 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stable anode performance of an Sb–carbon nanocomposite in lithium-ion batteries and the effect of ball milling mode in the course of its preparation†
Thrinathreddy
Ramireddy
,
Md Mokhlesur
Rahman
,
Tan
Xing
,
Ying
Chen
and
Alexey M.
Glushenkov
*
Institute for Frontier Materials, Deakin University, Waurn Ponds, 3216 Victoria, Australia. E-mail: alexey.glushenkov@deakin.edu.au
First published on 8th January 2014
Abstract
Materials that alloy with lithium (Si, Ge, Sn, Sb, and P) are considered as alternatives to graphitic anodes in lithium-ion batteries. Their practical use is precluded by large volume changes (200–370%) during cycling. Embedding nanoparticles into carbon is being investigated as a way to tackle that, and ball milling is emerging as a technique to prepare nanocomposites with enhanced capacity and cyclic stability. Using Sb as a model system, we investigate the preparation of Sb–carbon nanocomposites using a reconfigurable ball mill. Four distinctive milling modes are compared. The structure of the composites varies depending on the mode. Frequent strong ball impacts are required for the optimal electrochemical performance of the nanocomposite. An outstanding stable capacity of 550 mA h g−1 for 250 cycles at a current rate of 230 mA g−1 is demonstrated in a thin electrode (1 mg cm−2) and a capacity of ∼400 mA h g−1 can be retained at 1.15 A g−1. Some capacity fade is observed in a thicker electrode (2.5 mg cm−2), i.e. the performance is sensitive to mass loading. The electrochemical stability originates from the nanocomposite structure containing Sb nanoparticles (5–15 nm) dispersed in a carbon component.
Introduction
Li-ion batteries have successfully penetrated the markets of portable electronics and power tools and are becoming primary power sources in these applications.1 In addition, this type of battery is currently considered as a possible solution for electrochemical energy storage in other areas such as stationary storage of energy from intermittent renewable sources (solar and wind) and automotive industry (electric vehicles and hybrid electric vehicles).2–4 The wide range of applications available for Li-ion batteries is underpinned by their high energy and power densities, low self-discharge rate, wide operating temperature, the lack of memory effects and higher operating voltage than those of the preceding battery technologies.5 The ultimate battery performance is generally defined by its main components, a cathode, an anode and a suitable electrolyte. Graphite has long been used as the anode of choice in commercial Li-ion batteries due to its excellent cycling stability and a low volume change of only 12% during lithium intercalation–deintercalation.6 At the same time, this material has a major disadvantage of having a relatively low theoretical capacity of only 372 mA h g−1, much smaller than that of many other prospective anode materials for Li-ion batteries. Obviously, a low capacity of the anode limits the energy density of the whole battery,7 and the research community is actively investigating the possibility of replacing graphite with a higher capacity anode material. The primary candidates are the materials that can alloy and de-alloy with lithium in a reversible manner. The list of such materials includes Si, P, Ge, Sn and Sb as main candidates, and their theoretical capacities are 4200,8 2595,9 1600,10 994 (ref. 6) and 660 mA h g−1 (ref. 6) respectively, exceeding that of graphite by a wide margin. These materials can potentially become excellent alternatives to the graphitic anode owing to their high gravimetric capacities.The major problem preventing the practical use of the above materials is the enormous volume change they experience during the charge–discharge cycles in a battery. The volume variations for Si, P, Ge, Sn and Sb phases are estimated to be as high as 320%,6 220%, 370%,10 260% (ref. 6) and 200%,6 respectively. Such dramatic expansion and contraction of the materials result in pulverization and cracking of electrodes leading to the loss of contact in the electrode and/or detachment of the electrode materials from the current collector. Consequently, the capacity and cyclic stability of the battery are impacted.6 The anode materials that alloy and de-alloy with lithium will only find practical use if the problems associated with their dramatic volume variation are successfully resolved. One of the possible solutions to tackle the issue is to prepare alloying materials in the form of nanocomposites with carbon. In such nanocomposites, the alloying materials should be prepared as nanoparticles (that can tolerate high level of stress and volume change without disintegration) and these nanoparticles should be simultaneously dispersed within a carbon host material. Depending on the nature of the carbon component, a nanocomposite can be either active–active (the carbon component is able to store lithium as well)8,11 or active–inactive (where the carbon component is only responsible for providing the electron migration path between the current collector and active nanoparticles).12
Among other methods, mechanical ball milling is one of the promising practical techniques to prepare suitable nanocomposites incorporating some of the materials that alloy with lithium. Indeed, a range of nanocomposites (Sb–carbon, P–carbon, Sn–carbon and SnSb–carbon) prepared by ball milling have been evaluated by Park and Sohn.13–15 Practically interesting results and significant performance improvements with respect to conventionally prepared Sb, P, Sn, and SnSb electrodes have been reported by their group. The composite of black phosphorus and carbon has showed a stable capacity of more than 600 mA h g−1 at a current rate of 100 mA g−1 for 100 cycles14 while an Sb–C nanocomposite has demonstrated a stable capacity of about 350 mA h g−1.13 However, restricting potential windows for testing to 0.8–2 V and 0.7–2 V vs. Li/Li+ was required for these two materials in order to obtain stable cyclic performances, and the cyclic performances were inadequate if the anode materials were discharged down to 0 V vs. Li/Li+.13,14 An Sb–C nanocomposite with a more complex structure (involving two carbon precursors, Super P carbon black and graphite) has been shown to retain the capacity of 460 mA h g−1 over 100 cycles at a current rate of 100 mA g−1 (ref. 13) within the full potential window 0–2 V vs. Li/Li+. Finally, promising anode performance has been observed in the SnSb–C composite which showed a capacity of 550 mA h g−1 after 300 cycles at a current rate of 100 mA g−1,15 and 78% of the initial charge capacity was retained in the 300th cycle.
In our view, these results indicate that ball milling is a suitable technique for the preparation of nanocomposites of carbon with materials that alloy with lithium. We expect that optimisation of the ball milling procedure as well as better understanding of the structure of the nanocomposites and its relationship with the electrochemical properties will lead to the preparation of new anode materials with high capacity and excellent cyclic stability. Our analysis indicates that there is currently no existing knowledge on how different ball milling devices and/or particular ball milling modes in the same device affect the structure of a nanocomposite and its performance in the lithium-ion battery. Most of the preceding research was done by Park and Sohn,13–15 and the type of the ball milling device they used is typically not indicated in their papers. Further research is definitely warranted in order to adapt the ball milling method better to the preparation of advanced electrode materials for lithium-ion batteries.
Herein, we employ an easily reconfigurable ball mill to study the connection between the milling mode used, the structure and anode performance of nanocomposites. Sb–carbon nanocomposites prepared from Sb and graphite powders are selected as model systems for the study. Four milling modes involving shear action only, shear action with an occasional weak impact as well as two milling modes with a strong impact are evaluated. It is evident that the strong impact of balls during milling is critical to achieve stable electrochemical performance of an Sb–C composite. Excellent stable cyclic behaviour is observed in the optimal sample. The structural characteristics of the optimal sample are assessed by X-ray diffraction, advanced transmission electron microscopy and Raman spectroscopy. The operating mechanism of the nanocomposite is discussed.
