 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence7-Azaisoindigo as a new electron deficient component of small molecule chromophores for organic solar cells†
Nicholas M.
Randell
,
Amy F.
Douglas
and
Timothy L.
Kelly
*
Department of Chemistry, University of Saskatchewan, 110 Science Place, Saskatoon, SK, Canada. E-mail: tim.kelly@usask.ca; Fax: +1 306 966-4730; Tel: +1 306 966-4666
First published on 27th November 2013
Abstract
The use of 7-azaisoindigo as an electron accepting moiety in donor–acceptor chromophores is reported for the first time. Four chromophores were synthesized that contained either 2,2′-bithiophene or 3,3′-bis(dodecyloxy)-2,2′-bithiophene as an electron-donating sub-unit and either isoindigo or 7-azaisoindigo as an electron acceptor. The inclusion of a heteroatom in the isoindigo framework was designed to probe its effect on the LUMO, while 3,3′-bis(dodecyloxy)-2,2′-bithiophene was used as an electron rich alternative to 2,2′-bithiophene in order to similarly evaluate its effect on the HOMO level. The electronic properties of the resulting chromophores were fully characterized by UV/visible spectroscopy, cyclic voltammetry, and density functional theory. All four dyes were evaluated for their performance as electron donors in bulk heterojunction solar cells using PC61BM as the electron acceptor. Devices that used the isoindigo and 3,3′-bis(dodecyloxy)-2,2′-bithiophene-based dye displayed the best performance, reaching a maximum power conversion efficiency of 0.93%.
1. Introduction
Research in the area of organic photovoltaics (OPVs) has recently produced substantial gains in device efficiency.1–7 The current record for power conversion efficiency (PCE) in an OPV is a tandem, small molecule based cell with 12% PCE produced by the German company Heliatek.8 The use of oligomeric materials in this record breaking cell is an excellent example of how small molecule dyes can hold several advantages over their polymeric counterparts. These include their inherent monodispersity, greater reproducibility in their syntheses, and increased crystallinity. Polymeric OPV components often suffer from a broad distribution in molecular weights that can negatively affect cell performance, whereas small molecules can be readily obtained in extremely high purity using conventional synthetic organic techniques. Additionally, the increased crystallinity of small molecule chromophores can drive increases in charge carrier mobility, which in turn can lead to improvements in device efficiency.9,10Further increases in OPV efficiency will require the design of new dye molecules tailored to better overlap with the AM1.5G solar spectrum (which extends well into the near-infrared).11 In both polymer and small molecule materials the juxtaposition of an electron donor with an electron acceptor is a common design strategy used to produce low bandgap materials.3,12–16 The HOMOs of these molecules reside largely on the donor component, while in turn the LUMO is predominantly localized on the electron acceptor. This increases the charge transfer character of the HOMO to LUMO transition, leading to both a substantial red-shift of the absorption band and an increase in molar extinction coefficient.
Electron deficient sub-units bearing amide or imide functionalities, such as diketopyrrolopyrole3,17–19 and perylene bisimide,20–23 have been thoroughly explored as the acceptor portion of these donor–acceptor (DA) dyes. The electron deficient nature of these molecules lowers their frontier orbital energy levels, producing efficient low bandgap chromophores when coupled to appropriate electron donors. In 2010, Mei et al. first reported the use of isoindigo as a new electron deficient sub-unit in oligomeric DA chromophores.14 Since then, numerous research groups have made use of the isoindigo motif in new conjugated materials.24 These include the poly(isoindigo) homopolymer,25 DA oligomers26,27 and co-polymers,13,28–32 various n-type co-polymers,33 and dyes for metal-free dye sensitized solar cells.34 However, despite the extensive amount of research on isoindigo-containing compounds, relatively little work has been carried out on variants of the isoindigo core structure. While co-polymers containing chloro- and fluoro-substituted isoindigo have been studied for their ambipolar charge transport characteristics in organic field effect transistors,35,36 and as a component in polymer solar cells,37 little other work has been done in this area.
Here we report the use of 7-azaisoindigo as an electron deficient moiety in DA small molecule chromophores containing 2,2′-bithiophene donors. The presence of the heteroatom in the 7-azaisoindigo structure serves to fine-tune the energy of the LUMO, which is localized on the azaisoindigo sub-unit. In order to independently probe the effects of the electron withdrawing and donating components, a series of four chromophores were synthesized using a modular synthetic approach. Each contained either isoindigo or 7-azaisoindigo as the electron deficient sub-unit, and either 2,2′-bithiophene or 3,3′-bis(dodecyloxy)-2,2′-bithiophene as the electron donating component. The electronic structure of all four dyes was characterized by density functional theory (DFT) calculations, cyclic voltammetry, and electronic spectroscopy. Finally, the chromophores were combined with PC61BM and tested in prototype bulk heterojunction (BHJ) solar cells.
