuit voltages using:
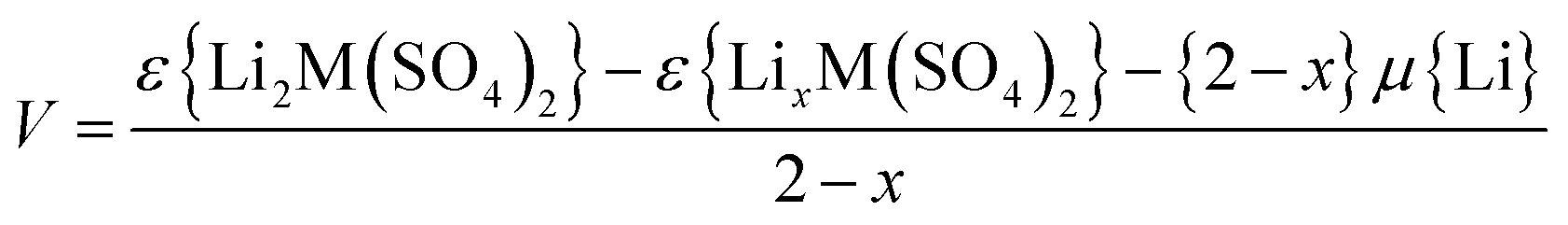 | (1) |
3. Results and discussion
3.1 Structures and intrinsic defects
The structures of the monoclinic Li2M(SO4)2 (M = Co, Mn, Fe) compounds have previously been reported,21,22 and the atomic positions and unit cell parameters are collected in Table S2.† This structure comprises isolated MO6 octahedra linked through shared oxygen vertices with the surrounding SO4 tetrahedra. Each octahedron is linked to six SO4 tetrahedra which are orientated in a star or pinwheel pattern when viewed along the b-axis. Conversely, each SO4 is only bound to three MO6 octahedra and the fourth unshared corner of the SO4 tetrahedron points into an open channel where the lithium resides, as shown in Fig. 1.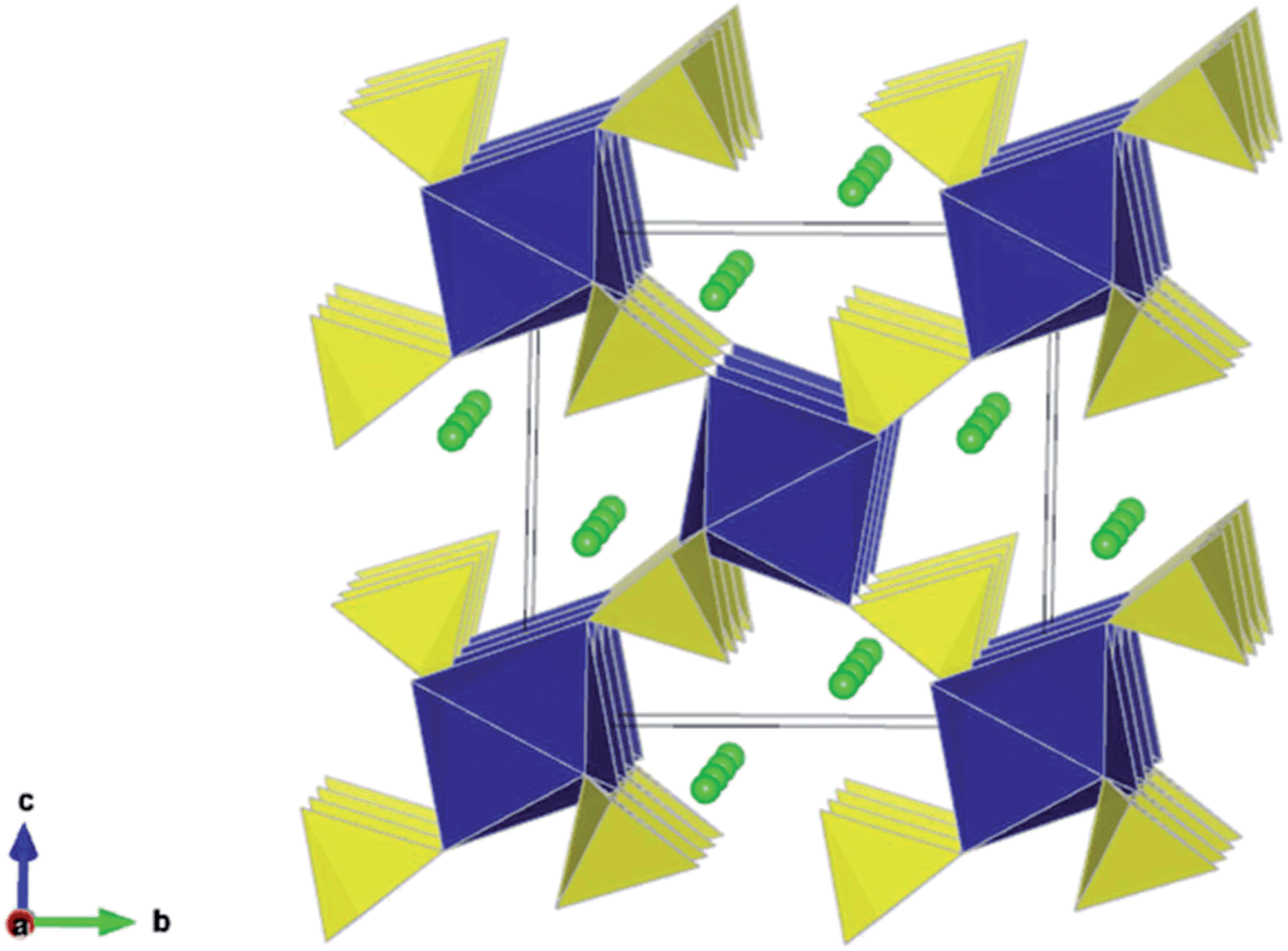 | ||
| Fig. 1 Li2M(SO4)2 (M = Fe, Mn, Co) crystal structure showing MO6 octahedra (blue), SO4 tetrahedra (yellow) and chains of Li ions (green) running parallel to the a-axis. [Experimental refinement data in Table S2.†] | ||
Despite the isostructural nature of the three Li2M(SO4)2 phases, only the Fe analogue is observed to be electrochemically active.21,22 Upon cycling, the Li2Fe(SO4)2 material transforms to the delithiated phase LiFe(SO4)2 through a biphasic mechanism, as suggested by the plateau observed at 3.83 V vs. Li/Li+ on the electrochemical curve and further confirmed by in situ XRD measurements.21 The delithiated phase LiFe(SO4)2 can also be obtained through chemical oxidation of Li2Fe(SO4)2. It was found that the delithiated phase maintains the general 3D framework of Li2Fe(SO4)2,22 but the FeO6 octahedra and SO4 tetrahedra undergo slight rotations upon lithium removal. As a result, each octahedron is still linked to six SO4 tetrahedra with the fourth unshared corner of the SO4 tetrahedra pointing at the open channels where the lithium resides. However, in contrast to the lithiated phase, the lithium ions of the oxidized LiFe(SO4)2 were localized at the centre of the channels, in a half-occupied general position in the vicinity of the (1/2 0 1/2) site.22 The lower panel of Table S2† presents the precise atomic positions obtained from a joint refinement of the synchrotron XRD and NPD data,22 and Fig. 2 shows the resulting crystal structure.
