 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchically porous graphene sheets and graphitic carbon nitride intercalated composites for enhanced oxygen reduction reaction†
Kaipei
Qiu
* and
Zheng Xiao
Guo
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: k.qiu.12@ucl.ac.uk; Tel: +44 (0)7869738380
First published on 18th December 2013
Abstract
The electrocatalytic activity of graphitic carbon nitride (GCN), a potential metal-free alternative to the conventional platinum-based catalysts for oxygen reduction reaction (ORR), is restricted by its poor electrical conductivity. Introducing conductive carbon substrates can enhance the ORR performance of GCN, but until now, none of the carbon-supported GCN catalysts has shown both excellent catalysis selectivity and fast ORR kinetics. Two-dimensional graphene sheets (GSs) may serve as suitable substrates for GCN due to their fast electron collection and transport properties, and their structural similarity to GCN as well. In this work, we present a facile method of producing intercalated GS/GCN composites with hierarchical porosity, which is experimentally achieved for the first time. The ORR activity is optimised by tuning the amount of active sites, electrical conductivity and mass transport. The obtained material possesses 100% catalysis selectivity towards the four-electron pathway, and its ORR activities outperform any other existing GCN-based catalysts. It also shows significantly improved tolerance against methanol and enhanced long-term stability, compared with the commercial platinum-loaded carbon catalysts. Thus it is expected that the hierarchically porous GS/GCN intercalated composite is a promising future ORR catalyst.
Introduction
The electrochemical performance of fuel cells and metal–air batteries depends greatly on the cathodic oxygen reduction reaction (ORR).1 Although conventional platinum-loaded carbon composites (Pt/C) have long been regarded as effective electrocatalysts for ORR, the large-scale practical application has been hampered by the prohibitive cost of Pt, as well as its limited reserve in nature.2 In addition Pt/C suffers from the weak long-term durability,2b methanol fuel crossover effect,3 and rapid degradation against carbon monoxide impurity.2a Therefore finding low-cost, efficient and stable alternatives is essential to mass-market the above two technologies. Over the past years, two main approaches have been taken to partly/completely replace Pt catalysts through the formation of Pt-based alloys2c,4 or non-precious metal compounds,5 but until now neither approach has fully met the requirements for commercialization. Very recently metal-free materials, such as nitrogen-doped carbon and its derivatives, have attracted considerable interest as the next generation ORR electrocatalysts, due to their comparable catalytic activity, reduced cost and improved durability, compared with the existing counterparts.6It is commonly accepted that introducing nitrogen into a carbon matrix is a key step to synthesize highly active metal-free ORR catalysts.6b Despite the controversy on the exact role of the nitrogen heteroatom in ORR, both quantum mechanical calculations7 and experimental observations8 reveal that incorporation of nitrogen, especially the pyridinic or/and graphitic species, induces a positive charge on the adjacent carbon, which can facilitate oxygen adsorption and subsequently weaken the O–O bond in the adsorbed oxygen molecules. In this regard, graphitic carbon nitride (GCN) – stacked two dimensional (2D) heptazine (C6N7) sheets connected by tertiary amines – may serve as a promising metal-free ORR catalyst owing to its high nitrogen content (60.9 wt% in theory and mainly of pyridinic/graphitic nature) and stable molecular structure.9
Unfortunately, the electrocatalytic activity of pristine GCN is restricted by its semi-conductive nature (<10−2 S cm−1), which obstructs electron transfer during the ORR process.10 Thus introducing electron-conductive carbon materials as substrates should be a general strategy to improve the ORR performance of GCN. First-principles studies on the ORR capability of GCN have shown that if sufficient electrons are available in the GCN–O2 system, the ORR could occur via a direct four-electron (4e−) pathway, rather than the two-step two-electron (2e−) pathway for the pristine GCN.11 The first experimental attempt of carbon-supported GCN composites was to mechanically blend GCN with carbon black (CB).12 The GCB/CB composites showed much improved onset potential and reduction current density, compared with pristine GCN, as a result of the increased conductivity and surface area derived from the carbon black support.12 To further clarify the role of substrates, ordered porous carbon materials, such as CMK-3, were then adopted as their morphology and structures were easy to control.11 It was shown that incorporation of GCN into CMK-3 led to a direct 4e− pathway, consistent with the theoretical prediction.11 However, the small mesopores in CMK-3 may not be capable of fast mass exchange during ORR and the catalytic activity of GCN@CMK-3 was thus worse than that of the commercial Pt/C. From this point of view, uniform GCN embedded porous carbon (GCN/C) composites with a tuneable pore size have been prepared.13 The ORR activity of macroporous GCN/C with a pore size of 150 nm was shown superior to that of the mesoporous GCN/C with a pore size of 12 nm obtained via a similar synthesis route. The macroporous GCN/C facilitated the oxygen diffusion, and led to a much smaller Tafel slope value in the high overpotential region than that of the mesoporous sample. However the overall electron transfer number for this macroporous composite was only three, indicating a combined 2e− and 4e− reaction pathway, which was probably due to the low specific surface area and the lack of exposed active sites.13 So far, none of the aforementioned carbon-supported GCN composites can show both fast ORR kinetics and excellent selectivity of 4e− catalysis pathway.