2. Experimental
2.1 Materials and methods
Prior to use, DMSO, DMF, toluene and THF were dried over activated 3 Å molecular sieves and stored under N2. Pd(PPh3)4 was stored in an inert atmosphere N2 glove box when not in use. All other solvents and reagents were used as received. NMR spectra were obtained using a Bruker Avance 500 MHz spectrometer. UV/visible spectroscopy measurements were performed in CHCl3 or as thin films on glass substrates using a Cary 6000 UV/visible spectrophotometer. AFM measurements were carried out using a Dimensions Hybrid Nanoscope system (Veeco Metrology Group). Mass spectra were recorded using a MALDI-TOF mass spectrometer, in positive ion reflectron mode, using a matrix of either α-cyano-4-hydroxycinnamic acid/trifluoroacetic acid (compounds 1–3) or 2,5-dihydroxybenzoic acid (compound 4).2.2 Cyclic voltammetry
Cyclic voltammetry was carried out in 0.05 mol L−1 tetrabutylammonium hexafluorophosphate dissolved in dry, degassed DCM. The working electrode was glassy carbon, the counter electrode was a Pt wire, and the reference electrode was a Ag wire. Voltammagrams were referenced to an internal Fc/Fc+ standard. All scans were acquired at a scan rate of 20 mV s−1.2.3 Device fabrication
Pre-cleaned ITO coated glass substrates (Delta Technologies) (Rs ≈ 6 Ω □−1) were UV–ozone cleaned for 15 min immediately prior to use. A layer of poly(3,4-ethylenedioxythiophene–polystyrene sulfonate (Clevios P VP AI 4083) was spin coated onto the ITO substrates and annealed at 150 °C before being placed in a N2-atmosphere glove box. The active layer solution (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 1–4:PC61BM by mass, total solid concentration of 18 mg mL−1 in chlorobenzene) was then deposited by spin coating. The samples were allowed to dry at ambient temperature for 2 hours, and then annealed at 100 °C for 20 min. Donor to acceptor ratios, concentrations, and annealing conditions were previously optimized for 1 by Mei et al.,14 and were left unchanged for 2–4. LiF (0.8 nm) and Al (100 nm) were then thermally evaporated onto the substrates at a base pressure of 10−6 mbar.
:40 1–4:PC61BM by mass, total solid concentration of 18 mg mL−1 in chlorobenzene) was then deposited by spin coating. The samples were allowed to dry at ambient temperature for 2 hours, and then annealed at 100 °C for 20 min. Donor to acceptor ratios, concentrations, and annealing conditions were previously optimized for 1 by Mei et al.,14 and were left unchanged for 2–4. LiF (0.8 nm) and Al (100 nm) were then thermally evaporated onto the substrates at a base pressure of 10−6 mbar.
2.4 Device characterization
Current–voltage measurements were made inside a N2-atmosphere glove box using a Keithley 2400 source-measure unit. The cells were illuminated by a 450 W Class AAA solar simulator equipped with an AM1.5G filter (Sol3A, Oriel instruments) at a calibrated intensity of 100 mW cm−2, as determined by a standard silicon reference cell (91150 V, Oriel Instruments). The cell area was defined by a non-reflective anodized aluminium mask to be 0.07065 cm2. Incident photon to current efficiency (IPCE) measurements were performed on the highest efficiency devices under ambient conditions using a QE-PV-SI system (Oriel Instruments) consisting of a 300 W Xe arc lamp, monochromator, chopper, lock-in amplifier and certified silicon reference cell. Measurements were made using a 30 Hz chop frequency.3. Results and discussion
3.1 Synthesis of donor–acceptor chromophores
The synthesis of DA chromophores is often carried out in a modular fashion, synthesizing the donor and acceptor sub-units separately and combining them late in the synthesis using metal-catalyzed cross-coupling reactions. This modular approach was adopted here, enabling the efficient synthesis of four dyes with an acceptor–donor–acceptor structure.7-Azaisoindigo was first synthesized in order to provide a more electron deficient analogue to isoindigo. While 7-azaisoindigo derivatives have been explored for their use in medicinal chemistry,38,39 no work has been done to evaluate their utility in optoelectronic applications. The synthesis started from the commercially available 7-azaindole, which was N-alkylated and then oxidized to the corresponding 1-alkyl-7-azaisatin. An aldol condensation with 6-bromo-2-oxindole was then used to form the 7-aza-6′-bromoisoindigo sub-unit.14 As shown by Mei et al.,14 due to the hydrolysable nature of the exocyclic double bond in the isoindigo backbone, upon exposure to base these molecules undergo a retro-aldol/aldol decomposition and reformation process. This same process was found to occur during alkylation of 7-aza-6′-bromoisoindigo, and produced a mixture of all three possible compounds, which were separated by chromatography.
The electron donor groups chosen for this work were 2,2′-bithiophene and 3,3′-bis(dodecyloxy)-2,2′-bithiophene. 2,2′-Bithiophene was chosen in order to provide a direct comparison to previous work.14 It was decided that in addition to using the more electron deficient azaisoindigo, an electron rich 3,3′-bis(dodecyloxy)-2,2′-bithiophene would also be synthesized. In this manner the relative effects of electron withdrawing and donating groups could be tested separately. Both 5,5′-bis(tributylstannyl)-2,2′-bithiophene41,42 and 5,5′-bis(tributylstannyl)-3,3′-bis(dodecyloxy)-2,2′-bithiophene15 were synthesized using previously reported methods.