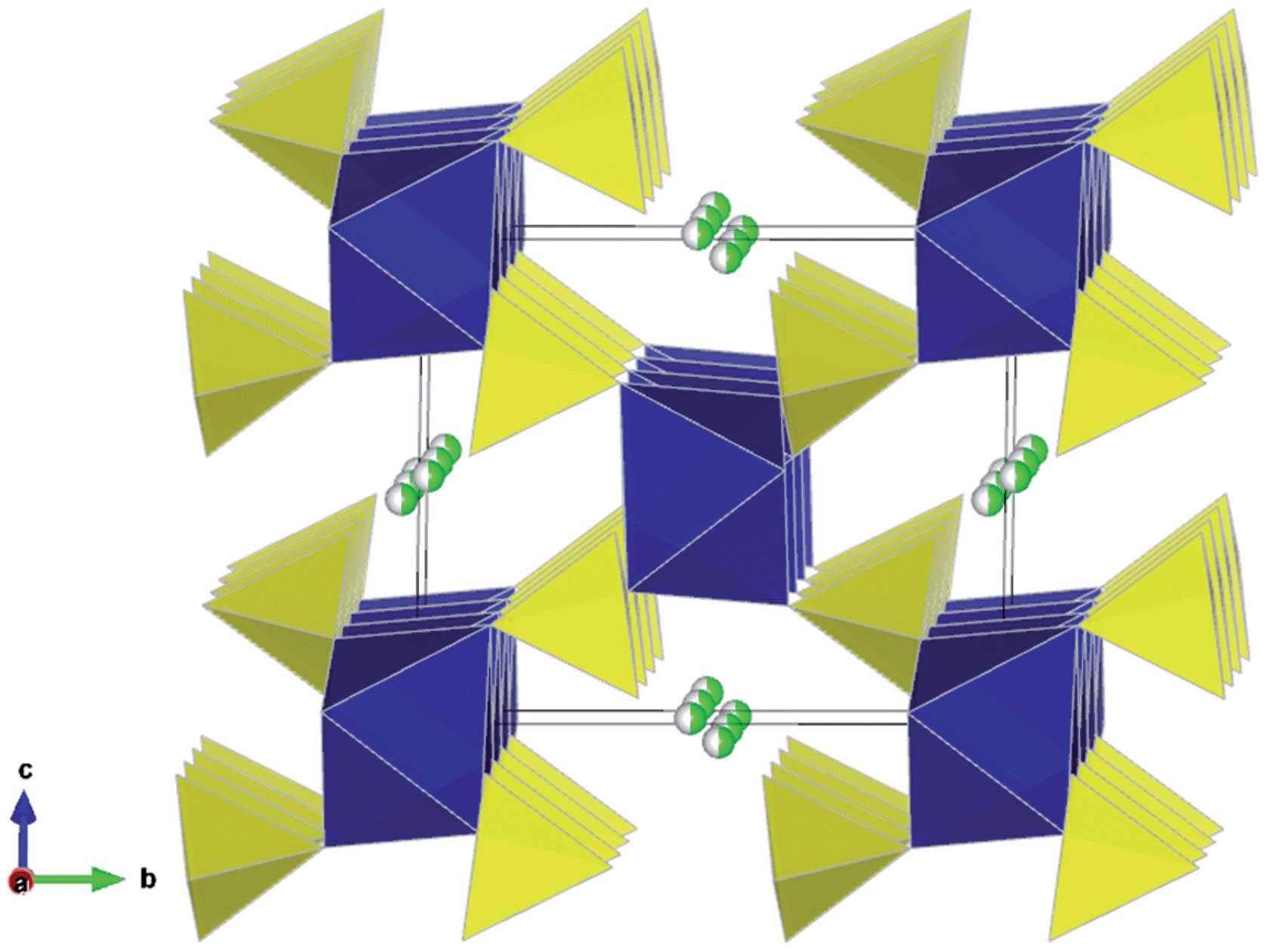 | ||
| Fig. 2 Crystal structure of the delithiated phase LiFe(SO4)2 showing FeO6 octahedra (blue), SO4 tetrahedra (yellow) and half occupied Li sites (green/white) running parallel to the a-axis. [Experimental refinement data in Table S2.†] | ||
The starting point of the simulation study was to reproduce the experimentally observed crystal structures exhibited by the Li2M(SO4)2 (M = Fe, Mn, Co) electrode materials. A direct comparison of their calculated and experimental structures is given in Table 1, which shows that the unit cell parameters calculated by interatomic potentials deviate from the experiment by at most 0.1 Å, and in most cases by much less; the same is found for the individual bond lengths with mean deviations less than 0.03 Å. The successful reproduction of these complex sulphate structures gives us confidence that the potential models can be used reliably for subsequent defect, diffusion and surface calculations.