Compared with carbon-based supports, 2D graphene sheets (GSs) are expected to be better substrates, given the excellent electron collection and transport properties of GSs and their structural similarity to GCN.6b,14 One latest theoretical investigation demonstrated that the π–π interaction at the interface of hybrid GS/GCN composites could force electrons to transfer from GS to GCN and accumulate on the active sites in GCN, which was predicted to significantly enhance the ORR catalytic activity.15 However the early investigation of immobilizing GCN on GS only showed limited improvement – the ORR occurred via a 3e− pathway with reduction current density slightly lower than that of the home-made platinum-loaded graphene composite.10 Such an improvement was below expectation because only the effect of conductivity was considered and the GCN loading ratio (the amount of active materials) was low (ca. 10 wt% nitrogen). In addition, the layered morphology was unfavourable for ORR due to the limited exposure of active sites and the inefficient mass transport and access.10 Inspired by that, here for the first time, we experimentally synthesized GS/GCN intercalated composites with hierarchical porosity. The ORR activity was first optimised by balancing two intrinsic parameters – active sites and conductivity. After further taking into account of mass transport, the obtained hierarchically porous GS/GCN intercalated composites showed fast reduction kinetics and 100% ORR catalysis selectivity, with better long-term stability and stronger tolerance against methanol than the commercial Pt/C catalysts. To the best of our knowledge, such excellent catalytic activities outperformed any other existing GCN-based ORR catalysts.10–13,16
Experimental details
Synthesis of GO
Graphite oxide (GO) was synthesized by a modified Hummer method, as previously reported.17 In a typical experiment, 5 g graphite (<20 micron, Sigma-Aldrich) was vigorously stirred with 115 ml concentrated sulfuric acid (95–97%, Merck KGaA) in an ice bath (0 °C) and 15 g potassium permanganate (99+%, Sigma-Aldrich) was slowly added to prevent a rapid temperature increase. The mixture was stirred in the ice bath for 30 minutes. After that the mixture was transferred to an oil bath (30 °C) and was further stirred for 2 hours. Then 115 ml DI water (18.2 MΩ, Barnstead Easypure RoDi) was slowly added and the temperature of the mixture was raised to ∼40 to 45 °C. The diluted mixture was kept stirring at that temperature for another 30 minutes. Subsequently 700 ml DI water was added into the mixture followed by 50 ml hydrogen peroxide aqueous solution (30 wt%, Sigma-Aldrich). The mixture was left overnight before being washed with 6–7 l DI water using a centrifuge (Heraeus Biofuge Primo). When the pH value of the drained water was 7, the obtained GO was freeze-dried (Virtis BT2KES) for about a week before further use.Synthesis of GCN
GCN was synthesized by the polymerization of cyanamide, as previously reported.18 In a typical experiment, cyanamide powder (99%, Sigma-Aldrich) was directly heated at 550 °C for 4 hours under nitrogen, with a ramping of 2.2 °C min−1.Synthesis of GSGCN
The GS/GCN intercalated composites were synthesized by wet chemical mixing of GO and cyanamide (GCN precursor) followed by thermal annealing. In a typical experiment, desirable mass ratios of cyanamide aqueous solution (50 wt%, Sigma-Aldrich) were added to 100 ml GO suspension (1 mg ml−1). The mixture was sonicated for 1 hour until there was no visible precipitate. After that, the sample was dried in a vacuum oven (80 °C) overnight. The obtained grey/black powder was then ground and transferred into a crucible with lid. For thermal annealing, the heating programme was the same as that for the synthesis of GCN – the sample was annealed at 550 °C for 4 hours under nitrogen, with a ramping of 2.2 °C min−1. The GS/GCN composites were labelled based on the mass ratio of cyanamide and GO, i.e. if 200 mg cyanamide was mixed with 100 mg GO, the obtained GS/GCN composite was marked as GSGCN_2×.Synthesis of hp-GSGCN_2×
Similar to the synthesis of GSGCN_2×, but an extra 5 ml colloidal silica (40 wt% suspension in water, Sigma-Aldrich) was added to the mixture of cyanamide and GO before sonication. After heating at 550 °C, the obtained material was washed with 10 g ammonium bifluoride (95%, Sigma-Aldrich) for 24 hours, followed by filtration and washing with 1 l DI water and 0.5 l ethanol (100%, Merck KGaA). The obtained hp-GSGCN_2× was dried in a vacuum oven (80 °C) overnight before characterization.Electrochemical measurement
The cyclic voltammetry, rotating voltammetry and chronoamperometric responses were measured in O2-saturated 0.1 M KOH. For electrode preparation, 4 mg catalyst was added to a mixture of 100 μl Nafion (5 wt% in alcohol and water, Sigma-Aldrich) and 900 μl DI water, followed by one-hour sonication so as to fully disperse the sample in the solvent. 5 μl catalyst suspensions were then drop cast on a glassy carbon (GC) working electrode (3 mm diameter, Metrohm) and dried at 60 °C. The ORR activity was tested in a three-electrode configuration. Ag/AgCl (sat. KCl, Sigma-Aldrich) was used as a reference electrode and a platinum sheet (Metrohm) was used as a counter electrode. Oxygen was purged into the electrolyte for an hour before measurement and kept bubbling during the tests, in order to make the electrolyte oxygen saturated. The scan rate for cyclic and rotating voltammetry was 100 and 10 mV s−1 respectively. Chronoamperometric responses for both methanol tolerance and long-term durability were measured at −0.3 V vs. Ag/AgCl. All the results were recorded by Metrohm Autolab 302N.Characterization