Experimental
Material synthesis
Antimony (325 mesh, 99.5% purity, Johnson Matthey Electronics) and graphite (Sigma Aldrich, 282863, <20 μm) powders were mixed in a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 weight ratio and 5 g of the mixture were loaded into a magneto-ball mill with four stainless steel balls (25.4 mm in diameter). The milling was performed under an Ar atmosphere at an excess gas pressure of 100 kPa. The same mixture was milled for a constant period of 100 hours using four different ball milling modes. An external magnet was used in two of the ball milling modes and removed in the other two ball milling modes. The mechanical milling without the magnet was conducted at rotation speeds of 75 and 360 rpm whereas the milling in the presence of the external magnet was carried out at 160 rpm and varied positions of the magnet (45 and 135° in relation to the vertical direction). The milled composites were removed from the ball milling containers in the presence of an inert Ar atmosphere. A detailed description of the ball milling device used can be found elsewhere.
:3 weight ratio and 5 g of the mixture were loaded into a magneto-ball mill with four stainless steel balls (25.4 mm in diameter). The milling was performed under an Ar atmosphere at an excess gas pressure of 100 kPa. The same mixture was milled for a constant period of 100 hours using four different ball milling modes. An external magnet was used in two of the ball milling modes and removed in the other two ball milling modes. The mechanical milling without the magnet was conducted at rotation speeds of 75 and 360 rpm whereas the milling in the presence of the external magnet was carried out at 160 rpm and varied positions of the magnet (45 and 135° in relation to the vertical direction). The milled composites were removed from the ball milling containers in the presence of an inert Ar atmosphere. A detailed description of the ball milling device used can be found elsewhere.
Material characterization
The powder samples were characterized by X-ray diffraction (XRD) using a PANalytical X'Pert Pro instrument with a Cu Kα radiation source (λ = 1.54181 Å) operated at 40 kV with 50 mA current. XRD patterns were collected over the range of 20–80° by keeping the step angle and dwell time at 0.02° and 2 s. The X'Pert High Score Plus software was used to identify the phases present in the powder. Raman spectroscopy measurements were performed using a Renishawin Via micro-spectroscopic system. The laser used had a wavelength of 514 nm. The power was 0.5 mW to prevent the laser damage of the material. 20× lens was used in the test. Each spectrum was accumulated from two scans. The transmission electron microscopy (TEM) characterisation of the nanocomposite was performed using a JEOL JEM 2100F instrument operated at 200 kV. A high-angle annular dark-field (HAADF) image was obtained in a scanned TEM (STEM) mode. The elastic bright-field image and energy filtered TEM (EFTEM) elemental maps of carbon and Sb were obtained using a Gatan Quantum ER 965 Imaging Filter. The three-window method was used for the acquisition of the elemental maps.Electrochemical characterization
To test the electrochemical performance, Sb powder or an Sb–carbon composite was mixed with Super P Li™ carbon black (Timcal Ltd.) and a carboxymethyl cellulose (CMC) binder (a weight ratio of 80:10:10) in de-ionized water to form a homogenous slurry. The slurry in each case was coated onto copper foils and the coated electrodes were dried in a vacuum at 90 °C overnight. The electrodes were then pressed using a stainless steel metal disc to enhance the contact between the material and the Cu foil. The size of the prepared electrodes was 1 × 1 cm2. CR 2032 coin cells were assembled inside the Ar-filled glove box (Innovative Technology, USA). Li foil was used as the counter electrode and a microporous polyethylene film (MTI Corporation, USA) was used as the separator. The commercial electrolyte (MTI Corporation, USA) with 1 M LiPF6 in a mixture of ethylene carbonate (EC), dimethyl carbonate (DMC), and diethyl carbonate (DEC) with a volume ratio of 1:1:1 was used. The cells were galvanostatically charged–discharged at suitable current rates between 2.0 and 0.01 V vs. Li/Li+ using the LAND battery system (Wuhan LAND Electronics, Ltd. China). The temperature in the laboratory was maintained within a range of 20–25 °C during the tests. Material loading was 1.07–1.11 mg cm−2 (including the weight of a nanocomposite, Super P Li™ carbon black and the binder) for the comparative tests of the composite samples obtained from four different ball milling modes and ∼1.35 mg cm−2 for the rate capability test (Fig. 4c). Additionally, an electrode with a material loading of ∼2.5 mg cm−2 was tested to evaluate the electrochemical performance of a thicker electrode. It is important to note that Super P Li™ carbon black was assigned no capacity during the calculation of the nanocomposite capacity presented in this paper and was considered an inert additive to improve the conductivity of the electrode. Strictly speaking, the real capacity of the carbon black should be subtracted from the reported capacity and the unknown adjustment value can be up to 46 mA h g−1 (corresponding to the situation where the full theoretical capacity can be displayed by the carbon black). The capacities reported in Fig. 3 and 4 may be therefore slightly overestimated.
Ex situ X-ray characterisation of discharged and charged electrodes
The discharged and charged cells (in the first and fourth cycles) were disassembled inside the Ar filled glove box. The extracted electrodes were washed with DMC (anhydrous, Sigma Aldrich) before taking them out of the box. Ex situ XRD was performed on the electrodes after attaching them to a diffraction peak-free silicon wafer using double-sided sticky tape. The same PANalytical X'Pert Pro instrument was used for the analysis.Results and discussion
Effect of the ball milling mode on the structure and electrochemical properties of Sb–carbon composites
In order to study the influence of various milling modes, an easily reconfigurable magneto-ball mill was used in this work.16,17 The device was originally developed at the Australian National University with the fundamental purpose to generate distinctly different ball milling modes and study the influence of milling parameters on the characteristics of the resulting ball milled powders.16 Subsequently, the ball milling vials were modified via attaching gas-tight valves that allow the mills to be filled with a desired atmosphere and conduct milling in the gas atmosphere of choice.17 In brief, this mill consists of a disc vial that rotates around a horizontal axis. Certain amounts of powder and stainless steel balls (we normally use four hardened steel balls with a diameter of 25.4 mm) are placed inside the vial and an external magnet is located outside the vial. The combination of gravity force, magnetic field and rotation makes the balls move in a particular way in the ball mill. The powder is processed via mechanical deformation when it is trapped between the surfaces of the balls or between a ball and the vial's wall. The advantage of the magneto-ball mill is the flexibility in selecting the best suitable milling mode. By varying the rotation speed and the position of the external magnet, distinctive ball milling modes can be chosen, and a number of ball movement patterns possible in the milling devices are analysed in the original paper by Calka and Radlinski.16 These ball milling modes can provide predominantly shear or impact action or a combination of these two types of mechanical treatment and also vary the strength of the impact (i.e., the speed of colliding balls).We chose four different ball milling modes in this study, and they are schematically represented in Fig. 1. The ball movement pattern is shown using solid arrows. Two of the milling modes (Fig. 1a and b) are conducted without the presence of the external magnet. The rotation of the vial at a rate of 360 rpm (Fig. 1a) causes four balls to roll back and forth at the bottom of the vial (milling mode A). This milling mode involves processing of the powder by shear force only, and is similar to the low energy, low rotation frequency mode described by Calka and Radlinski16 (Fig. 2b in ref. 16) but the shear force in our case is weaker as an optional external magnet at the bottom of the vial is not used. The second milling mode is caused by the rotation of the vial at a rate of 75 rpm (milling mode B, Fig. 1b). In this mode the ball movement pattern involves not only rolling of balls at the bottom of the vial but also occasional weak impacts. The rightmost ball is getting released from time to time and slides along the sides of the other three balls before hitting the opposite wall of the vial. Two video clips portraying the patterns of ball movement in milling modes A and B are presented in the ESI (Videos A and B†).