| Parameter | a (Å) | b (Å) | c (Å) | β (°) |
|---|---|---|---|---|
| Li2Fe(SO4)2 | ||||
| Calc. | 4.9518 | 8.2212 | 8.9293 | 121.86 |
| Expt.21 | 4.9886 | 8.2062 | 8.8293 | 121.75 |
| Δ | −0.0368 | 0.0150 | 0.1000 | 0.11 |
| Li2Mn(SO4)2 | ||||
| Calc. | 4.9478 | 8.2930 | 8.9456 | 121.58 |
| Expt.22 | 4.9920 | 8.3396 | 8.8614 | 121.23 |
| Δ | −0.0442 | −0.0466 | 0.0842 | 0.35 |
| Li2Co(SO4)2 | ||||
| Calc. | 4.9596 | 8.0875 | 8.8142 | 121.37 |
| Expt.21 | 4.9787 | 8.1113 | 8.7831 | 121.81 |
| Δ | 0.0191 | 0.0238 | 0.0311 | −0.44 |
As noted, insight into the defect properties of cathode materials is crucial to the full understanding of their electrochemical behaviour. Isolated point defect (vacancy and interstitial) energies were calculated for these sulphate systems, which were combined to determine the formation energies for Frenkel- and Schottky-type intrinsic defects. The following equations represent the reactions involving these defects (using Kröger–Vink notation and where M = Fe, Mn or Co):
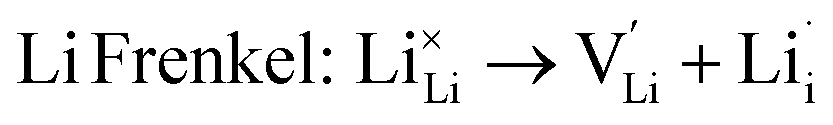 | (2) |
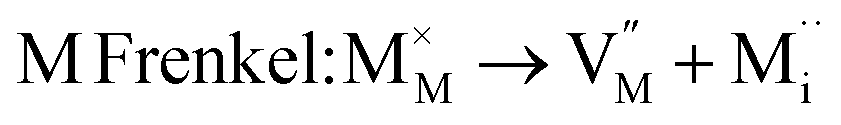 | (3) |
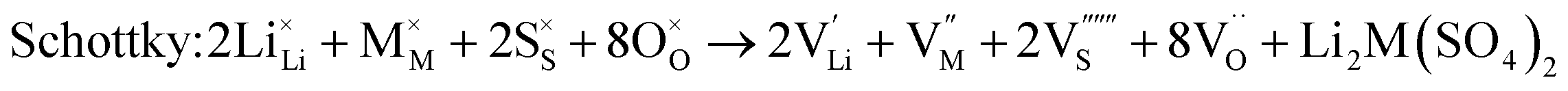 | (4) |
We also examined the Li/M “anti-site” pair defect, which involves the exchange of an Li+ ion (radius 0.76 Å) with an M2+ ion (Fe2+ radius 0.78 Å, Mn2+ radius 0.83 Å, and Co2+ radius 0.75 Å), according to:
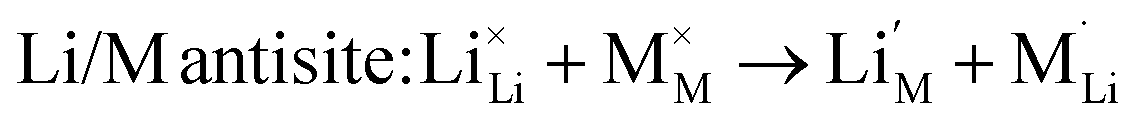 | (5) |
This type of defect is worth investigating since Li/M anti-site or cation exchange effects have been found in other polyanionic systems including LiMPO4 (M = Mn, Fe, Co, Ni)23,24 and Li2FeP2O7.29
Examination of the resulting defect energies listed in Table 2 reveals two main predictions. First, the formation of all Frenkel and Schottky defects is unfavourable in all Li2M(SO4)2 structures. This suggests that the creation of vacancies (especially oxygen vacancies) and interstitial defects are unfavourable on energetic grounds. Second, the antisite energies are also relatively high, which suggests that there would be no significant concentration of M on Li sites at typical operating temperatures in these fluorine-free sulphate materials, and is consistent with the experimental data. This result contrasts with the LiMPO4 (M = Mn, Fe, Co and Ni) materials,23,53,54 which exhibit cation exchange behaviour between M and Li sites. The contrast may be related to differences in structure and in the bonding of the polyanion groups. Therefore these results suggest that conduction “blocking” effects involving Fe, Mn or Co on Li sites are much less likely in the Li2M(SO4)2 cathode materials.
3.2 Lithium diffusion paths
To fully understand charge/discharge rates of potential cathodes, it is important to examine the energy barriers and dimensionality of lithium diffusion. Using atomistic simulation techniques, it is possible to examine various possible Li+ transport paths, which are often difficult to probe on the atomic scale by experiment alone.Five possible migration paths in the sulphate structures were examined, including all possible jumps between adjacent lithium sites, which are symmetry equivalent; these paths are labelled L1–L5 in Fig. 3. Activation energies for these migration paths can be derived by calculating the energy of the migrating ion along the diffusion path, and are listed in Table 3.
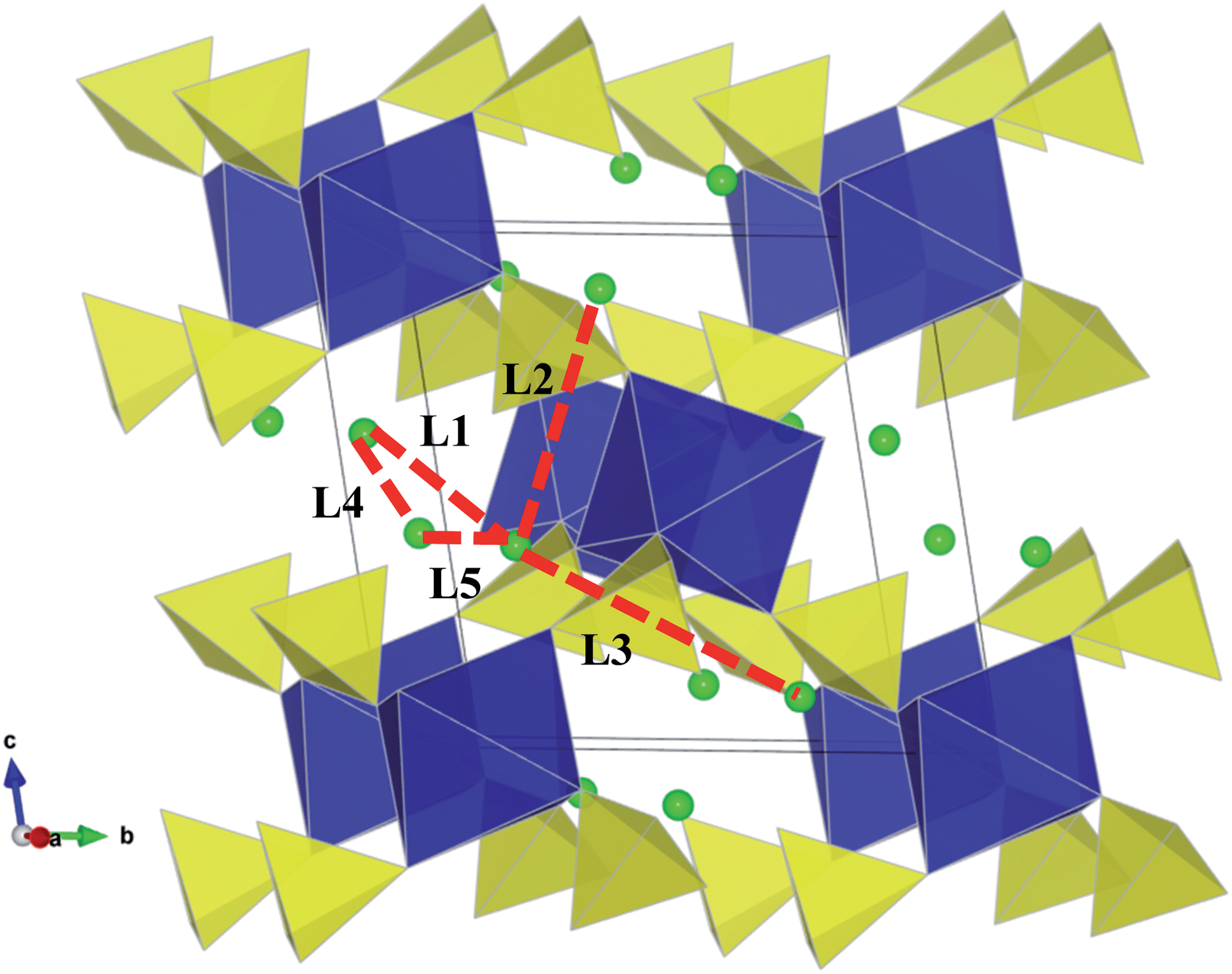 | ||
| Fig. 3 Lithium ion migration paths in Li2M(SO4)2 (M = Fe, Mn, Co) between adjacent Li sites (with initial Li positions found in Table S2†). Li–Li jumps considered are labelled L1–L5 (with Li–Li separations found in Table S3†). | ||
| Path | Activation energy (eV) | ||
|---|---|---|---|
| Li2Fe(SO4)2 | Li2Mn(SO4)2 | Li2Co(SO4)2 | |
| L1 | 0.33 | 0.39 | 0.32 |
| L2 | 1.61 | 1.58 | 1.20 |
| L3 | 1.72 | 1.93 | 1.77 |
| L4 | 0.47 | 0.54 | 0.42 |
| L5 | 1.79 | 1.74 | 1.98 |
The calculated activation energies reveal two main points. First, for all sulphates, the paths with lowest migration energies are found to be the L1 and L4 jumps. The results indicate that combinations of the L1 and L4 pathways consists of continuous diagonal or zigzag jumps enabling long-range diffusion along the [100] direction, i.e. along the a-axis channels (Fig. 4a and b). The net diffusion barriers are 0.47 eV, 0.54 eV and 0.42 eV for Li2Fe(SO4)2, Li2Mn(SO4)2, and Li2Co(SO4)2 respectively. We note that as with other battery cathode materials, there is no clear correlation between the Li–Li jump distances (Table S3†) and the migration barriers are largely due to the different local structural and steric factors at the saddle-point configuration.