The morphology and microstructure of the samples were investigated by scanning electron microscopy (JSM6301F, Japan), transmission electron microscopy (JEOL 2100, Japan) and X-ray diffraction (STOE Stadi P). Nitrogen sorption isotherms and BET surface areas were measured at 77 K with Quantachrome Autosorb iQ-c. The chemical composition was analyzed by Fourier transform infrared spectroscopy (Thermo Scientific Nicolet iS10), X-ray photon spectroscopy (Thermo Scientific K-Alpha, UK), and thermogravimetric analysis (Setaram Setsys 16/18). Electric conductivity was measured by the four-probe technique (Jandel RM3) with the pressed pellet of materials as samples. Typically, 200 mg sample was pressed by 10 tonnes for 5 minutes to form the pellet.Results and discussion
It is first shown under a scanning electron microscope (SEM) that all GS/GCN composites possess a layered structure (Fig. 1a and S1†), similar to the morphology of GCN (Fig. 1b) and GS (Fig. 1c). The X-ray diffraction (XRD) patterns display that the peak positions of the GS/GCN composites are at 26.9°, corresponding to an interlayer distance of 3.31 Å, which can be further confirmed from the high-resolution transmission electron microscopy image (HRTEM, Fig. 1d). The interlayer distances for GS and GCN are 3.27 and 3.44 Å, according to the peak positions at 25.9° and 27.3° respectively (Fig. 1e). Note that the XRD peak positions of GS/GCN composites are between those of GS and GCN (highlighted by the dashed lines in Fig. 1e). Hence, the change in the interlayer distance suggests successful intercalation of GS into GCN layers.10 The Brunauer–Emmett–Teller specific surface area (BET SSA) of GS/GCN composites (Fig. 1f) derived from the nitrogen isotherm at 77 K (Fig. S2†) is within the range of 50–75 m2 g−1, with a slight rise from GSGCN_1× to GSGCN_1.5× then drops as the GCN content in the composites further increases. The SSA of the intercalated GS/GCN composites is lower than the previously reported value for graphene-based materials19 due to the restacking of graphene sheets during the formation of intercalated layered structures. | ||
| Fig. 1 (a–c) SEM images of GSGCN_2×, GCN and GS. The layered morphology of GSGCN_2×, GCN and GS is highlighted by the white circles in the SEM images; (d) HRTEM image of GSGCN_2×; (e) XRD of GO, GS, GCN and GS/GCN composites. Those three dashed lines exhibit the difference in the peak positions of GS, GCN and GS/GCN composites; and (f) BET SSA of GS/GCN composites. | ||
Chemical constitutions of the intercalated GS/GCN composites are then examined by Fourier transform infrared spectroscopy (FTIR). Results (Fig. 2) show that the out-of-plane bending vibration characteristics of heptazine rings20 (∼800 cm−1, highlighted by the ellipse) for GCN are preserved in all GS/GCN composites, while the typical stretching vibration modes of heptazine-derived repeating units20 (∼1200–1650 cm−1, highlighted by the straight lines in the inserted small plot) appear in GSGCN_2×, GSGCN_2.5× and GSGCN_3× but are not evident in GSGCN_1× and GSGCN_1.5×, implying either an incomplete polymerization or partial decomposition of the GCN component for the latter two composites.20c What is more, it is noticed that GO has been successfully reduced to GS via thermal annealing as the non-sp2 carbon bonds (i.e. C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO) of GO significantly diminish in all GS/GCN composites21 (highlighted by the straight lines in the main plot), which could be further supported by the X-ray photoelectron spectroscopy (XPS) results.
O and O–CO) of GO significantly diminish in all GS/GCN composites21 (highlighted by the straight lines in the main plot), which could be further supported by the X-ray photoelectron spectroscopy (XPS) results.
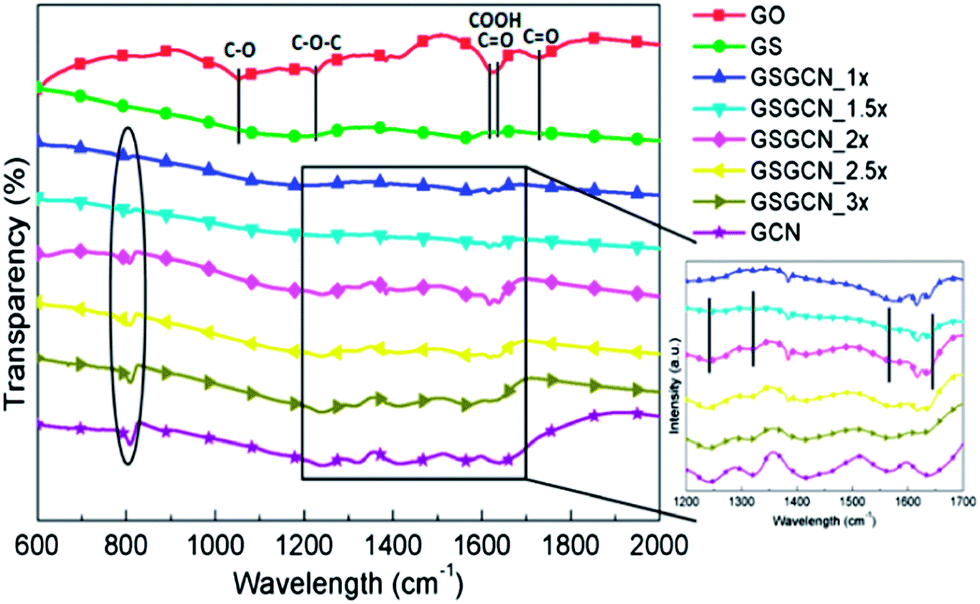 | ||
| Fig. 2 FTIR of GO, GS, GCN and GS/GCN composites. The ellipse and box (and also the lines in the inserted plot) represent the stretching mode of GCN; those lines in the main plot represent the vibration modes of epoxides, hydroxyls, carboxyls and ketones, respectively. | ||
It is shown in XPS C1s spectra (Fig. 3a) that the intensities of the C–O bond (∼286.5 eV) and the CO bond (∼288 eV) are largely reduced in GS and GS/GCN composites and the atomic ratio of Csp2/Csp3 increases from 0.9 for GO to 2.7 for GS.22 Also the N–CN bond (∼288.3 eV) from GCN22 could hardly been observed in GSGCN_1× and GSGCN_1.5×, in line with the FTIR results. It is worth noting that the C–C bond (∼284.7 eV in GS/GCN)22 and the N–CN bond (∼288.1 eV in GS/GCN, if applicable) of GS/GCN composites show a right/left shift of ca. 0.1 eV compared with that of the pure GS and GCN respectively (highlighted by the dashed lines in Fig. 3a), which should be attributed to the strong electron transfer between the intercalated GS/GCN interfaces.14 The relative ratios of pyridinic nitrogen bonding (∼399 eV)22d derived from XPS N1s spectra for the GSGCN_1× and GSGCN_1.5× are much lower than that of pure GCN (Fig. 3b), confirming the incomplete polymerization or partial decomposition of GCN discovered in the FTIR.