Finally, the donor and acceptor portions of the dyes were combined via a metal-catalyzed cross-coupling. Because of the sensitivity of the isoindigo framework to basic conditions, a Stille coupling (rather than a Suzuki coupling) was chosen to couple the donor and acceptor sub-units (Scheme 1). The effect of coupling the donor and acceptor was immediately visible, with the red (aza)isoindigo solutions turning a deep purple when combined with 2,2′-bithiophene and dark blue when coupled to 3,3′-bis(dodecyloxy)-2,2′-bithiophene.
 | ||
| Scheme 1 Synthesis of compounds 1–4via the Stille coupling of stannylated bithiophene donor groups with brominated isoindigos. | ||
3.2 Orbital energy calculations
Density functional theory calculations at a B3LYP/6-31G(d,p) level of theory were used to determine the optimized geometries and frontier orbital energy levels for each of the synthesized oligomers; the results are summarized in Table 1. It was anticipated that the nitrogen-containing azaisoindigo would lower the LUMO energy of the dye, while adding alkoxy groups to the bithiophene core was expected to raise the energy of the HOMO with respect to vacuum. Both of these alterations were expected to lower the HOMO–LUMO gap of the oligomers.| Compound | E HOMO (calc.a) (eV) | E LUMO (calc.a) (eV) | ΔE (calc.) (eV) | λ max (soln) (nm) | ε/104 (soln) (M−1 cm−1) | E g (opt.) (eV) | E HOMO (elec.) (eV) | E LUMO (elec.) (eV) | ΔE (elec.) (eV) |
|---|---|---|---|---|---|---|---|---|---|
| a Carried out at the B3LYP/6-31G(d,p) level of theory. To reduce the complexity of the calculations 2-ethylhexyl groups were replaced with ethyl groups, dodecyloxy groups were replaced with methoxy groups and the calculations were performed in vacuum. b Optical bandgaps were taken from the absorption edge of thin film spectra. c Voltammagrams were referenced to the Fc/Fc+ redox couple and subsequently referenced to vacuum (the Fc/Fc+ redox couple was assumed to be 5.1 eV with respect to vacuum).40 HOMO and LUMO values were taken from the onset of oxidation and reduction respectively. | |||||||||
| 1 | 5.10 | 2.85 | 2.25 | 571 | 2.5 | 1.77 | 5.64 | 3.86 | 1.78 |
| 2 | 5.25 | 3.00 | 2.25 | 569 | 1.6 | 1.74 | 5.68 | 3.92 | 1.76 |
| 3 | 4.68 | 2.78 | 1.90 | 653 | 5.4 | 1.50 | 5.23 | 3.95 | 1.28 |
| 4 | 4.80 | 2.93 | 1.88 | 663 | 4.1 | 1.46 | 5.25 | 3.92 | 1.33 |
The effect of aza-substitution can be seen in the lower lying energy levels of 2 and 4 relative to their isoindigo counterparts; however, it appears that the inductive effect of the nitrogen atom affects both the HOMO and LUMO equally. There is no difference in the HOMO–LUMO gap between compounds 1 and 2 and only a minor change between compounds 3 and 4. The absolute energy of these orbitals, however, is dependent on the presence of the nitrogen heteroatom. Comparing 1 to 2 there is a 150 meV shift towards lower energies for both the HOMO and the LUMO. The effect on the 3,3′-bisalkoxybithiophene-based dyes is similar. In comparing 3 and 4, the LUMO level decreases by 150 meV while the HOMO energy drops by 120 meV, leading to a 30 meV reduction in HOMO–LUMO gap.
The effect of alkoxy-substitution on the frontier orbital energy levels and bandgap is more pronounced. In the isoindigo-based dyes (1 and 3) the addition of alkoxy groups to the bithiophene core increases the HOMO energy by 420 meV, while the LUMO energy is only raised by 70 meV, leading to a red-shift in the HOMO–LUMO gap of 350 meV. A similar shift is seen in the azaisoindigo-based dyes (2 and 4). The HOMO level increases by 450 meV while the LUMO increases by 70 meV, lowering the HOMO–LUMO gap energy by 370 meV.
Fig. 1 depicts the frontier molecular orbitals for 1–4. As expected, the HOMO is predominantly localized on the electron-rich bithiophene core, while the LUMO is more localized on the (aza)isoindigo sub-units. One notable difference in the structure of the frontier orbitals is that they are much more localized in 3 and 4 than in 1 and 2. This is attributed to the presence of the dodecyloxy groups in 3 and 4. In these compounds the relative energies of the bithiophene π-orbitals are substantially higher than those of the (aza)isoindigo sub-unit, limiting the amount of orbital mixing that can occur. This increase in localization agrees well with the fact that there was no change in the HOMO–LUMO gap for 1 and 2 but that there is a 20 meV difference in the gap energy for 3 and 4.