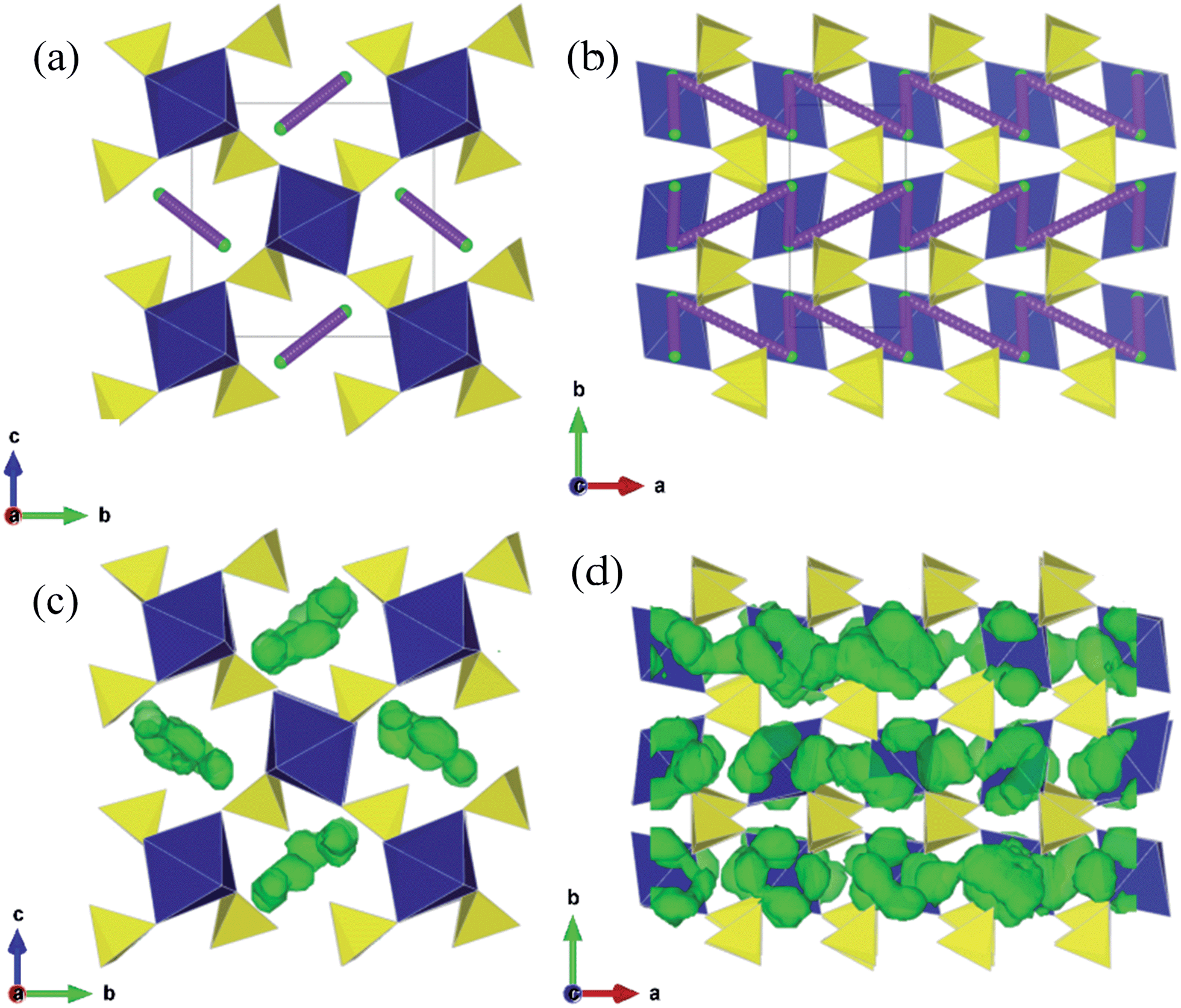 | ||
| Fig. 4 Low energy Li diffusion paths within [100] channels of Li2M(SO4)2 (M = Fe, Mn, Co). (a) and (b) Static lattice calculations of low energy Li migration path combining L1 and L4 jumps. (c) and (d) Li density plots as calculated using MD at 473 K. (a), (c) and (b), (d) are equivalent views along the a-axis and c-axis respectively. | ||
Secondly, we note that the remaining Li migration paths considered (L2, L3 and L5) were found to have high and unfavourable activation energies (≥1.2 eV); this is probably due to the migrating Li ion coming into close proximity of the MO6 octahedra. Hence it would appear that the Li2M(SO4)2 materials exhibit effective 1D transport of Li ions. We have determined experimentally the conduction activation energy in Li2Fe(SO4)2 by AC-conductivity performed on a sintered pellet using blocking electrodes within the 200–350 °C temperature range and found from linear extrapolation a value of ∼1.1 eV. This corresponds to the total conductivity of pristine stoichiometric Li2Fe(SO4)2 and will comprise both ionic and electronic conductivity terms. Hence direct comparison between this measured conductivity value and the calculated migration energies for Li ion conduction (using Li vacancy sites) is not straightforward. Despite the difference, our simulations have elucidated the pathways and dimensionality of Li ion diffusion, which are the main aims of this study.
To complement the energy minimisation calculations, molecular dynamics (MD) simulations were performed to examine the Li ion diffusion pathways and dimensionality within the sulphate structures. Scatter or density plots of Li ion coordinates over the simulation timescale enable the migration paths and the regions of the structure most frequently traversed to be visualized.