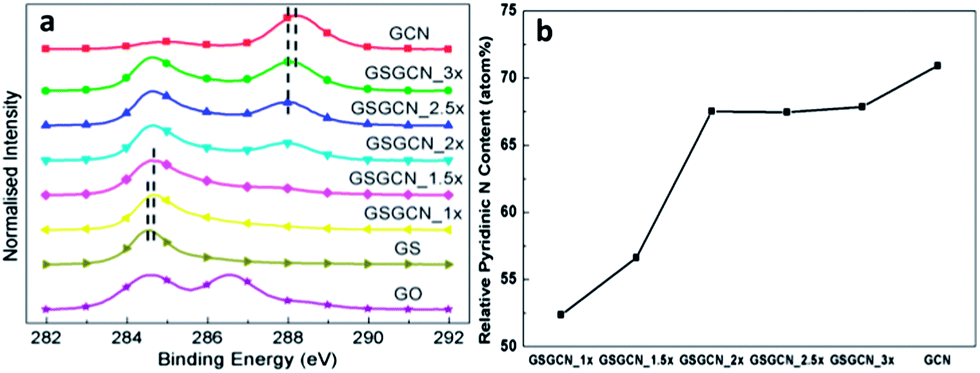 | ||
| Fig. 3 (a) XPS C1s spectra for GO, GS, GCN and GS/GCN composites; (b) the relative pyridinic nitrogen content in GCN and GS/GCN. | ||
The residual mass of the GS/GCN composites after thermogravimetric (TG) analysis (Fig. 4a) could be used to estimate the mixing ratio of GS/GCN in the composites23 since GCN has almost completely decomposed after heating at 800 °C while no significant weight change could be detected for GS. The decrease of the GS content in the composites, from GSGCN_1× to GSGCN_3×, is in agreement with the increasing nitrogen content derived from XPS (Fig. 4b). Note that the intercalation of GCN into the GS layer lowers the GCN decomposition temperature of ca. 30 °C (Fig. 4a), indicating that this intercalated hybrid structure decreases the decomposition temperature of GCN. Hence the lack of GCN pattern in GSGCN_1× and GSGCN_1.5× should be attributed to the partial decomposition of GCN.
 | ||
| Fig. 4 (a) TG patterns for GS, GCN and GS/GCN composites; (b) GS content in GS/GCN composites derived from the TG results and nitrogen content derived from XPS. | ||
The electrocatalytic behaviours of intercalated GS/GCN composites are examined by cyclic voltammetry (CV) and the current density presented here equals the measured current divided by the geometric surface area of the GC electrode. As shown in Fig. 5 that the ORR performances of all the GS/GCN composites with different GCN ratios are superior to that of pure GCN, in terms of both onset/peak reduction potential and current density. More importantly, as the GCN content in the composites increases initially, from GSGCN_1× to GSGCN_2×, the reduction current density increases while the onset/peak reduction potential remains the same; but when the GCN content further increases, from GSGCN_2× to GSGCN_3×, both the reduction current density and potential decrease considerably and gradually show a pattern similar to that of pure GCN. Such phenomena could also be confirmed by linear sweep voltammetry (LSV) measurement (Fig. S3†).