 | ||
| Fig. 1 Frontier molecular orbitals of 1–4 calculated from optimized geometries at the B3LYP/6-31G(d,p) level of theory. | ||
3.3 Cyclic voltammetry
The effects of both alkoxy and aza functionalities on the frontier orbital energy levels were experimentally studied by cyclic voltammetry. Both 1 and 3 show partially symmetrical peaks for both oxidation and reduction processes, with >59 mV separation between the corresponding anodic and cathodic peaks. This is consistent with quasi-reversible behaviour. By contrast, the aza-substituted dyes have less pronounced features. Both the oxidation and reduction processes observed in 2 appear to be largely irreversible, and while 4 undergoes an oxidation process consistent with quasi-reversible behaviour, the reversibility of its first reduction peak is poor. Since this same reduction process appears to be poorly reversible in both 2 and 4, it suggests that the reduced azaisoindigo moiety is chemically reactive. Compound 4 also shows an additional reduction process at more negative potential; however, the reversibility of the first reduction was not improved by changing sweep direction immediately after the first peak, before the second reduction occurred.Analysis of the peak positions reveals a dramatic shift in the potential of the peak anodic current when the bithiophene is substituted with alkoxy groups. The oxidation potential is shifted to lower potentials by 0.40 V between 1 and 3, and by 0.50 V between 2 and 4. The replacement of isoindigo with azaisoindigo, however, has little effect on the position of the reduction peak. The differences in reduction potentials between 1 and 2 and between 3 and 4 are within experimental error.
The HOMO and LUMO energies measured using cyclic voltammetry exhibit qualitatively the same trends as those determined using density functional theory. Placement of alkoxy groups on the bithiophene raises the HOMO energy level by 0.4–0.5 eV, while replacing isoindigo with azaisoindigo has only a relatively minor effect on the LUMO energy. The differences in ΔE between the CV and DFT results are likely the result of the DFT calculations being carried out in the absence of a dielectric continuum or solvent model.
3.4 UV/Visible spectroscopy
Solution and solid state UV/visible spectroscopy were carried out on all four compounds to determine their suitability as OPV dyes. Extinction coefficients and λmax values, as well as solid state optical bandgaps, are listed in Table 1.One of the most pronounced differences in the UV/visible spectra is that the addition of alkoxy groups to the bithiophene donor more than doubles the extinction coefficient of the broad, low energy absorption band. Since the oscillator strength of a transition is proportional to the transition dipole moment, this dramatic change in extinction coefficient is likely due to an increase in the degree of charge transfer character associated with the transition. This is in agreement with the increased localization observed in the frontier orbitals (Fig. 1).
The inclusion of alkoxy substituents also red-shifts the λmax by 82 nm in the isoindigo dyes (1 and 3) and by 94 nm in the azaisoindigo dyes (2 and 4). The presence of azaisoindigo has a much smaller effect, very slightly blue-shifting the λmax from 571 to 569 between 1 and 2 and providing a slight bathochromic shift of 10 nm between 3 and 4. The bathochromic shift between 3 and 4 is further evidence of increased orbital localization in these two dyes (Fig. 1). Localization of the frontier orbitals would increase the inductive effect of the nitrogen on the LUMO, while decreasing its effect on the primarily bithiophene-centred HOMO.
When the chromophores are spin cast as thin films on glass slides, the features of the UV/visible spectra change dramatically (Fig. 2). All four dyes develop a shoulder on the low energy side of the most prominent absorption band, indicative of intermolecular interactions in the solid state. This feature is most prominent in 1. The calculated optical bandgaps for the chromophores are summarized in Table 1. In all cases the band edge in the solid state occurs at lower energy than the solution phase HOMO–LUMO gap. In the case of 2, the λmax value shifts to slightly higher energy in the thin film than in solution; however, the growth of a prominent shoulder feature nonetheless red-shifts the absorption edge. In the solid state both 3 and 4 absorb well into the near- infrared, which is highly promising for their application in bulk heterojunction solar cells.
 | ||
| Fig. 2 Cyclic voltammetry of 1–4 performed in 0.05 M TBAPF6 in DCM and referenced to an internal Fc/Fc+ standard. | ||
3.5 Photovoltaic cell performance
The frontier orbital energy levels of 1–4 may make them suitable for use as either the electron accepting or donating component of the active layer in a bulk heterojunction solar cell. The most electron deficient dyes (1 and 2) were first evaluated for their ability to act as electron acceptors by blending them with poly(3-hexylthiophene) (P3HT) and fabricating OPV devices. These devices performed very poorly (cf. ESI†), with maximum PCEs of 0.016% and 0.002% for 1 and 2, respectively. Since the more electron-rich alkoxy-substituted compounds 3 and 4 are unlikely to perform better as electron acceptors, this suggests that these compounds are best utilized as the electron donors in BHJ devices.In order to assess the performance of 1–4 as electron donors, OPV devices were fabricated by blending each of the four dyes with PC61BM, and the current–voltage characteristics of the highest performing devices are shown in Fig. 4. The performance of OPVs incorporating 1 and PC61BM is in good agreement with the previous report of Mei et al.14Table 2 summarizes the device performance parameters of the cells based on each of the four oligomers.