Fig. 4c and d show the location of the Li density found from the simulation runs; this confirms the prediction of our static lattice calculations that Li diffusion will occur via a combination of the L1 and L4 jumps and thus allow long-range 1D diffusion along the [100] direction. As noted, Li/M antisite disorder is energetically unfavourable in these sulphates, and hence the 1D Li+ diffusion is much less likely to be hindered by such defects. The MD simulations also confirm that Li diffusion involves conventional hopping between Li sites with no evidence of cooperative ‘knock-on’ mechanisms.
3.3 Surfaces and particle morphology
The particle morphology is important because for facile lithium insertion/extraction the predicted 1D conduction channels must terminate at a low energy surface facet. However, direct experimental measurements on surface structures of the sulphates have not been carried out.We initially focus on the Fe-based material as a model system and due to its superior electrochemical activity. Thirteen low index crystal facets and their symmetry related equivalent surfaces of Li2Fe(SO4)2 have been modelled for the first time and the resulting surface energies are listed in Table 4. Due to the highly interconnected nature of the FeO6 and SO4 polyhedra, there are no simple planar cuts that are non-polar. In all cases the layers must be reconstructed in order to remove the dipole.
| Surface | Surface energy (J m−2) | Surface area in morphology (%) |
|---|---|---|
| {011} | 0.68 | 65.5 |
| {102} | 0.86 | 2.2 |
| {112} | 0.87 | 0.4 |
| {111} | 0.88 | 1.5 |
| {001} | 0.90 | 1.0 |
| {210} | 0.90 | 27.1 |
| {121} | 0.92 | 2.3 |
| {122} | 0.93 | 0.0 |
| {211} | 0.93 | 0.0 |
| {101} | 0.95 | 0.0 |
| {100} | 0.95 | 0.0 |
| {010} | 1.01 | 0.0 |
| {221} | 1.03 | 0.0 |
Using these calculated surface energies a Wulff construction of the equilibrium morphology has been made which reveals a rhombohedral-like shape (Fig. 5). For small crystals where rearrangement of the crystal at each stage of the growth process is possible, a morphology close to the equilibrium form would be expected. However, there are no published indexed TEM images of Li2Fe(SO4)2 for direct comparison; indeed, such comparison is not trivial due to different experimental synthesis methods and possible non-equilibrium conditions. Nevertheless, our focus here is to reveal new information on the surface facets that are exposed, which are significant for lithium extraction/insertion processes.
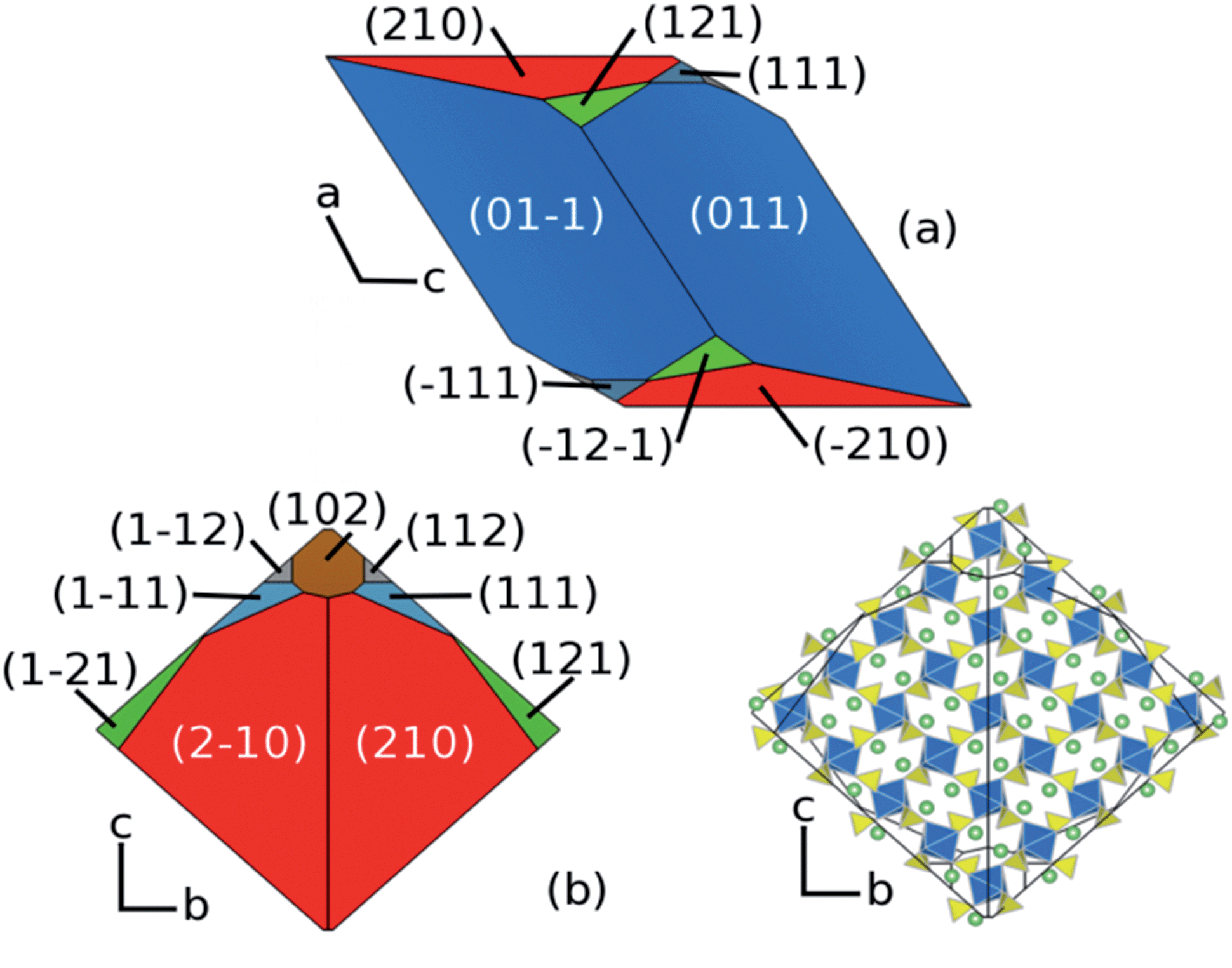 | ||
| Fig. 5 Calculated particle morphology of Li2Fe(SO4)2; as viewed along (a) the b-axis, (b) the a-axis. In (c) the atomic structure is overlaid upon the a-axis view showing the availability of access to the 1D migration channels. | ||
The {011} facet has the lowest surface energy at 0.68 J m−2 and makes up two thirds (65.5%) of the overall surface area of the particle (shown in blue in Fig. 5a). The {210} surface is the next most dominant surface comprising 27% of the surface area (shown in red in Fig. 5b). Earlier in this study we showed that the facile direction for lithium migration is along the channels in the [100] direction. Both the {210} and {102} facets expose these channels (Fig. 5c). It is well known that the growth environment can have a significant effect upon the morphology of a particle and it may be possible to increase the surface area that allows facile Li access by alteration of the synthesis conditions.