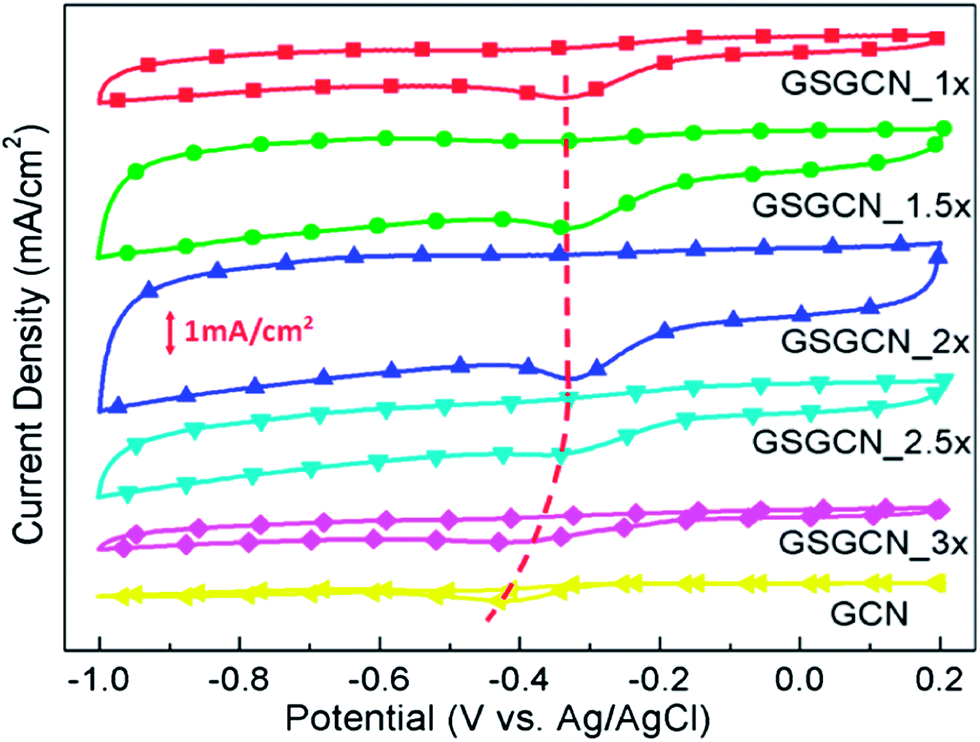 | ||
| Fig. 5 CV of GS/GCN composites and GCN in 0.1 M KOH (scan rate: 100 mV s−1). The red curve highlights the shift of peak reduction potential for GS/GCN composites. | ||
To explain the above observations on the ORR properties of GS/GCN intercalated composites, both structural characteristics and chemical composition need to be considered. Particularly the BET SSA (m2 g−1) and total nitrogen content from XPS (atom%) should be taken into account, as the surface nitrogen heteroatom is the origin of ORR catalytic activity.6a Here we introduce a new parameter called ‘effective-SSA’ (E-SSA), which is defined as the product of SSA and nitrogen content. In this sense, E-SSA could represent the amount of active sites, and E-SSA normalized peak reduction current thus represents the conductivity, assuming no major difference in the mass transport capability for GS/GCN composites as they all possess layered structures, a similar macro-morphology. It is shown in Fig. 6, when the GCN content in the composites is low, the E-SSA normalized current is almost constant, indicating sufficient conductivity supplied by the intercalated GS. As a result, the ORR performances, similar reduction potential but increasing current density from GSGCN_1× to GSGCN_2×, are dependent on the amount of active sites. However when the GCN content is in excess, though the E-SSA of GSGCN_2.5× and GSGCN_3× is similar to that of GSGCN_2×, the E-SSA normalized current decreases significantly, in accordance with the rapid decline in both reduction potential and peak current density. This suggests that the amount of GS in the composites no longer affords to provide enough conductivity and the ORR activity turns into conductivity dependent. Accordingly, GSGCN_2× shows the best ORR performance among all five GS/GCN intercalated composites because it possesses the largest amount of active sites while retains sufficient conductivity. The increase in electrical conductivity, which is led by the intercalation of GS into GCN layers, is one of the key requirements for the improved ORR activities. Such an enhancement can be confirmed by the four-probe conductivity measurement (Table S1†). Typically, the powder conductivity for GCN is under the detection limitation of the instrument, and thus must be smaller than 10−2 S cm−1, similar to the previous report.10 For GS/GCN intercalated composites, the conductivity is about 0.5–4.5 S cm−1, i.e. 3.06 S cm−1 for GSGCN_2×, at least two magnitudes greater than that of pristine GCN.
 | ||
| Fig. 6 E-SSA and E-SSA normalized peak current for GS/GCN composites, calculated from the CV, BET and XPS nitrogen content. | ||
Mass transport is another crucial factor for ORR besides active sites and electrical conductivity.24 Though macropores are capable of fast oxygen exchange during ORR, the relatively low surface area leads to a lack of exposed active sites.13,25 It is thus expected that hierarchical porosity with combined macropores and mesopores may solve the dilemma of both high accessibility and high surface area, as macropores can promise efficient mass transport while mesopores offer sufficient accessible active sites. Here we demonstrate that such a hierarchically porous GS/GCN intercalated composite (hp-GSGCN_2×) can be easily fabricated by the addition of excess silica nanospheres before the polymerization of GCN. The macroporosity of the obtained hp-GSGCN_2× is confirmed by SEM (Fig. 7a) and the mesopores with an average size close to the diameter of individual silica sphere template (ca. 10 nm) are shown in a HRTEM image (Fig. 7b). Note that the interlayer spacing of hp-GSGCN_2× (∼0.33 nm, as shown in the HRTEM image) is close to that of GSGCN_2×, which means that the hp-GSGCN_2× also retains the intercalated structure. The XPS patterns of hp-GSGCN_2× are similar to those of GSGCN_2× with a slight decrease in nitrogen content, but the SSA of hp-GSGCN_2× is ∼50% greater than that of GSGCN_2× (Fig. S2†), making the overall E-SSA roughly 30% higher.