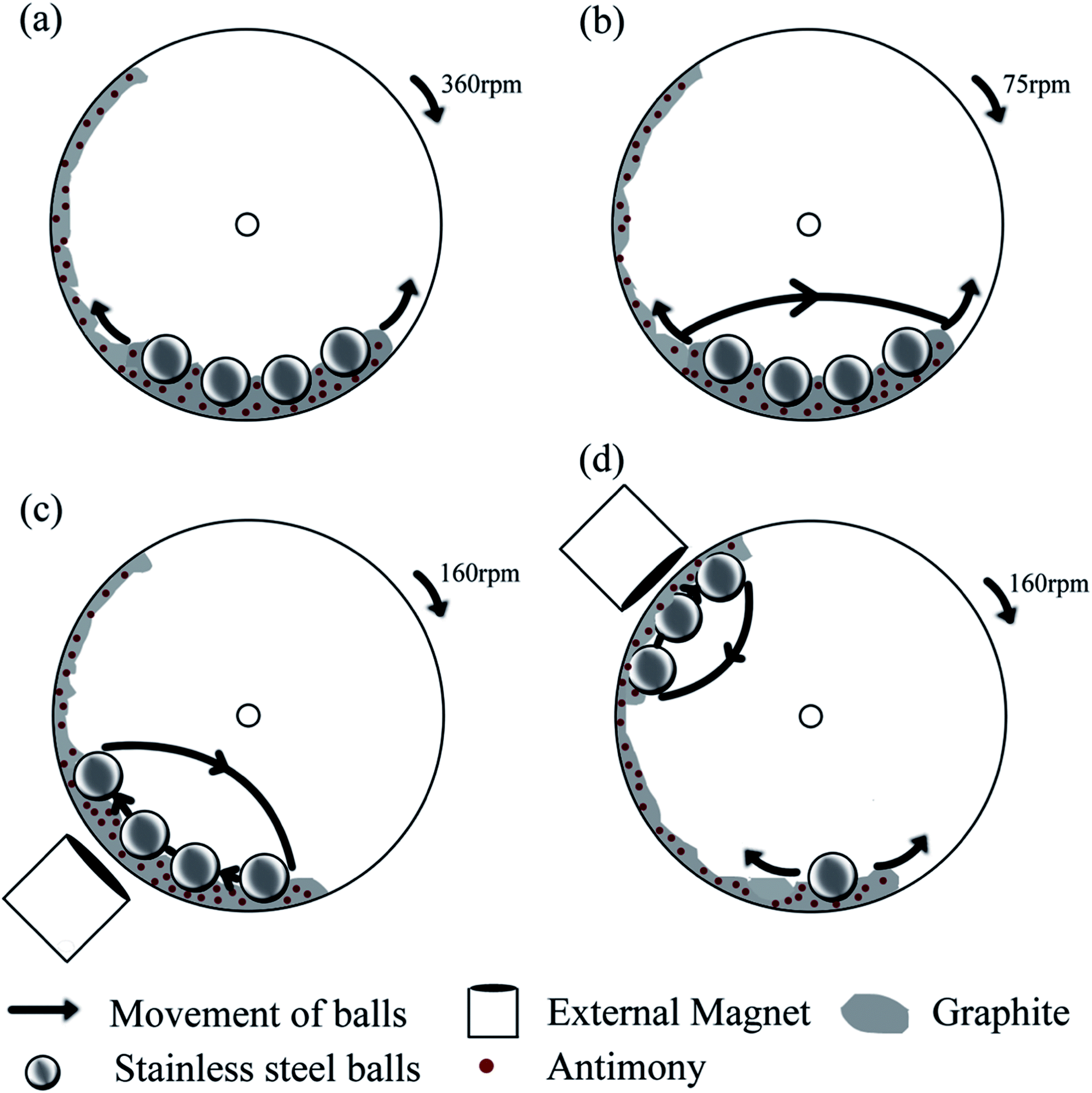 | ||
| Fig. 1 A schematic representation of the ball milling device and the four ball milling modes used in this study: (a–d) ball milling modes A, B, C and D. The ball milling modes A and B do not involve an external magnet. The ball milling modes C and D use an external magnet at two different positions (45 and 135° with respect to the vertical direction) and are capable of creating strong ball impacts in the mill. | ||
The other two ball milling modes involve a strong ball impact caused by the installation of a magnet outside of the vial. The ball movement patterns corresponding to the presence of a magnet at either 45° or 135° position with respect to the vertical direction and the milling vial's constant rotation speed of 160 rpm are shown in Fig. 1c and d. When the magnet is installed at a 45° position (milling mode C, Fig. 1c), the balls are elevated along the left wall of the rotating vial and fall towards the bottom of it, creating impacts with each other and the bottom of the vial. This milling mode is close to the high-energy, two points of equilibrium mode described by Calka and Radlinski (Fig. 2c in ref. 16). When the magnet is installed at the 135° position (milling mode D, Fig. 1d), three balls appear to be localised near the magnet while the remaining ball is rolling at the bottom of the vial. The balls adjacent to the magnet periodically fall onto the wall of the vial creating a strong impact with the wall while they do not collide with each other much during their movement. In addition to the presence of ball-to-ball and ball-to-wall impacts, some processing by shear mode is also expected during the milling modes C and D. The corresponding video clips are presented in the ESI (Videos C and D†).
XRD patterns of the original Sb powder and the four Sb–carbon composites prepared by ball milling are shown in Fig. 2. Most of the visible peaks in all patterns belong to the antimony phase (JCPDS no. 01-085-1324). Fig. 2a shows the XRD pattern of the pure Sb material with a set of strong peaks indexed in accordance with the standard diffraction card. The XRD patterns of the composites prepared using the ball milling modes A and B are shown in Fig. 2b and c. It is obvious that there are no dramatic structural changes in the Sb phase in these two composites as the patterns still show well-pronounced sets of narrow and sharp diffraction peaks of Sb. In addition to the Sb peaks, a single weak and broad (002) peak of graphite is also visible in the patterns (labelled with a “star” symbol in Fig. 2b and c). Other diffraction peaks of graphite are not visible. This type of pattern indicates that some long-range graphitic structures still remain in these samples while, at the same time, significant structural modification and disordering of graphite were introduced by the ball milling process. We can conclude that the ball milling conducted in modes A and B does alter the structure of the graphitic component while it does not affect the Sb phase to a significant extent.