Having predicted the dominance of the {011} and {210} surfaces in the morphology, we present details of both relaxed surface structures in Fig. 6. A useful quantity here is the relaxation energy, which is the difference between the unrelaxed surface energy of the crystal cleaved from the bulk and the final relaxed surface energy. This quantity gives a measure of the degree of structural change in the surface region as the surface is formed. Structural relaxations in the {011} surface are small which is reflected in the relaxation energy of 0.16 J m−2. The largest relaxations occur in the topmost layer and these are associated with a small rotation of the terminal SO4 units, which causes an inward displacement of 0.43 Å of the oxygen ion at the outer vertex and an inward displacement of 0.13 Å of the Li atom that is adjacent to this. There are 50% lithium vacancies in the topmost layer and half of the SO4 tetrahedra are absent which gives rise to a degree of rumpling.
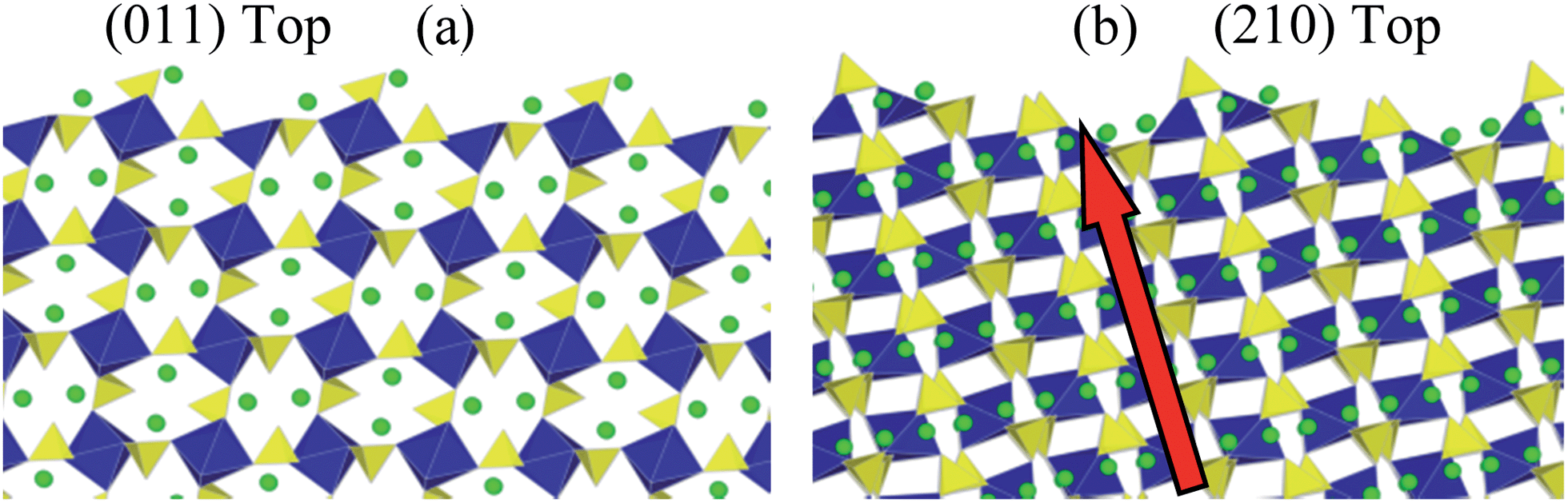 | ||
| Fig. 6 Side view of relaxed (a) {011} and (b) {210} surfaces of Li2Fe(SO4)2. The red arrow indicates the Li migration direction for {210} towards the outermost surface. For the {011} surface Li migration is into the page. | ||
By contrast there is significant relaxation at the {210} surface with a large relaxation energy of 1.79 J m−2. Some loss of coordination is present in all of the terminal FeO6 octahedra and the terminal SO4 units undergo significant tilting. The near surface Li move outwards by up to 1 Å to form part of the terminal layer. As indicated in Fig. 6b, the prominence of the {210} surface is significant since it exposes the facile pathway for lithium ion conduction (along the [100] channel), and hence is important for the reversible insertion/de-insertion of lithium ions.
3.4 Cell voltage trends
As with the potentials-based calculations, the DFT simulations have reproduced the Li2M(SO4)2 (M = Fe, Mn, Co) experimental structures and also the delithiated LiFe(SO4)2 observed structure with a high level of accuracy (Tables S4 and S5†). The computed cell voltage relies on a knowledge of the structure of the delithiated end member LiFe(SO4)2. Typically for cell voltage calculations the relevant lithium atoms are removed directly from the lithiated structure, which is then energy minimised to form the delithiated structure. Numerous configurations of differing lithium ordering schemes were considered, and in our calculations we used the lowest energy structure. However, this energy minimisation may only find the local energy minimum and it is entirely possible that a phase change can occur on lithium extraction.The cell voltage computed directly in this way for Li2Fe(SO4)2 is 3.99 V which is in reasonable agreement with the measured voltage of 3.83 V.21 However, using the joint Synchrotron XRD/neutron refined LiFe(SO4)2 structure (Table S2†), it is clear that the main difference between the lithiated and delithiated materials is that in the Li2Fe(SO4)2 structure there are two rows of lithium in a zig-zag arrangement whereas in the LiFe(SO4)2 structure (Fig. 2) there are half-occupied Li sites in the channel. This subtle change in the structure results in a computed cell voltage of 3.91 V, which is in better agreement with that measured experimentally (Table 5). This result highlights the effect of structural changes on lithium removal and their importance in cell voltage computations.
| Process | Expt.21 (V) | Calc. (V) |
|---|---|---|
| Li2Fe(SO4)2 ↔ LiFe(SO4)2 | 3.83 | 3.91 |
| Li2Mn(SO4)2 ↔ LiMn(SO4)2 | — | 4.54 |
| Li2Co(SO4)2 ↔ LiCo(SO4)2 | — | 5.20 |
Having isolated the delithiated (LiFe(SO4)2) structure we have subsequently calculated voltages for the Mn- and Co-based sulphates by setting both LiMn(SO4)2 and LiCo(SO4)2 initial structures equivalent to LiFe(SO4)2 prior to structural relaxation. The voltages calculated for Li2M(SO4)2 (M = Fe, Mn, Co) are presented in Table 5.
Our calculations suggest that both Li2Mn(SO4)2 and Li2Co(SO4)2 have higher cell voltages than that of Li2Fe(SO4)2 as typically observed for polyanion cathodes. The value for the Co-based sulphate is above the electrolyte stability window. Such a significant shift upwards in voltage is found for other Co-polyanion compounds, such as LiCoPO4 for which a voltage of 4.8 V vs. Li/Li+ has been observed3,55 in comparison to 3.4 V for LiFePO4.