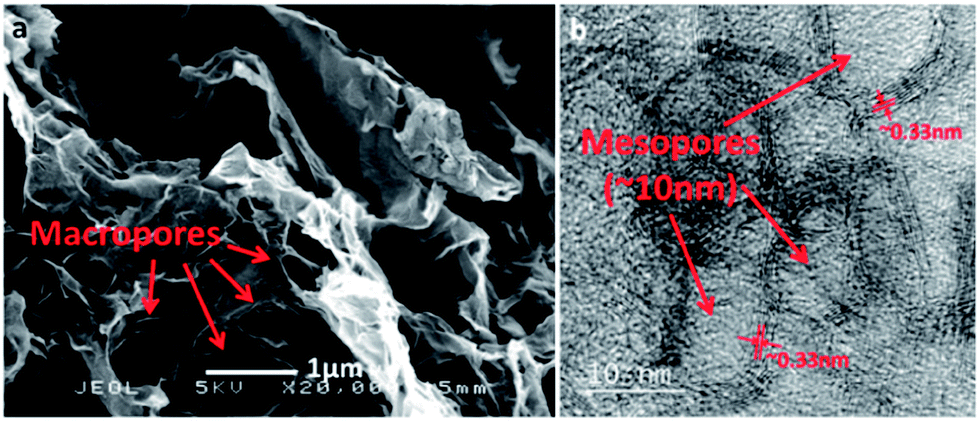 | ||
| Fig. 7 (a) SEM and (b) HRTEM of hp-GSGCN_2×. | ||
The ORR activities of hp-GSGCN_2× are first assessed by CV. As shown in Fig. 8a, the peak current density of hp-GSGCN_2× is almost 1.5 times higher than that of GSGCN_2× and the onset/peak reduction potential also increases by ∼30 mV, which is further confirmed by LSV (Fig. 8b and S4†). Surprisingly, though the onset reduction potential of hp-GSGCN_2× is still lower than that of Pt/C, the current density surpasses Pt/C at a potential of only −0.24 V vs. Ag/AgCl and the half wave potential is only 90 mV lower. The electron transfer numbers for GSGCN_2× and hp-GSGCN_2× are calculated by the Koutecky–Levich equation:3a
| 1/J = 1/JL + 1/JK = 1/0.62nFC0(D0)2/3v−1/6ω1/2 + 1/JK |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 483 sA mol−1), C0 is the bulk saturated concentration of O2 in 0.1 M KOH (1.2 × 10−3 mol l−1), D0 is the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 mol l−1), ν is the kinematic viscosity of the 0.1 M KOH (0.01 m2 s−1), and ω is the angular velocity of the disk (rad s−1). According to the Koutecky–Levich plots (K–L plots, 1/J vs. ω1/2, Fig. 8c), the number of electrons exchanged during ORR can be calculated from the slope, 1/0.62nFC0(D0)2/3v−1/6.3a It is seen from Fig. 8c that the K–L plot of h-GSGCN_2× at −0.4 V vs. Ag/AgCl is parallel to the straight line for the theoretical 4e− pathway, and the electron transfer number is calculated to be 3.98 (Fig. 8d);3 on the other hand, the electron transfer number for GSGCN_2× is only 2.25, close to an indirect 2e− pathway. In addition, the efficient 4e− transfer for hp-GSGCN_2× can also be observed within a broad potential range from −0.3 to −0.6 V vs. Ag/AgCl, as shown in Fig. 8d. Such excellent catalysis selectivity and fast ORR kinetics in a relatively high potential region should be attributed to the synergistic effect of the balanced active sites and electrical conductivity and so as the pre-designed hierarchically porous structure, especially those macropores for facile mass transport. The enhancement in ORR performance derived from the hierarchical porosity can be further confirmed by the significantly reduced Tafel slope in the high potential range of −0.15 to −0.2 V vs. Ag/AgCl (70.6 and 108.9 mV dec−1 for hp-GSGCN_2× and GSGCN_2× respectively, Fig. S5†).13 As far as we know, none of the existing metal-free GCN-based ORR catalysts possesses both the aforementioned excellent catalysis selectivity and fast ORR kinetics in such a relatively high potential region (Table S2†). However, it is also noted that although the catalytic activity of the hp-GSGCN_2× has been dramatically improved, it still cannot compete with the commercial Pt/C, particularly in terms of the onset/half-wave reduction potential. Considering the overall energy efficiency of metal–air batteries highly depends on the over-potential, our continuous effort is to introduce other non-precious metal based catalysts with higher onset reduction potential (i.e. iron or cobalt based) into the current hierarchically porous GS/GCN intercalated composites. It is expected that the strong interaction between the obtained metal/metal oxide and nitrogen-doped carbon, as well as the synergistic effect of two different active sites, can lead to much higher onset/peak reduction potential while retaining the excellent catalysis selectivity and fast kinetics.
483 sA mol−1), C0 is the bulk saturated concentration of O2 in 0.1 M KOH (1.2 × 10−3 mol l−1), D0 is the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 mol l−1), ν is the kinematic viscosity of the 0.1 M KOH (0.01 m2 s−1), and ω is the angular velocity of the disk (rad s−1). According to the Koutecky–Levich plots (K–L plots, 1/J vs. ω1/2, Fig. 8c), the number of electrons exchanged during ORR can be calculated from the slope, 1/0.62nFC0(D0)2/3v−1/6.3a It is seen from Fig. 8c that the K–L plot of h-GSGCN_2× at −0.4 V vs. Ag/AgCl is parallel to the straight line for the theoretical 4e− pathway, and the electron transfer number is calculated to be 3.98 (Fig. 8d);3 on the other hand, the electron transfer number for GSGCN_2× is only 2.25, close to an indirect 2e− pathway. In addition, the efficient 4e− transfer for hp-GSGCN_2× can also be observed within a broad potential range from −0.3 to −0.6 V vs. Ag/AgCl, as shown in Fig. 8d. Such excellent catalysis selectivity and fast ORR kinetics in a relatively high potential region should be attributed to the synergistic effect of the balanced active sites and electrical conductivity and so as the pre-designed hierarchically porous structure, especially those macropores for facile mass transport. The enhancement in ORR performance derived from the hierarchical porosity can be further confirmed by the significantly reduced Tafel slope in the high potential range of −0.15 to −0.2 V vs. Ag/AgCl (70.6 and 108.9 mV dec−1 for hp-GSGCN_2× and GSGCN_2× respectively, Fig. S5†).13 As far as we know, none of the existing metal-free GCN-based ORR catalysts possesses both the aforementioned excellent catalysis selectivity and fast ORR kinetics in such a relatively high potential region (Table S2†). However, it is also noted that although the catalytic activity of the hp-GSGCN_2× has been dramatically improved, it still cannot compete with the commercial Pt/C, particularly in terms of the onset/half-wave reduction potential. Considering the overall energy efficiency of metal–air batteries highly depends on the over-potential, our continuous effort is to introduce other non-precious metal based catalysts with higher onset reduction potential (i.e. iron or cobalt based) into the current hierarchically porous GS/GCN intercalated composites. It is expected that the strong interaction between the obtained metal/metal oxide and nitrogen-doped carbon, as well as the synergistic effect of two different active sites, can lead to much higher onset/peak reduction potential while retaining the excellent catalysis selectivity and fast kinetics.