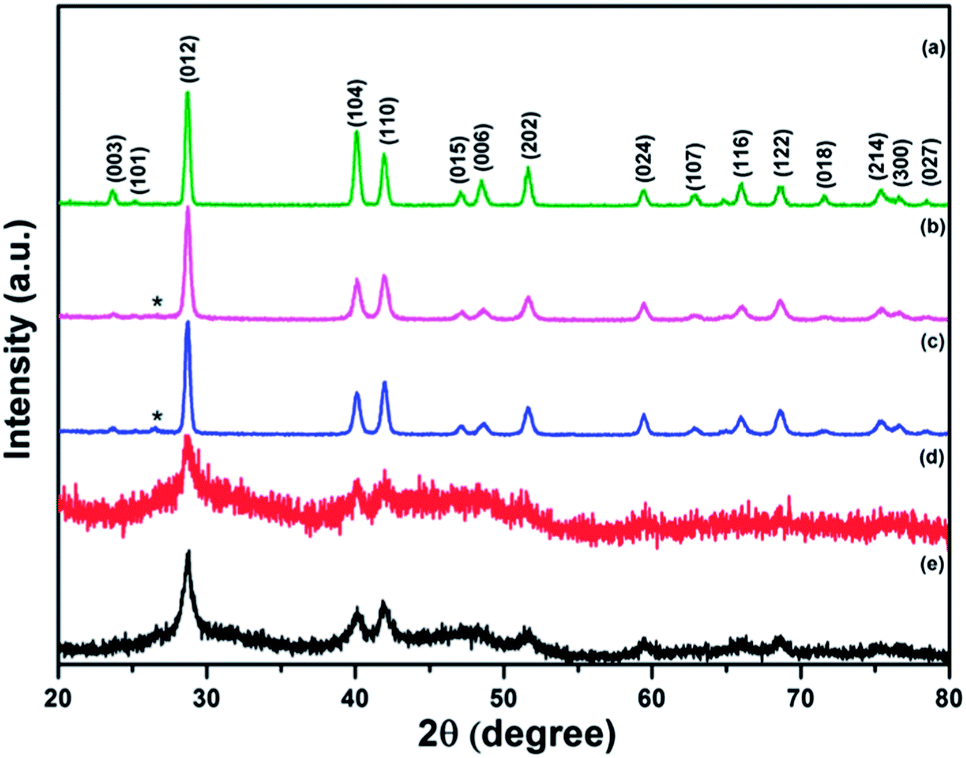 | ||
| Fig. 2 X-ray diffraction patterns of Sb powder and the nanocomposites: (a) pure Sb and (b–e) Sb–carbon nanocomposites prepared using ball milling modes A, B, C and D. The diffraction peaks of the Sb phase are indexed and the position of the (002) peak of graphite is marked with a star symbol. | ||
The XRD patterns from the composite samples obtained in the ball milling modes C and D are quite different (Fig. 2d and e). The (002) graphitic peak is no longer observed, indicating that the long-range graphitic structure is lost in these samples. At the same time, the diffraction peaks of the Sb phase have a low intensity with respect to the noise level and are significantly broadened, highlighting a small crystallite size of Sb. For example, the crystallite size of 15.5 nm was estimated from the width of the (012) peaks using the Scherrer equation in the composite prepared by the ball milling mode D. In addition, the diffraction patterns in Fig. 2d and e show new broad low-intensity features located within the ranges of 25–35 and 38–55 degrees. Further in-depth discussion of the structural features of the composite prepared using the ball milling mode D is presented in the next section of this paper. Here, we conclude that the ball milling modes involving the strong impact (C and D) lead to the significant changes in the structural features of the composites while the ball milling modes A and B (shear action with and without the presence of weak ball impacts) alter the graphitic component of the composite only and do not seem to modify the Sb component of the composite much.
The electrochemical performance of the four Sb–carbon composites is shown in Fig. 3. The testing was performed for 100 cycles using a galvanostatic discharge–charge technique at a current rate of 0.4 C (230 mA g−1). In order to estimate the theoretical capacity of the composites and select appropriate C-rates, we used the theoretical values for Sb and graphite (660 and 372 mA h g−1, respectively) as references. Assuming that the lithium incorporation into the composite occurs via the conventional mechanisms for Sb and graphite, the theoretical capacity of a Sb–graphite mixture with a 7:3 weight ratio equals 573.6 mA h g−1. The level of this theoretical capacity is shown as a bold interrupted line in all graphs in Fig. 3. The level of the highest discharge capacity demonstrated by each composite is also shown with dotted lines in each graph. Capacity retention after 100 cycles is compared and the capacity drop with respect to both the theoretical capacity and the highest discharge capacity is shown in the graphs. The composites obtained using ball milling modes A and B generally show deteriorating cyclic behaviour (Fig. 3a and b). The composite prepared by the ball milling mode A shows the highest capacities (both discharge and charge ones) in the 4th cycle, with the highest value of the discharge capacity being 571 mA h g−1. The corresponding electrode loses 27% of the highest discharge capacity (and shows 72.7% of the theoretical capacity of a mixture of Sb and graphite) after 99 cycles. The highest discharge capacity of the composite obtained by the ball milling mode B is 437 mA h g−1 in the second cycle, and only 46% of this capacity is retained after 100 cycles (Fig. 3a). This represents approximately 35.1% of the theoretical capacity of the mixture of Sb and graphite. The deteriorating cyclic performance of the composites produced by milling modes A and B is attributed to the large size of Sb particles remaining in the structure. During the lithiation and delithiation processes these particles cannot cope with the volume expansion and associated stress and, as a result, the electrodes gradually disintegrate.
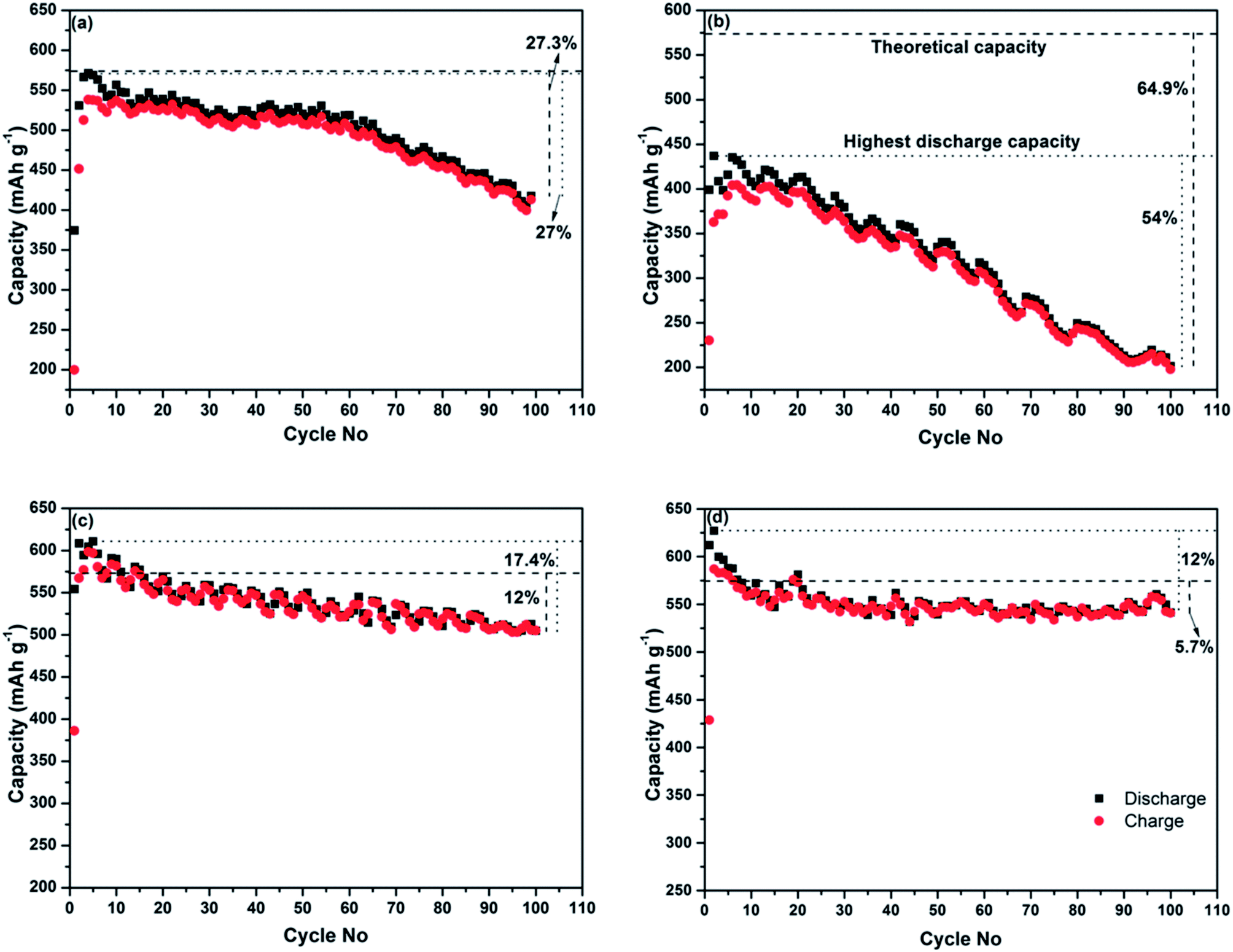 | ||
| Fig. 3 The plots of discharge and charge capacities vs. cycle number for the electrodes prepared from the nanocomposites synthesised using (a) ball milling mode A, (b) ball milling mode B, (c) ball milling mode C, and (d) ball milling mode D. The test was conducted in the potential range of 2.0–0.01 vs. Li/Li+ at a current rate of 0.4 C (230 mA g−1). | ||
The cyclic behaviour of the two composites produced by the ball milling modes involving strong ball impacts (C and D) is significantly better, with well-preserved electrochemical reactivity with lithium after 100 cycles. The highest discharge capacity of the sample prepared using the ball milling mode C is 610 mA h g−1 (during the fifth cycle), and the sample retains about 82.6% of its maximal discharge capacity (and 88% of theoretical capacity) at the end of 100 cycles (Fig. 3c). The composite prepared using the ball milling D (Fig. 3d) shows the initial discharge and charge capacities of 612 and 428 mA h g−1, respectively. The sample loses about 12% of the initial discharge capacity after 100 cycles. Importantly, the capacity value after 100 cycles represents 94.3% of the theoretical capacity still retained after this extended number of cycles. The structure and good electrochemical performance of the composite samples prepared using the ball milling modes C and D are obviously of interest for further investigation. Therefore, the structural characteristics and operating mechanism of the sample prepared by the ball milling mode D are further discussed in the next section.