The Mn-based sulphate is known to be electrochemically inactive. As the system is delithiated, Mn3+ (d4) forms which is a Jahn–Teller active valence state. It has been debated whether Jahn–Teller distortion of the MnO6 octahedra could lead to non-uniform structural distortions which will render the delithiated phase unstable and thus unable to form. Such effects are well known in other Mn-based battery materials.56,57 GGA + U calculations provide a useful probe of local structural distortions that are not affected by thermal motion and statistical averaging over many unit cells. To check for Jahn–Teller effects we have examined the MO6 distortion in the Fe, Co and Mn based sulphates, which is presented in Table 6. For this we have used the Baur58 distortion coefficient, D, which is defined as:
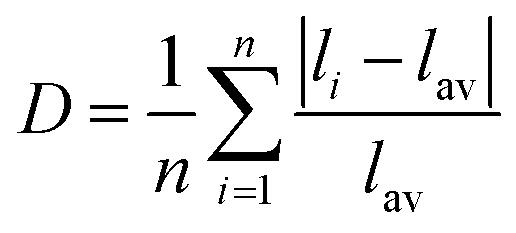 | (6) |
| Material | Mean M–O bond length (Å) | MO6 distortion coefficient (×10−2) |
|---|---|---|
| Li2Fe(SO4)2 | 2.15 | 1.8 |
| LiFe(SO4)2 | 2.01 | 1.4 |
| Li2Mn(SO4)2 | 2.21 | 2.0 |
| LiMn(SO4)2 | 2.08 | 7.2 |
| Li2Co(SO4)2 | 2.12 | 1.6 |
| LiCo(SO4)2 | 2.01 | 2.0 |
The data in Table 6 indicate that all the lithiated structures Li2Fe(SO4)2, Li2Mn(SO4)2 and Li2Co(SO4)2 possess a similar low degree of distortion. Upon delithiation there is a large increase in the MnO6 distortion with a coefficient of 7.2 × 10−2 (compared to 1.4 × 10−2 and 2.0 × 10−2 for FeO6 and CoO6 respectively). Since the Mn-based sulphate is electrochemically inactive, the calculated voltage presented in Table 5 assumes the same delithiated starting structure as the Co and Fe based sulphates, but contains cooperative Jahn–Teller distortion after structure relaxation. Further work to determine if such structural distortion in this phase leads to mechanical instability is on-going including phonon spectrum simulations.
A possible source of the different cell voltages for Li2M(SO4)2 (M = Fe, Mn, Co) is in the relative strength of the corresponding Fe2+/Fe3+, Mn2+/Mn3+ and Co2+/Co3+ redox couples. As with other systems involving M2+/M3+ redox couples we would expect the respective M–O bond lengths to shorten upon Li removal (oxidation) within these sulphate systems. This is indeed found with the Fe–O bond lengths contracting by ∼6.5% (from 2.15 Å to 2.01 Å), the Mn–O bond lengths contracting by ∼6% (from 2.21 Å to 2.08 Å) and the Co–O bond lengths contracting by ∼5% (from 2.12 Å to 2.01 Å).
However, distinguishing among the most reliable parameters to account for voltage variation is not an easy task. The voltage also depends upon other factors such as: inductive effects of SO4, PO4 and SiO4; the degree of ionic or covalent character of the cation–anion bonds; and the Madelung energy of the ionic component of the bonding. As discussed previously,2,59 another possibility relates to the inductive effect played by lithium. There are obviously hidden issues which remain to be unravelled, and are part of on-going studies.
4. Conclusions
This systematic survey of the Li2M(SO4)2 (M = Fe, Mn, Co) cathode materials has used a combination of atomistic simulation and DFT techniques to provide insights into the defect chemistry, Li diffusion paths, crystal morphologies and voltage trends. The key results can be summarized as follows:(1) The atomistic simulations of Li2M(SO4)2 (M = Fe, Mn, Co) show good reproduction of their observed structures. The defect calculations indicate that the formation of all Frenkel, Schottky and Li/M anti-site intrinsic defects is unfavourable for all phases. In particular, the results suggest there would be no significant intrinsic anti-site defect concentration of Fe, Mn or Co on Li sites at typical operating temperatures within these sulphates. This is in contrast to the LiFePO4 material, which has a small amount of Fe on Li sites.
(2) Lithium diffusion simulations reveal low migration energies (∼0.4–0.5 eV) along the a-axis channels of Li2Fe(SO4)2, Li2Mn(SO4)2 and Li2Co(SO4)2 suggesting 1D diffusion and good Li mobility which is important for favourable rate capability. MD calculations verify the 1D migration pathways along the a-axis channels, with no evidence of cooperative mechanisms.
(3) The calculated equilibrium morphology of Li2Fe(SO4)2 adopts a rhombohedral-like shape with {011} and {210} surfaces dominating. The prominence of the {210} surface is significant since it allows access to the facile pathway for lithium ion conduction (along the [100] channel), and hence is important for the reversible insertion/de-insertion of lithium ions.
(4) The calculated cell voltage (3.91 V) for Li2Fe(SO4)2 is in excellent agreement with that determined experimentally (3.83 V). Characterisation of the experimental structure of the delithiated LiFe(SO4)2 phase was found to be important in this agreement. Li2Mn(SO4)2 is known to be electrochemically inactive and the predicted voltage for Li2Co(SO4)2 was found to be 5.20 V, which may be beyond the stability range of the current electrolyte.
Acknowledgements
This work was funded by the EPSRC Supergen programme and made use of the high-performance computing service HECToR, via the HPC Materials Chemistry Consortium. M.R. acknowledges the French “Ministère de l'Enseignement Supérieur et de la Recherche” for her Ph.D. Grant, and thanks Loïc Dupont and Charles Delacourt for fruitful discussions.Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587 CrossRef CAS.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691 CrossRef CAS.
- M. R. Palacin, Chem. Soc. Rev., 2009, 38, 2565 RSC.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 CAS.
- Z. L. Gong and Y. Yang, Energy Environ. Sci., 2011, 4, 3223 CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CrossRef CAS PubMed.
- A. Nyten, A. Abouimrane, M. Armand, T. Gustafsson and J. O. Thomas, Electrochem. Commun., 2005, 7, 156 CrossRef CAS PubMed.
- M. S. Islam, R. Dominko, C. Masquelier, C. Sirisopanaporn, A. R. Armstrong and P. G. Bruce, J. Mater. Chem., 2011, 21, 9811 RSC.
- A. Yamada, N. Iwane, Y. Harada, S. Nishimura, Y. Koyama and I. Tanaka, Adv. Mater., 2010, 22, 3583 CrossRef CAS PubMed.
- N. Recham, J. N. Chotard, L. Dupont, C. Delacourt, W. Walker, M. Armand and J. M. Tarascon, Nat. Mater., 2010, 9, 68 CrossRef CAS PubMed.
- P. Barpanda, M. Ati, B. C. Melot, G. Rousse, J. N. Chotard, M. L. Doublet, M. T. Sougrati, S. A. Corr, J. C. Jumas and J. M. Tarascon, Nat. Mater., 2011, 10, 772 CrossRef CAS PubMed; R. Tripathi, T. N. Ramesh, B. L. Ellis and L. F. Nazar, Angew. Chem. Int. Ed., 2010, 49, 8738 CrossRef PubMed.