 | ||
| Fig. 8 (a) CV of GSGCN_2× and hp-GSGCN_2× (scan rate: 100 mV s−1); (b) LSV of GSGCN_2×, hp-GSGCN_2× and Pt/C at 1500 rpm (scan rate: 10 mV s−1); (c) K–L plots of GSGCN_2× and hp-GSGCN_2× at −0.4 V (dashed lines represent the K–L plots for the theoretical 2e− and 4e− pathway); (d) number of electrons transferred for GSGCN_2× and hp-GSGCN_2× at −0.3 to −0.6 V. | ||
Since safety is the first priority for the widespread application of electric vehicles,26 the tolerance against the methanol crossover effect should be of particular importance to the direct methanol fuel cells (DMFCs), and so as the long-term durability. The methanol tolerance is evaluated by the current–time response at −0.3 V vs. Ag/AgCl. Oxygen is purged into the electrolyte from 500 s and 3 M methanol is added after 3500 s. It is shown in Fig. 9a that the ORR of Pt/C switches to a methanol oxidation reaction (MOR) after the addition of 3 M methanol, as indicated by the shift to a positive current density;3b however the ORR activity of hp-GSGCN_2× is almost unaffected. The long-term durability of hp-GSGCN_2× and commercial Pt/C is also assessed through the chronoamperometric response at −0.3 V vs. Ag/AgCl. The ten-hour test only causes a slight activity loss for hp-GSGCN_2×, whereas Pt/C loses nearly 40% of its initial activity. These two measurements confirm that the hp-GSGCN_2× in this work is much more stable in the alkaline electrolyte and against the methanol cross-over effect, compared with the commercial Pt/C.
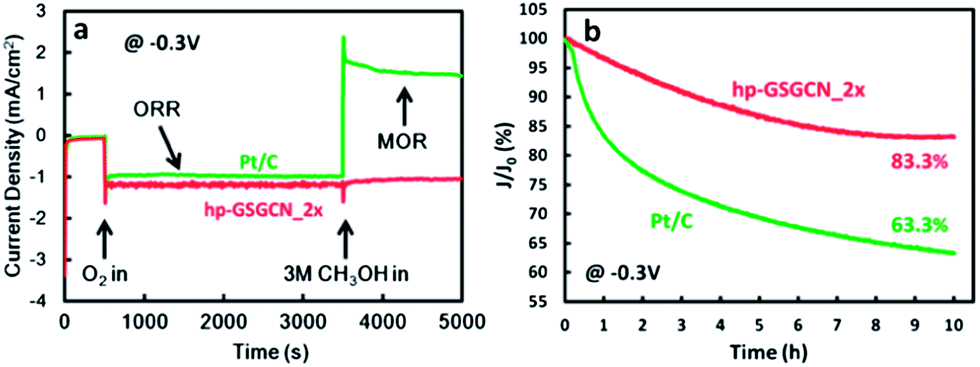 | ||
| Fig. 9 Chronoamperometric response of hp-GSGCN_2× and Pt/C at −0.3 V vs. Ag/AgCl for (a) tolerance against methanol and (b) long-term stability. | ||
Conclusions
In this work, a facile synthesis method of hierarchically porous GS/GCN intercalated composites is presented, and the composites display much better ORR activities than the existing GCN-based catalysts and also improved durability and methanol tolerance compared with commercial Pt/C. Firstly, the electrochemical properties of GS/GCN intercalated composites without hierarchical porosity are optimised, by changing the mixing ratio of GS/GCN to balance the amount of active sites and electrical conductivity. Based on that, a hierarchically macro/mesoporous structure is designed, so as to achieve both efficient mass transport and sufficient exposed active sites. The obtained hp-GSGCN_2× shows a smaller half wave potential difference with commercial Pt/C than any other GCN-based ORR catalysts, so as the highest potential when current density outperforms Pt/C. In addition, it possesses 100% 4e− catalysis pathway selectivity within a broad potential region of −0.3 to −0.6 V vs. Ag/AgCl. The long-term stability and tolerance to the methanol crossover effect are also significantly improved, compared with the commercial Pt/C. In this regard, it is reasonable to anticipate that such a hierarchically porous GS/GCN intercalated composite could be a promising future catalyst for both fuel cells and metal–air batteries.Acknowledgements
This work is supported by the Engineering and Physical Sciences Research Council (EPSRC) grant (Ref: EP/K002252/1).Notes and references
- (a) B. C. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS PubMed; (b) F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172 RSC.
- (a) M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS; (b) X. Yu and S. Ye, J. Power Sources, 2007, 172, 133 CrossRef CAS PubMed; (c) B. Lim, M. J. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. M. Lu, Y. M. Zhu and Y. N. Xia, Science, 2009, 324, 1302 CrossRef CAS PubMed.