The results of additional electrochemical characterisation of the nanocomposite prepared using ball milling mode D are shown in Fig. 4. Fig. 4a shows the dependency of the discharge capacity and the Coulombic efficiency of the electrode on the cycle number in the course of an extended cycling for 250 cycles. The Coulombic efficiency of the first cycle was approximately 70% (a discharge capacity of 612 mA h g−1 and a charge capacity of 428.6 mA h g−1) but quickly reached 98% in the fifth cycle and stayed above that level until the completion of the experiment. The discharge capacity stabilised at the level of 550 mA h g−1 and stayed around this value with minor fluctuations until the end of the experiment (250 cycles). The discharge and charge profiles are shown in Fig. 4b. As can be seen from Fig. 4b, the discharge and charge curves retain the same shape and are situated within a narrow band of lines for 250 cycles. The minor fluctuations can be explained by subtle variations of temperature in our laboratory during the test.
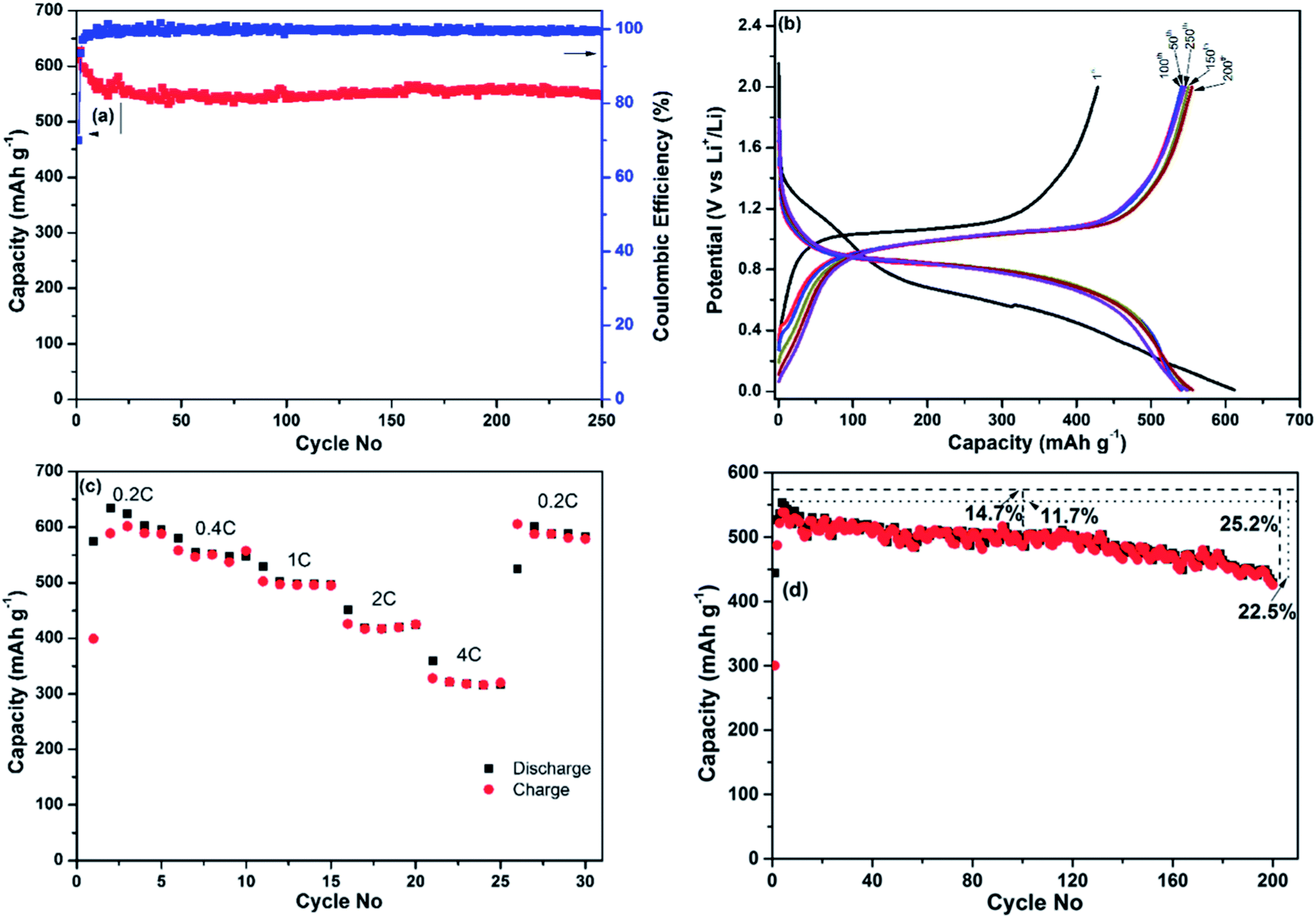 | ||
| Fig. 4 Additional electrochemical characterisation of the Sb–carbon nanocomposite prepared using ball milling mode D (strong ball impact): (a) discharge capacity and Coulombic efficiency vs. cycle number at a 0.4 C current rate (230 mA g−1), with a total material loading of 1.1 mg cm−2; (b) discharge and charge profiles for various cycles from the same test; (c) charge and discharge capacities obtained at varied current rates, with a total material loading of 1.35 mg cm−2 in the electrode; (d) discharge and charge capacities vs. cycle number in a thicker electrode, with a total material loading of 2.5 mg cm−2. Levels of the theoretical capacity and the highest discharge capacity are also indicated in (d). | ||
A separate coin cell (electrode's material loading of ∼1.35 mg cm−2) was used to test the rate capability of the nanocomposite and the results are shown in Fig. 4c. The current rate was sequentially set to 0.2 C, 0.4 C, 1 C, 2 C and 4 C (that is, 115, 230, 575, 1150 and 2300 mA g−1, respectively). The cell was discharged and charged five times at each current rate, and the capacities were recorded from the fifth cycle at each current rate. The discharge capacities obtained were 595, 547, 498, 424 and 317 mA h g−1, respectively. Importantly, the material is able to demonstrate a capacity exceeding the theoretical capacity of graphite at a high current rate of 1.15 A g−1. To complete the experiment, the current rate was returned to 0.2 C for five cycles and a capacity of 589 mA h g−1 was measured in the last cycle, indicating that the variation of the current rates during the test did not noticeably alter the capacity of the nanocomposite.