- M. Ati, B. C. Melot, J. N. Chotard, G. Rousse, M. Reynaud and J. M. Tarascon, Electrochem. Commun., 2011, 13, 1280 CrossRef CAS PubMed.
- G. Rousse and J. M. Tarascon, Chem. Mater., 2014, 26, 394 CrossRef CAS.
- B. L. Ellis, W. R. M. Makahnouk, Y. Makimura, K. Toghill and L. F. Nazar, Nat. Mater., 2007, 6, 749 CrossRef CAS PubMed.
- S. Nishimura, M. Nakamura, R. Natsui and A. Yamada, J. Am. Chem. Soc., 2010, 132, 13596 CrossRef CAS PubMed.
- C. V. Subban, M. Ati, G. Rousse, A. M. Abakumov, G. Van Tendeloo, R. Janot and J. M. Tarascon, J. Am. Chem. Soc., 2013, 135, 3653 CrossRef CAS PubMed.
- M. A. Reddy, V. Pralong, V. Caignaert, U. V. Varadaraju and B. Raveau, Electrochem. Commun., 2009, 11, 1807 CrossRef PubMed.
- M. A. Gomez, G. Ventruti, M. Celikin, H. Assaaoudi, H. Putz, L. Becze, K. E. Lee and G. P. Demopoulos, RSC Adv., 2013, 3, 16840 RSC.
- M. Reynaud, M. Ati, B. C. Melot, M. T. Sougrati, G. Rousse, J. N. Chotard and J. M. Tarascon, Electrochem. Commun., 2012, 21, 77 CrossRef CAS PubMed.
- M. Reynaud, G. Rousse, J. N. Chotard, J. Rodriguez-Carvajal and J. M. Tarascon, Inorg. Chem., 2013, 52, 10456 CrossRef CAS PubMed.
- M. S. Islam, D. J. Driscoll, C. A. J. Fisher and P. R. Slater, Chem. Mater., 2005, 17, 5085 CrossRef CAS.
- C. A. J. Fisher, V. M. H. Prieto and M. S. Islam, Chem. Mater., 2008, 20, 5907 CrossRef CAS.
- G. R. Gardiner and M. S. Islam, Chem. Mater., 2010, 22, 1242 CrossRef CAS.
- C. A. J. Fisher and M. S. Islam, J. Mater. Chem., 2008, 18, 1209 RSC.
- A. R. Armstrong, N. Kuganathan, M. S. Islam and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 13031 CrossRef CAS PubMed.
- R. Tripathi, G. R. Gardiner, M. S. Islam and L. F. Nazar, Chem. Mater., 2011, 23, 2278 CrossRef CAS.
- J. M. Clark, S. Nishimura, A. Yamada and M. S. Islam, Angew. Chem., Int. Ed., 2012, 51, 13149 CrossRef CAS PubMed.
- C. Eames, A. R. Armstrong, P. G. Bruce and M. S. Islam, Chem. Mater., 2012, 24, 2155 CrossRef CAS.
- F. Zhou, M. Cococcioni, C. A. Marianetti, D. Morgan and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 23 Search PubMed.
- H. Zhou, S. Upreti, N. A. Chernova, G. Hautier, G. Ceder and M. S. Whittingham, Chem. Mater., 2011, 23, 293 CrossRef CAS.
- A. R. Armstrong, C. Lyness, P. M. Panchmatia, M. S. Islam and P. G. Bruce, Nat. Mater., 2011, 10, 223 CrossRef CAS PubMed.
- D. A. Tompsett and M. S. Islam, Chem. Mater., 2013, 25, 2515 CrossRef CAS.
- A. Walsh, A. A. Sokol and C. R. A. Catlow, in Computational Approaches to Energy Materials, Wiley-Blackwell, 2013 Search PubMed.
- M. S. Islam and C. A. J. Fisher, Chem. Soc. Rev., 2014, 43, 185 RSC.
- W. Koch and M. C. Holthausen, in A Chemist's Guide to Density Functional Theory, Wiley-VCH Verlag GmbH, Weinheim, 2001 Search PubMed.
- N. L. Allan, A. L. Rohl, D. H. Gay, C. R. A. Catlow, R. J. Davey and W. C. Mackrodt, Faraday Discuss., 1993, 95, 273 RSC.
- S. E. Redfern and S. C. Parker, J. Chem. Soc., Faraday Trans., 1998, 94, 1947 RSC.
- G. Sastre and J. D. Gale, Chem. Mater., 2005, 17, 730 CrossRef CAS.
- L. Gomez-Hortiguela, F. Cora, C. R. A. Catlow and J. Perez-Pariente, J. Am. Chem. Soc., 2004, 126, 12097 CrossRef PubMed.
- B. G. Dick and A. W. Overhauser, Phys. Rev., 1958, 112, 90 CrossRef.
- J. D. Gale and A. L. Rohl, Mol. Simul., 2003, 29, 291 CrossRef CAS.
- W. Smith and T. R. Forester, J. Mol. Graphics, 1996, 14, 136 CrossRef CAS.
- G. W. Watson, E. T. Kelsey, N. H. deLeeuw, D. J. Harris and S. C. Parker, J. Chem. Soc., Faraday Trans., 1996, 92, 433 RSC.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- T. Mueller, G. Hautier, A. Jain and G. Ceder, Chem. Mater., 2011, 23, 3854 CrossRef CAS.
- G. Ceder, M. K. Aydinol and A. F. Kohan, Comput. Mater. Sci., 1997, 8, 161 CrossRef CAS.
- C. Arrouvel, S. C. Parker and M. S. Islam, Chem. Mater., 2009, 21, 4778 CrossRef CAS.
- J. Maier and R. Amin, J. Electrochem. Soc., 2008, 155, A339 CrossRef CAS PubMed.
- J. J. Chen, M. J. Vacchio, S. J. Wang, N. Chernova, P. Y. Zavalij and M. S. Whittingham, Solid State Ionics, 2008, 178, 1676 CrossRef CAS PubMed.
- K. Amine, H. Yasuda and M. Yamachi, Electrochem. Solid State Lett., 2000, 3, 178 CrossRef CAS PubMed.
- A. Van der Ven, C. Marianetti, D. Morgan and G. Ceder, Solid State Ionics, 2000, 135, 21 CrossRef CAS.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mat. Res. Bull., 1983, 18, 461 CrossRef CAS.
- W. H. Baur, Acta Crystallogr., Sect. B: Struct. Sci., 1974, 30, 1195 CrossRef CAS.
- B. C. Melot, D. Scanlon, M. Reynaud, M. Ati, G. Rousse, J. N. Chotard, M. Henry and J. M. Tarascon, ACS Appl. Mater. Interfaces, 2014 DOI:10.1021/am405579h.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ta15064j |
| ‡ Current address: CIC Energigune, Parque Tecnológico de Álava, Albert Einstein 48, 01510 Miñano, Spain |
| This journal is © The Royal Society of Chemistry 2014 |