- (a) R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565 CrossRef CAS PubMed; (b) W. Yang, T. P. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2010, 133, 206 CrossRef PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Norskov, Nat. Chem., 2009, 1, 552 CrossRef CAS PubMed.
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71 CrossRef CAS PubMed.
- (a) K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed; (b) Y. Zheng, Y. Jiao, M. Jaroniec, Y. Jin and S. Z. Qiao, Small, 2012, 8, 3550 CrossRef CAS PubMed; (c) L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132 CrossRef CAS PubMed; (d) Y. Zhao, L. Yang, S. Chen, X. Wang, Y. Ma, Q. Wu, Y. Jiang, W. Qian and Z. Hu, J. Am. Chem. Soc., 2013, 135, 1201 CrossRef CAS PubMed.
- (a) S. F. Huang, K. Terakura, T. Ozaki, T. Ikeda, M. Boero, M. Oshima, J. I. Ozaki and S. Miyata, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 235410 CrossRef; (b) X. Hu, Y. Wu, H. Li and Z. Zhang, J. Phys. Chem. C, 2010, 114, 9603 CrossRef CAS; (c) L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170 CrossRef CAS.
- (a) S. Yang, X. Feng, X. Wang and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 5339 CrossRef CAS PubMed; (b) S. Chen, J. Bi, Y. Zhao, L. Yang, C. Zhang, Y. Ma, Q. Wu, X. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593 CrossRef CAS PubMed; (c) T. Sharifi, G. Hu, X. Jia and T. Wagberg, ACS Nano, 2012, 6, 8904 CrossRef CAS PubMed; (d) L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936 RSC.
- (a) A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893 RSC; (b) P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763 CrossRef CAS; (c) Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717 RSC.
- Y. Sun, C. Li, Y. Xu, H. Bai, Z. Yao and G. Shi, Chem. Commun., 2010, 46, 4740 RSC.
- Y. Zheng, Y. Jiao, J. Chen, J. Liu, J. Liang, A. Du, W. Zhang, Z. Zhu, S. C. Smith, M. Jaroniec, G. Q. Lu and S. Z. Qiao, J. Am. Chem. Soc., 2011, 133, 20116 CrossRef CAS PubMed.
- S. M. Lyth, Y. Nabae, S. Moriya, S. Kuroki, M. A. Kakimoto, J. I. Ozaki and S. Miyata, J. Phys. Chem. C, 2009, 113, 20148 CAS.
- J. Liang, Y. Zheng, J. Chen, J. Liu, D. Hulicova-Jurcakova, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 3892 CrossRef CAS PubMed.
- S. Pei and H. M. Cheng, Carbon, 2012, 50, 3210 CrossRef CAS PubMed.
- A. Du, S. Sanvito, Z. Li, D. Wang, Y. Jiao, T. Liao, Q. Sun, Y. H. Ng, Z. Zhu, R. Amal and S. C. Smith, J. Am. Chem. Soc., 2012, 134, 4393 CrossRef CAS PubMed.
- K. Kwon, Y. J. Sa, J. Y. Cheon and S. H. Joo, Langmuir, 2012, 28, 991 CrossRef CAS PubMed.
- J. W. Burress, S. Gadipelli, J. Ford, J. M. Simmons, W. Zhou and T. Yildirim, Angew. Chem., Int. Ed., 2010, 49, 8902 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- (a) M. D. Stoller, S. J. Park, Y. W. Zhu, J. H. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498 CrossRef CAS PubMed; (b) Y. Zhu, S. Murali, M. D. Stoller, A. Velamakanni, R. D. Piner and R. S. Ruoff, Carbon, 2010, 48, 2118 CrossRef CAS PubMed; (c) S. R. C. Vivekchand, C. S. Rout, K. S. Subrahmanyam, A. Govindaraj and C. N. R. Rao, J. Chem. Sci., 2008, 120, 9 CrossRef CAS PubMed.
- (a) M. J. Bojdys, J. O. Müller, M. Antonietti and A. Thomas, Chem. – Eur. J., 2008, 14, 8177 CrossRef CAS PubMed; (b) S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397 CrossRef CAS PubMed; (c) F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171 RSC; (d) J. Liu, T. Zhang, Z. Wang, G. Dawsona and W. Chen, J. Mater. Chem., 2011, 21, 14398 RSC.
- Y. Zhang, T. Mori, L. Niu and J. Ye, Energy Environ. Sci., 2011, 4, 4517 CAS.
- (a) X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939 CrossRef CAS PubMed; (b) C. Zhang, L. Fu, N. Liu, M. Liu, Y. Wang and Z. Liu, Adv. Mater., 2011, 23, 1020 CrossRef CAS PubMed; (c) Z. Sheng, L. Shao, J. Chen, W. Bao, F. Wang and X. Xia, ACS Nano, 2011, 5, 4350 CrossRef CAS PubMed; (d) H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781 CrossRef CAS.
- X. Li, J. Chen, X. Wang, J. Sun and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 8074 CrossRef CAS PubMed.
- (a) M. Mirzaeian and P. J. Hall, Electrochim. Acta, 2009, 54, 7444 CrossRef CAS PubMed; (b) G. O. Shitta-Bey, M. Mirzaeian and P. J. Hall, J. Electrochem. Soc., 2012, 159, A315 CrossRef CAS PubMed.
- J. Liang, X. Du, C. Gibson, X. Du and S. Z. Qiao, Adv. Mater., 2013, 25, 6226 CrossRef CAS PubMed.
- (a) J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587 CrossRef CAS; (b) M. Mirzaeian and P. J. Hall, J. Power Sources, 2010, 195, 6817 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ta14158f |
| This journal is © The Royal Society of Chemistry 2014 |