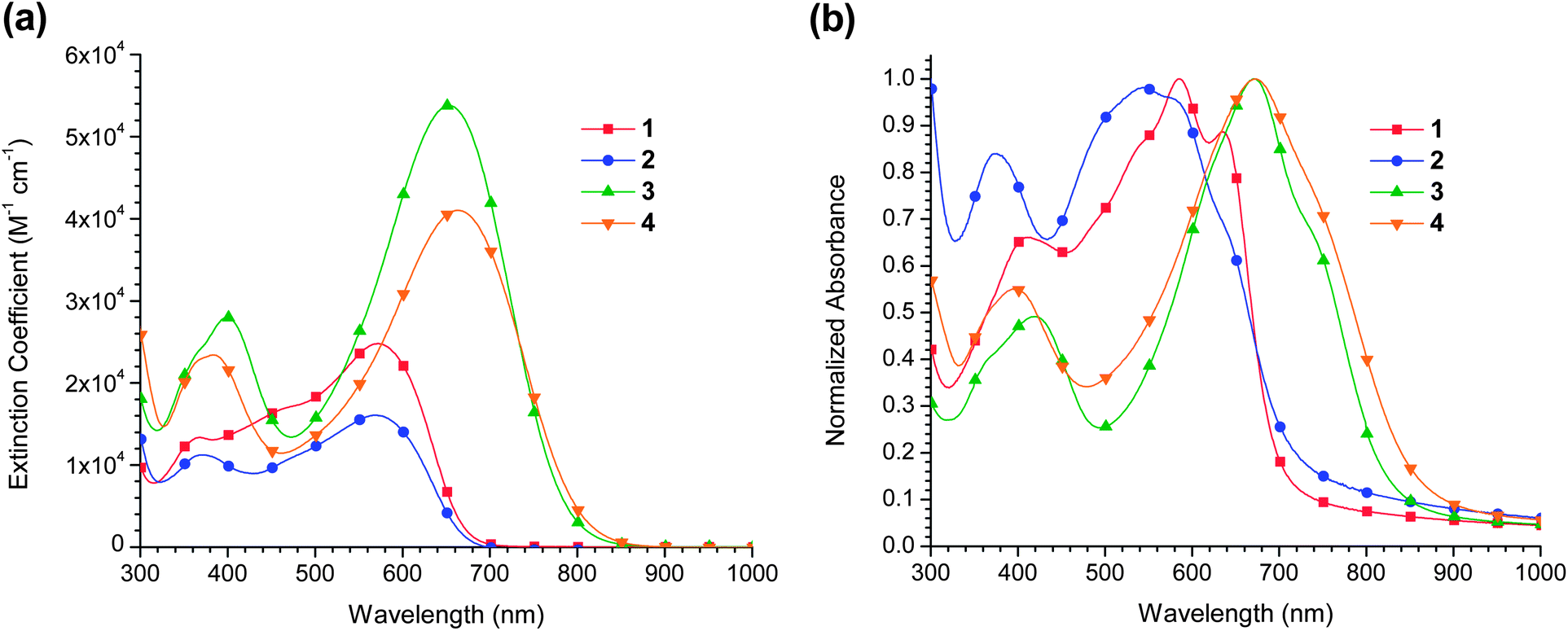 | ||
| Fig. 3 UV/visible spectra of 1–4: (a) in chloroform solution, and (b) as thin films on glass substrates. | ||
 | ||
| Fig. 4 Current–voltage curves for the most efficient BHJ solar cells based on active layers of 1–4:PC61BM (60:40 by mass). All devices were based on the ITO/PEDOT:PSS/1–4:PC61BM/LiF/Al architecture. | ||
:40 1–4:PC61BM/LiF/Al bulk heterojunction solar cells
| Compound | V oc (V) | J sc (mA cm−2) | Fill factora (%) | PCEa (%) | Best PCE (%) |
|---|---|---|---|---|---|
| a Averages and standard deviations for each data set were determined from measurements on a minimum of 23 separate devices. | |||||
| 1 | 0.66 ± 0.05 | 2.7 ± 0.3 | 31 ± 3 | 0.6 ± 0.1 | 0.74 |
| 2 | 0.28 ± 0.09 | 0.22 ± 0.04 | 28 ± 2 | 0.018 ± 0.006 | 0.03 |
| 3 | 0.67 ± 0.01 | 2.5 ± 0.9 | 40 ± 3 | 0.7 ± 0.2 | 0.93 |
| 4 | 0.3 ± 0.1 | 0.6 ± 0.2 | 22 ± 2 | 0.04 ± 0.02 | 0.07 |
From the data in Table 2, two major trends can be observed. The first is that alkoxy substitution of the bithiophene core (compound 3 compared to 1) results in a net improvement in PCE. The average data suggests that this increase in efficiency is primarily due to an increase in the fill factor; however, from the J–V curves of the most efficient cells, it can also be observed that an increase in Jsc also plays a role. Further evidence for this can be found by comparing the IPCE spectra of the most efficient devices (Fig. 5). The IPCE of cells containing 3 is substantially higher across the entire spectrum than the IPCE of cells containing 1, reflecting the larger extinction coefficient of 3.Furthermore, the IPCE spectrum of the device containing 3 extends substantially further into the near-infrared. Both spectra match well to the absorption spectra of thin films of 1 and 3.