As the initial testing was performed in a rather thin electrode (material loading of ∼1.1 mg cm−2), its performance may not reflect that of thicker, “real life” electrodes. For that reason we have conducted an additional cyclic stability test using a thicker electrode with a material loading of ∼2.5 mg cm−2 and the same specific current density. The results are shown in Fig. 4d. The capacity of this thicker electrode is somewhat lower and stabilises at the level of approximately 500 mA h g−1 between 50th and 120th cycles. The capacity recorded in the 100th cycle was approximately 85% of the theoretical capacity and 88% of the maximum discharge capacity (recorded in the 3rd cycle). The capacity starts to deteriorate after 120 cycles, indicating that the overall performance of the Sb–carbon nanocomposite can be sensitive to the level of material loading in the electrode. Approximately 75% of theoretical capacity was measured in this electrode after 200 cycles. The performance is attractive for an electrode material that participates in an alloying reaction with lithium and experiences significant volume change.
The structural characteristics and operating mechanism of the nanocomposite
The phase composition and the structure of the nanocomposite (prepared using ball milling mode D) were further evaluated by an additional analysis of the XRD pattern, Raman spectroscopy, dark-field STEM and EFTEM. Fig. 5a shows the magnified XRD pattern of the nanocomposite. All visible peaks in the pattern belong to Sb (JCPDS no. 01-085-1324), and the average Sb crystallite size calculated using the Scherrer equation is 15.5 nm. The absence of the graphitic peaks indicates that the long-range crystalline order in the graphitic component is essentially lost in the nanocomposite after ball milling. In addition to the pronounced diffraction peaks of Sb, two broad features located at 24–36° and 43–55° can also be seen in the pattern, highlighting the presence of an additional amorphous phase. One possible explanation for this amorphous phase can be the presence of tiny clusters of Sb with a much smaller size than that of the typical Sb nanoparticles in the sample. An alternative explanation is the presence of amorphous antimony oxide. A pattern with a similar broad feature around the position of the (012) peak of Sb was previously described by Yang et al.18 for Sb-based nanoparticles. These authors have assigned the phase to the amorphous antimony oxide and were able to confirm this assignment via post annealing in an Ar flow. After the annealing the amorphous phase was converted into a mixture of Sb2O3 and Sb in their experiment.18 We have conducted a similar post-synthesis annealing experiment. The XRD pattern of the material after annealing is shown in the ESI (Fig. S1†). Indeed, the intensity of the amorphous features was significantly reduced and, instead, two families of Sb and Sb2O3 peaks were observed. We propose that the broad amorphous peaks visible in Fig. 5a may belong to an amorphous antimony oxide and are formed as a result of the exposure of the sample to air. In either case, as it is demonstrated later, the amorphous phase is electrochemically active and is capable of reacting with lithium.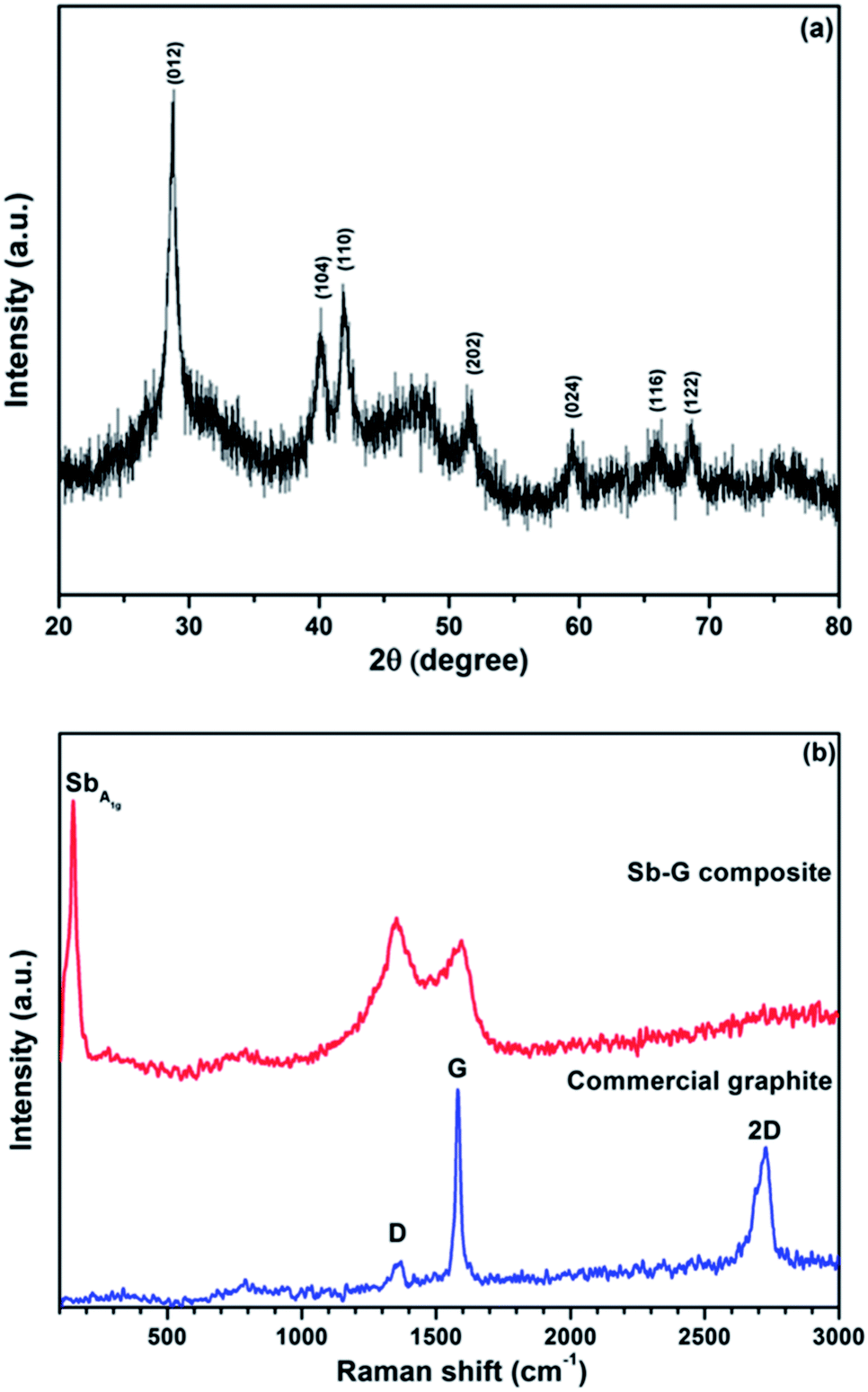 | ||
| Fig. 5 X-ray diffraction pattern (a) and a Raman spectrum (b) of the Sb–carbon nanocomposite prepared using ball milling mode D. A Raman spectrum of a sample of commercial graphite is also shown in (b) as a reference. | ||
Fig. 5b shows the Raman spectra of a sample of commercial graphite and the nanocomposite. Three typical bands, D band (1369 cm−1), G band (1581 cm−1) and 2D band (2728 cm−1),19 can be seen in the spectrum of the commercial graphite. The G band is the E2g vibration mode, which corresponds to the relative motion of sp2 carbon atoms. The D band represents the A1g ring-breathing mode of the aromatic six-member ring system of graphite; the latter band is however symmetrically forbidden in the perfect graphitic structure and arises due to the contribution from defect and boundary sites.20 Because of the nature of the G and D bands, the relative intensity ID/IG is widely used as an indication of the degree of disordering in graphite. The 2D band is the overtone of the D band. The D and G bands still exist in the spectrum of the nanocomposite after ball milling, while the 2D band is very wide and not distinguishable. As the nanocomposite also contains antimony, there is an additional A1g band from the Sb phase at about 149 cm−1.21 The graphitic ID/IG ratio in the nanocomposite is above 1, much higher than that in the commercial graphite (0.16). At the same time, both D and G bands are significantly broadened with respect to those of the commercial graphite. This type of Raman fingerprint indicates a disordered graphitic structure.22,23 The Raman result complements the XRD data. It is clear that, although the long-range graphitic order is significantly lost in the nanocomposite (no visible graphitic peaks in the XRD pattern), a short-range graphitic structure still remains in the material. The high intensity of the D band in the Raman spectrum confirms the presence of the graphitic ring structure in the sample.