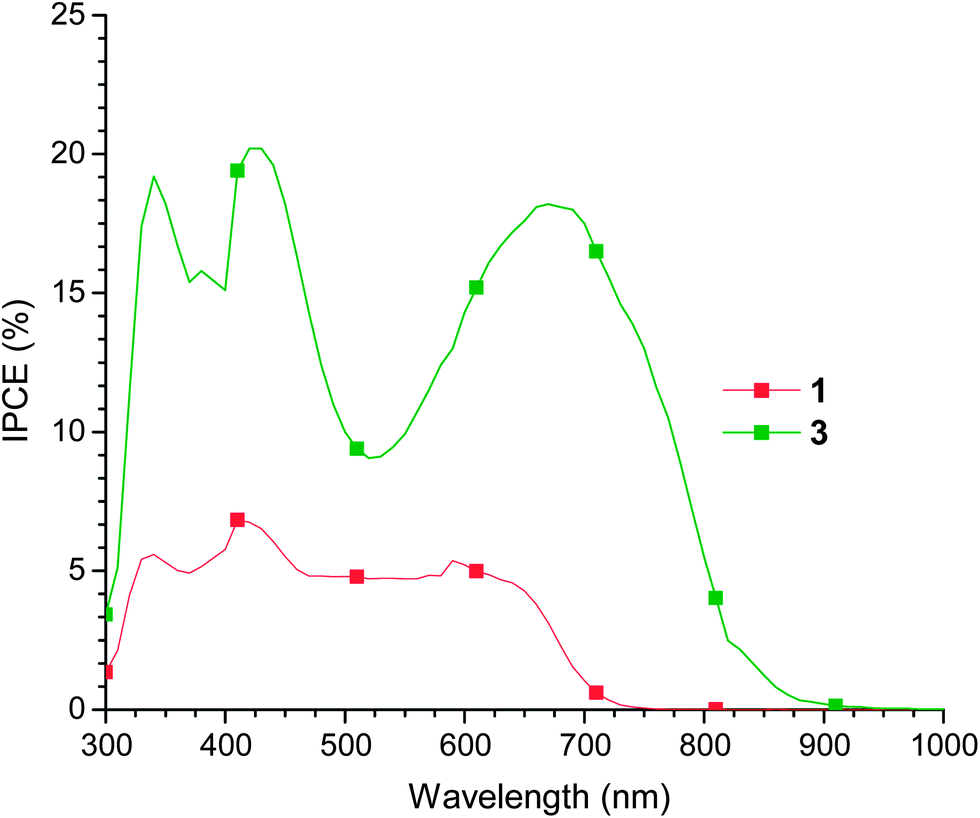 | ||
| Fig. 5 IPCE spectra of the most efficient BHJ solar cells based on active layers of 1 or 3:PC61BM (60:40 by mass). All devices were based on the ITO/PEDOT:PSS/1 or 3:PC61BM/LiF/Al architecture. | ||
The second, and most notable, trend in device performance is that the devices incorporating the azaisoindigo-based dyes (2 and 4) performed very poorly when compared to 1 and 3. The devices that used the azaisoindigo-based dye 2 were over an order of magnitude less efficient than those that used the isoindigo-based analogue 1. Similar results are obtained when the performance of devices based on compounds 3 (0.67%) and 4 (0.04%) is compared. The IPCE spectra of devices based on 2 and 4 are shown in Fig. S3,† and reflect the poor Jsc values of both devices. Given the very similar frontier orbital energy levels and solid state UV/visible spectra of compounds 1 and 2, and compounds 3 and 4, these results are somewhat surprising.
From Table 2, it can be seen that one of the main causes of the poor efficiency for the devices that use the azaisoindigo-based dyes is a poor fill factor. From the slope of the J–V curve as it approaches Voc (Fig. 4) it can be seen that this is in turn caused by a low shunt resistance. The low shunt resistance also explains the very low values of Voc, which are much lower than would be predicted from the difference between the donor HOMO and the LUMO of PC61BM. This suggests that recombination within the active layer may be a substantial obstacle to the use of 2 and 4 in OPV devices. 4 performed slightly better than 2 in test devices, owing to its more intense and red-shifted absorption bands (Fig. 3b), which produced slightly higher values of both IPCE (Fig. S3†) and Jsc.
3.6 Atomic force microscopy
One possible explanation for the poor performance of 2 and 4 is that the presence of the nitrogen atom in the isoindigo structure negatively affected the morphology of the bulk heterojunction. In order to determine if this was the case, AFM images were recorded of films of dyes 1–4 blended with PC61BM. Measurements were conducted on OPV substrates in areas lacking the metal cathode, and the height contrasted AFM images for each active layer blend are shown in Fig. 6. The film morphology of 1 and PC61BM closely resembles that of previous reports.14 The morphologies of the blends containing 2 and 4 were not substantially different from those of 1 and 3, and no definite correlations could be drawn between the blend morphologies of the azaisoindigo-based dyes and their poor photovoltaic performance. The root-mean-square (RMS) roughness of all films is similar, again suggesting that the morphology is not responsible for the observed decrease in performance. Additionally, since the energy levels of 2 and 4 are quite similar to those of 1 and 3 (Table 1), it is unlikely that a decrease in the amount of exciton dissociation is responsible for the lower efficiencies. | ||
| Fig. 6 Tapping mode AFM height images of 60:40 blends of: (a) 1:PC61BM, (b) 2:PC61BM, (c) 3:PC61BM, and (d) 4:PC61BM. Blends were spin coated from chlorobenzene onto thin films of PEDOT:PSS and annealed at 100 °C for 20 min. RMS roughness values: (a) 4.6 nm, (b) 1.8 nm, (c) 3.8 nm, (d) 2.0 nm. All images have dimensions of 1 μm × 1 μm. | ||
After ruling out morphological effects, it is most likely that the poor performance of the OPVs incorporating 2 and 4 is due to a reduced hole mobility in the electron donor. In their work on benzothiadiazole-based polymers, Blouin et al. also synthesized a pyridinothiadiazole-based system43 and found that the pyridino analogue had a hole mobility two orders of magnitude lower than the benzothiadiazole-based parent polymer. Steinberger et al. have also developed similar DA oligomers based on pyridinothiadiazole,44 and found that while the compounds worked well when incorporated into planar junction solar cells, when incorporated into a BHJ the performance dropped dramatically. This is consistent with the findings of Blouin et al., since there should be an increased dependence on charge carrier mobility in the BHJ device. These precedents lend credence to the hypothesis that a low hole mobility is the driving force behind the relatively poor efficiency of 2 and 4 in OPV devices.
4. Conclusions
In conclusion, we have presented the use of azaisoindigo in optoelectronic applications for the first time. Using a modular synthetic strategy, a series of acceptor–donor–acceptor dyes incorporating either azaisoindigo or isoindigo as the electron acceptor and either 2,2′-bithiophene or 3,3′-bis(dodecyloxy)-2,2′-bithiophene as the electron donor have been synthesized. The electronic structure of these dyes was investigated using DFT calculations, cyclic voltammetry and electronic spectroscopy. It was discovered that the dyes incorporating 3,3′-bis(dodecyloxy)-2,2′-bithiophene had large extinction coefficients and also absorbed well into the near-infrared, meeting two important criteria for their use in OPV applications. The replacement of isoindigo with 7-azaisoindigo induced a slight red-shift in the absorption spectrum of dyes bearing the bis(alkoxythiophene) donors; however, the effects of aza-substitution of isoindigo were small compared with the electron donating effects of the dodecyloxy groups.All four dyes were subsequently used as the electron donor in the active layer of bulk heterojunction solar cells. Devices that used the isoindigo/bis(alkoxythiophene) dye (3) performed the best, with PCEs reaching 0.93%. Both compounds based on 7-azaisoindigo performed poorly relative to their isoindigo counterparts, likely due to a reduction in hole mobility. These results clearly demonstrate a number of important structure–property relationships in the design of small molecule chromophores for organic photovoltaic devices.
Acknowledgements
The National Science and Engineering Research Council of Canada (NSERC) and the University of Saskatchewan are acknowledged for financial support. T.L.K. is a Canada Research Chair in Photovoltaics. This research was undertaken, in part, thanks to funding from the Canada Research Chairs Program. N.M.R. thanks NSERC and the Government of Saskatchewan (Saskatchewan Opportunity and Innovation scholarship) for scholarship funding.Notes and references
- Z. B. Henson, G. C. Welch, T. van der Poll and G. C. Bazan, J. Am. Chem. Soc., 2012, 134, 3766–3779 CrossRef CAS PubMed.
- Y. Sun, G. C. Welch, W. L. Leong, C. J. Takacs, G. C. Bazan and A. J. Heeger, Nat. Mater., 2011, 11, 44–48 CrossRef PubMed.
- A. B. Tamayo, B. Walker and T.-Q. Nguyen, J. Phys. Chem. C, 2008, 112, 11545–11551 CAS.
- M. Weidelehner, C. Wessendorf, J. Hanisch, E. Ahlswede, G. Götz, M. Linden, G. Schulz, E. Mena-Osteritz, A. Mishra and P. Bäuerle, Chem. Commun., 2013, 49, 10865–10867 RSC.
- S. Haid, M. Marszalek, A. Mishra, M. Wielopolski, J. Teuscher, J. E. Moser, R. Humphry-Baker, S. M. Zakeeruddin, M. Grätzel and P. Bäuerle, Adv. Funct. Mater., 2012, 22, 1291–1302 CrossRef CAS.
- S. Steinberger, A. Mishra, E. Reinold, E. Mena-Osteritz, H. Müller, C. Uhrich, M. Pfeiffer and P. Bäuerle, J. Mater. Chem., 2012, 22, 2701–2712 RSC.
- G. C. Welch, L. A. Perez, C. V. Hoven, Y. Zhang, X.-D. Dang, A. Sharenko, M. F. Toney, E. J. Kramer, T.-Q. Nguyen and G. C. Bazan, J. Mater. Chem., 2011, 21, 12700–12709 RSC.
- http://www.heliatek.com/wp-content/uploads/2013/01/<?ccdc_no 130116?>130116<?ccdc END?>_PR_Heliatek_achieves_record_cell_effiency_for_OPV.pdf, retrieved October 8, 2013.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- H. Shang, H. Fan, Y. Liu, W. Hu, Y. Li and X. Zhan, Adv. Mater., 2011, 23, 1554–1557 CrossRef CAS PubMed.
- M. Wan, H. Zhu, H. Deng, L. Jin, J. Guo and Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3477–3485 CrossRef CAS.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS PubMed.
- X. Guo and M. D. Watson, Org. Lett., 2008, 10, 5333–5336 CrossRef CAS PubMed.
- J. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS PubMed.