The structure of the nanocomposite was visualised using TEM. A HAADF STEM image is shown in Fig. 6. The advantage of the HAADF technique is its good sensitivity to the average atomic numbers. As a result, areas with heavier elements tend to have much brighter contrast in this type of image while areas with lighter elements (such as carbon) appear to have less pronounced, darker contrast. Indeed, Fig. 6 presents a larger number of bright spots with a typical diameter below 10 nm surrounded by the material producing only weak contrast in the image that corresponds to the nanocomposite consisting of Sb nanoparticles dispersed (quite homogeneously) in the carbon component. Micro-sized aggregates of Si nanoparticles and carbon with a similar structure were recently reported to have stable cyclic stability as anode materials of Li-ion batteries.24,25
 | ||
| Fig. 6 A high-angle annular dark-field (HAADF) STEM image of the nanocomposite. As a result of the sensitivity of the contrast to the atomic number Z, Sb nanoparticles dispersed in the carbon material are visible as bright spots. | ||
The structure was further confirmed by means of energy-filtered TEM. The EFTEM images are shown in Fig. 7, including an elastic bright-field image (Fig. 7a) and an overlay of energy-filtered elemental maps of carbon and antimony (Fig. 7b). The distribution of the Sb element is shown in blue while the distribution of the C element is shown in yellow. As can be seen from Fig. 7b, numerous Sb-containing nanoparticles with the characteristic sizes of 5–15 nm are distributed within the large particle of the nanocomposite depicted in Fig. 7. The distribution of the Sb-containing nanoparticles is in good correlation with the areas of darker contrast in the bright-field image (Fig. 7a).
 | ||
| Fig. 7 Energy-filtered TEM images of a typical particle in the nanocomposite: (a) an elastic bright-field image and (b) an overlay of energy-filtered elemental maps of carbon and Sb (colour scheme: C – yellow; Sb – blue). Note the correlation between the distribution of Sb particles in the elemental map (b) and the areas with darker contrast in (a). | ||
It can be concluded on the basis of XRD, Raman and TEM data that the nanocomposite consists of Sb nanoparticles finely dispersed in the carbon component. The carbon component still has some short-range graphitic structures. In addition, Sb nanoparticles may get oxidised to some extent during the exposure of the sample to air.
XRD patterns of an unused electrode containing Sb–carbon nanocomposites as well as the patterns of the electrodes after the discharge and charge in the first and fourth cycles are shown in Fig. 8. Three strong Cu peaks visible in Fig. 8 come from the copper current collectors. The pattern of the fresh electrode (Fig. 8a) shows the peaks corresponding to the Sb phase as well as a broad diffuse feature at about 24–36° that we attribute to the presence of an amorphous antimony oxide or tiny Sb clusters in the sample. Ex situ XRD patterns in Fig. 8b–e demonstrate the reversible formation of the Li3Sb phase (JCPS 03-065-3011) upon the discharge of the cell to 0.01 V vs. Li/Li+. This phase forms after the discharge on both the first and fourth cycles and converts back to Sb after the charge of the cell in both cycles. The results are in line with the previous data by Park et al.13 for Sb–carbon nanocomposites, who observed similar reversible phase changes on charge and discharge.
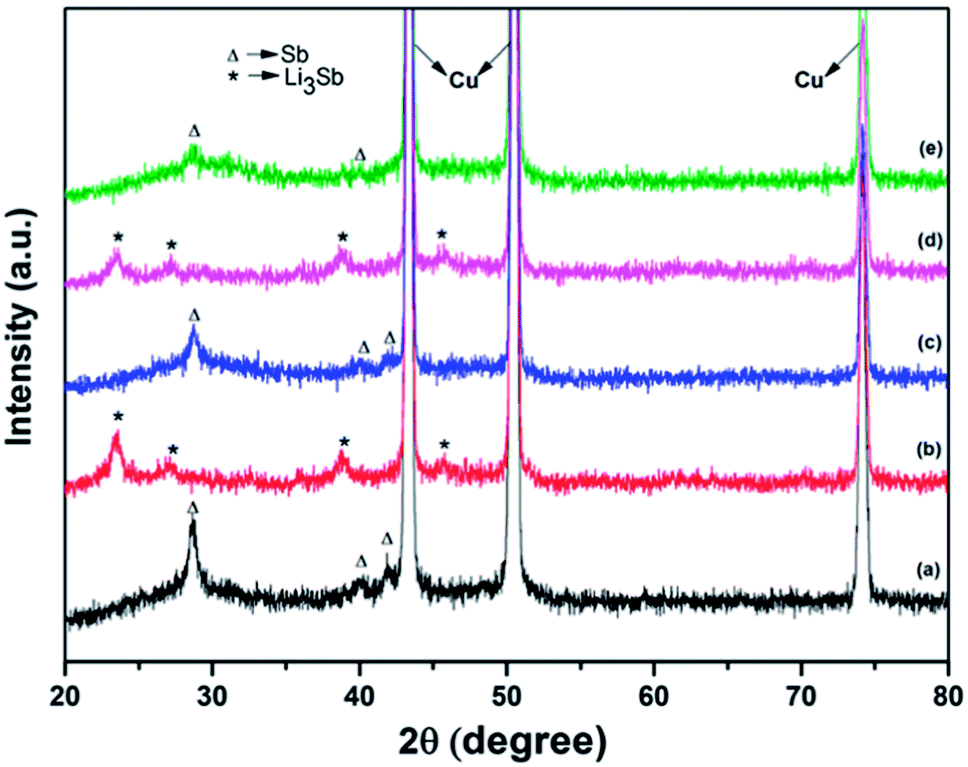 | ||
| Fig. 8 Ex situ X-ray diffraction patterns of the electrodes containing the Sb–carbon nanocomposite: (a) fresh unused electrode; (b) after the first discharge to 0.01 V vs. Li/Li+; (c) after the first charge to 2 V vs. Li/Li+; (d) after the fourth discharge; (e) after the fourth charge. | ||
It is also interesting to note that the amorphous phase is also electrochemically active, and this may be a common phenomenon in ball milled Sb–carbon composite electrodes, including those reported previously. Sb–carbon nanocomposites prepared by ball milling were previously studied by Park and Sohn13,15 and Qian et al.,26 who evaluated the performance of these nanocomposites in lithium-ion and sodium-ion batteries, respectively. It may be noted that the XRD patterns shown in Fig. 1a in ref. 15 and in Fig. 1a in ref. 26 display a similar broad peak of an amorphous phase around the position of the (012) peak of Sb. It appears that the presence of this amorphous phase is a common feature of all the samples, and it is practically important to understand its electrochemical reactivity with lithium. Our ex situ X-ray data (Fig. 8) indicate that the amorphous phase is electrochemically active and can store lithium reversibly. Indeed, the broad hump at 24–36° is present in the original (unused electrode) and in the electrodes after charging of the cell in the first and the fourth cycles (Fig. 8a, c and e). At the same time, this feature is absent in the XRD patterns of the electrodes after the discharge of the cell in the first and fourth cycles (Fig. 8b and d). It is concluded that the amorphous phase present in the sample reacts with lithium in a reversible manner during the cell operation.
The nanocomposites presented in this paper are active–active composites, i.e. both the Sb and carbon components participate in the lithium storage, which leads to the higher capacity displayed by our nanocomposites than that of the Sb component. Although it is difficult to define how lithium is stored exactly in the ball milled carbon component at this stage, there are grounds to propose that at least some of that storage happens via lithium intercalation between the layers, similarly to the classical intercalation mechanism in graphite. Indeed, the capacities demonstrated by the composites prepared using ball milling modes C and D are close to the theoretical capacity of a combination of Sb and graphite mixed in the same ratio of components, which may be indirect evidence of such a mechanism. In addition, our Raman spectroscopy data indicate that the short-range graphitic structure is retained in the nanocomposites and, correspondingly, lithium can intercalate between the layers of the local graphitic environment. Nevertheless, the exact mechanism for the lithium reactivity with the carbon component is currently not known and speculative. It needs to be investigated separately.
Conclusions
A reconfigurable magneto-ball mill was used to evaluate the influence of various ball milling modes on the structure and electrochemical properties of Sb–carbon nanocomposites. Four milling modes involving shear action only, shear action with an occasional weak impact as well as two milling modes with a strong ball impact were evaluated. It is found that ball milling modes without a ball impact or with a weak impact modify the structure of the carbon component significantly but do not affect the Sb component to a large extent. This results in an inferior performance of the resulting Sb–carbon nanocomposite and the significant drop in the capacity over multiple cycles. In contrast, if a ball milling mode involves strong ball impacts, the crystallite size of Sb is significantly reduced and a stable electrochemical performance can be obtained from a nanocomposite electrode.An electrode based on the optimal Sb–carbon nanocomposite can exhibit attractive cyclic performance, which is sensitive to the electrode's thickness. A thinner electrode (∼1 mg cm−2 material loading) exhibits outstanding stability for 250 cycles, displaying a steady capacity of 550 mA h g−1 at a current rate of 230 mA g−1. Furthermore, a capacity of ∼400 mA h g−1 is retained at a current rate of 1.15 A g−1. A thicker electrode (2.5 mg cm−2) displays some capacity fade after 200 cycles. The attractive cyclic performance is related to the structure of the nanocomposite. Sb nanoparticles with typical sizes of 5–15 nm (TEM data) are homogeneously dispersed in the carbon component. The carbon component does not possess a long-range graphitic structure (absence of graphitic peaks in XRD) after ball milling but still retains a local short-range graphitic environment, producing typical D and G bands in a Raman spectroscopy test. All of the Sb, and carbon components contribute to the charge storage. In accordance with the accepted mechanism, Sb reversibly transforms into Li3Sb during the cell operation.
Acknowledgements
The authors thank Mr Robert Lovett for his valuable help and Timcal Ltd. for providing a sample of Super P Li™ carbon black.Notes and references
- G. Jeong, Y.-U. Kim, H. Kim, Y.-J. Kim and H.-J. Sohn, Energy Environ. Sci., 2011, 4, 1986 CAS
.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419 CrossRef CAS PubMed
- A. Manthiram, J. Phys. Chem. Lett., 2011, 2, 176 CrossRef CAS
-
C. Grey, in Energy, Transport, & the Environment, ed. O. Inderwildi and S. D. King, Springer London, 2012, p. 253 Search PubMed
-
Hand Book of Batteries, ed. D. Linden, McGRAW-Hill INC, 1994, 36.2 Search PubMed
- W.-J. Zhang, J. Power Sources, 2011, 196, 13 CrossRef CAS PubMed
- L. Ji, Z. Lin, M. Alcoutlabi and X. Zhang, Energy Environ. Sci., 2011, 4, 2682 CAS
- Y. Zhang, X. G. Zhang, H. L. Zhang, Z. G. Zhao, F. Li, C. Liu and H. M. Cheng, Electrochim. Acta, 2006, 51, 4994 CrossRef CAS PubMed
- L. Wang, X. He, J. Li, W. Sun, J. Gao, J. Guo and C. Jiang, Angew. Chem., Int. Ed., 2012, 51, 9034 CrossRef CAS PubMed
- C. K. Chan, X. F. Zhang and Y. Cui, Nano Lett., 2008, 8, 307 CrossRef CAS PubMed
- J. R. González, R. Alcántara, F. Nacimiento and J. L. Tirado, Electrochim. Acta, 2011, 56, 9808 CrossRef PubMed
- G. Derrien, J. Hassoun, S. Panero and B. Scrosati, Adv. Mater., 2007, 19, 2336 CrossRef CAS
- C.-M. Park, S. Yoon, S.-I. Lee, J.-H. Kim, J.-H. Jung and H.-J. Sohn, J. Electrochem. Soc., 2007, 154, A917 CrossRef CAS PubMed
- C. M. Park and H. J. Sohn, Adv. Mater., 2007, 19, 2465–2468 CrossRef CAS
- C.-M. Park and H.-J. Sohn, Electrochim. Acta, 2009, 54, 6367 CrossRef CAS PubMed
- A. Calka and A. P. Radlinski, Mater. Sci. Eng., A, 1991, 134, 1350 CrossRef
- Y. Chen, T. Halstead and J. S. Williams, Mater. Sci. Eng., A, 1996, 206, 24 CrossRef
- Y. Yang, W. Huang, J. Zheng and Z. Li, J. Solid State Electrochem., 2012, 16, 803 CrossRef CAS PubMed
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter, 2000, 61, 14095 CrossRef CAS
- S. Reich and C. Thomsen, Philos. Trans. R. Soc., A, 2004, 362, 2271 CrossRef CAS PubMed
- X. Wang, K. Kunc, I. Loa, U. Schwarz and K. Syassen, Phys. Rev. B: Condens. Matter, 2006, 74, 134305 CrossRef
- T. Xing, L. H. Li, L. Hou, X. Hu, S. Zhou, R. Peter, M. Petravic and Y. Chen, Carbon, 2013, 57, 515 CrossRef CAS PubMed
- A. C. Ferrari, Solid State Commun., 2007, 143, 47 CrossRef CAS PubMed
- R. Yi, F. Dai, M. L. Gordin, S. Chen and D. Wang, Adv. Energy Mater., 2013, 3, 295 CrossRef CAS
- R. Yi, F. Dai, M. L. Gordin, H. Sohn and D. Wang, Adv. Energy Mater., 2013, 3, 1507 CrossRef CAS
- J. Qian, Y. Chen, L. Wu, Y. Cao, X. Ai and H. Yang, Chem. Commun., 2012, 48, 7070 RSC
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns and video clips. See DOI: 10.1039/c3ta14643j |
| This journal is © The Royal Society of Chemistry 2014 |