- B. P. Karsten, J. C. Bijleveld and R. A. J. Janssen, Macromol. Rapid Commun., 2010, 31, 1554–1559 CrossRef CAS PubMed.
- M. M. Wienk, M. Turbiez, J. Gilot and R. A. J. Janssen, Adv. Mater., 2008, 20, 2556–2560 CrossRef CAS.
- J. C. Bijleveld, A. P. Zoombelt, S. G. J. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. J. Janssen, J. Am. Chem. Soc., 2009, 131, 16616–16617 CrossRef CAS PubMed.
- I. A. Howard, F. Laquai, P. E. Keivanidis, R. H. Friend and N. C. Greenham, J. Phys. Chem. C, 2009, 113, 21225–21232 CAS.
- X. Zhang, Z. Lu, L. Ye, C. Zhan, J. Hou, S. Zhang, B. Jiang, Y. Zhao, J. Huang, S. Zhang, Y. Liu, Q. Shi, Y. Liu and J. Yao, Adv. Mater., 2013, 25, 5791–5797 CrossRef CAS PubMed.
- Z. Chen, Y. Zheng, H. Yan and A. Facchetti, J. Am. Chem. Soc., 2009, 131, 8–9 CrossRef CAS PubMed.
- J. L. Segura, H. Herrera and P. Bäuerle, J. Mater. Chem., 2012, 22, 8717–8733 RSC.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2013 DOI:10.1021/cm402219v.
- R. Stalder, J. Mei, J. Subbiah, C. Grand, L. A. Estrada, F. So and J. R. Reynolds, Macromolecules, 2011, 44, 6303–6310 CrossRef CAS.
- T. Wang, Y. Chen, X. Bao, Z. Du, J. Guo, N. Wang, M. Sun and R. Yang, Dyes Pigm., 2013, 98, 11–16 CrossRef CAS PubMed.
- M. Yang, X. Chen, Y. Zou, C. Pan, B. Liu and H. Zhong, J. Mater. Sci., 2012, 48, 1014–1020 CrossRef.
- K. Cao, Z. Wu, S. Li, B. Sun, G. Zhang and Q. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2012, 51, 94–100 CrossRef.
- E. Wang, Z. Ma, Z. Zhang, K. Vandewal, P. Henriksson, O. Inganäs, F. Zhang and M. R. Andersson, J. Am. Chem. Soc., 2011, 133, 14244–14247 CrossRef CAS PubMed.
- C. Wang, B. Zhao, Z. Cao, P. Shen, Z. Tan, X. Li and S. Tan, Chem. Commun., 2013, 49, 3857–3859 RSC.
- K. Mahmood, Z.-P. Liu, C. Li, Z. Lu, T. Fang, X. Liu, J. Zhou, T. Lei, J. Pei and Z. Bo, Polym. Chem., 2013, 4, 3563–3574 RSC.
- W. Sun, Z. Ma, D. Dang, W. Zhu, M. R. Andersson, F. Zhang and E. Wang, J. Mater. Chem. A, 2013, 1, 11141–11144 CAS.
- F. Grenier, P. Berrouard, J.-R. Pouliot, H.-R. Tseng, A. J. Heeger and M. Leclerc, Polym. Chem., 2013, 4, 1836–1841 RSC.
- W. Ying, F. Guo, J. Li, Q. Zhang, W. Wu, H. Tian and J. Hua, ACS Appl. Mater. Interfaces, 2012, 4, 4215–4224 CAS.
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-H. Yao, C.-J. Liu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2012, 134, 20025–20028 CrossRef CAS PubMed.
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-J. Liu, J.-Y. Wang and J. Pei, Chem. Sci., 2013, 4, 2447–2452 RSC.
- Y. Deng, J. Liu, J. Wang, L. Liu, W. Li, H. Tian, X. Zhang, Z. Xie, Y. Geng and F. Wang, Adv. Mater., 2013 DOI:10.1002/adma.201303586.
- J. Tatsugi, T. Zhiwei and Y. Izawa, ARKIVOC, 2001, i, 67–73 CrossRef.
- M. Kritsanida, P. Magiatis, A.-L. Skaltsounis, Y. Peng, P. Li and L. P. Wennogle, J. Nat. Prod., 2009, 72, 2199–2202 CrossRef CAS PubMed.
- P. M. Beaujuge, S. V. Vasilyeva, D. Y. Liu, S. Ellinger, T. D. McCarley and J. R. Reynolds, Chem. Mater., 2012, 24, 255–268 CrossRef CAS.
- R. Wu, J. S. Schumm, D. L. Pearson and J. M. Tour, J. Org. Chem., 1996, 61, 6906–6921 CrossRef CAS PubMed.
- J. Huang, J. Miragliotta, A. Becknell and H. E. Katz, J. Am. Chem. Soc., 2007, 129, 9366–9376 CrossRef CAS PubMed.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS PubMed.
- S. Steinberger, A. Mishra, E. Reinold, J. Levichkov, C. Uhrich, M. Pfeiffer and P. Bäuerle, Chem. Commun., 2011, 47, 1982–1984 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fabrication procedures and performance data for P3HT:1–2 devices, as well as IPCE spectra and detailed synthetic procedures. See DOI: 10.1039/c3ta14263a |
| This journal is © The Royal Society of Chemistry 2014 |