 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct visualisation of carbon dioxide adsorption in gate-opening zeolitic imidazolate framework ZIF-7†
Pu
Zhao
a,
Giulio I.
Lampronti
a,
Gareth O.
Lloyd‡
b,
Emmanuelle
Suard
c and
Simon A. T.
Redfern
*a
aDepartment of Earth Sciences, University of Cambridge, Downing Street, Cambridge, CB2 3EQ, UK. E-mail: satr@cam.ac.uk
bDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
cInstitut Laue-Langevin, BP 156, 6, rue Jules Horowitz, Grenoble, Cedex 9, 38042, France
First published on 26th November 2013
Abstract
The crystal structures of zeolitic imidazolate framework 7 (ZIF-7) under various CO2 pressures were studied by high-resolution neutron powder diffraction. CO2 adsorption in ZIF-7 is visualised and demonstrated to be primarily controlled by the benzimidazolate ligands via a gate-opening mechanism. Our results highlight the importance of pressure on the CO2 adsorption and the related structural framework responses in ZIF-7.
Zeolitic imidazolate frameworks (ZIFs) are important members of the family of metal organic framework (MOF) materials. They can act as host frameworks for CO2 adsorption, with important potential applications in low-temperature CO2 separation and sequestration technology. ZIFs are so-called because of their zeolite-like structures, but instead of aluminosilicate frameworks they are composed of transition metal cations coordinated by imidazolate ligands. There has been considerable interest in ZIFs' structure–property relations and the influences of modifications of its zeolite-like structures. ZIF-7 (Zn(PhIm)2, PhIm = benzimidazolate) is a typical ZIF with sodalite (SOD) framework topology.1,2 Due to the phenyl group in its benzimidazolate ligands, the structure and properties of ZIF-7 are distinct from its more commonly-considered cousin, ZIF-8 (ref. 1 and 3–10) and aluminosilicate sodalites which share similar cubic crystalline topology. ZIF-7 has previously been shown to exhibit gate-opening behaviour during CO2 sorption.11,12 Its framework flexibility was shown to be related to a reversible structural phase transition upon loading and unloading of CO2 guest molecules into the nanoporous host structure.13 Theoretical calculations indicate that the benzimidazolate ligands in ZIF-7 may be the key to its framework flexibility and related unique CO2 sorption behaviour.11,12,14
If we are to develop a better understanding of the CO2 adsorption process in ZIF-7 and the functionality of the benzimidazolate ligands, it is necessary to elucidate the guest molecular configurations and framework response of ZIF-7 during CO2 incorporation, ideally by direct crystal structural study. To this end, we have employed high-resolution neutron powder diffraction to monitor the structural effects associated with CO2 adsorption in ZIF-7 under industrial CO2 pressures. Our results show the dominant role of benzimidazolate ligands in the CO2 adsorption process in ZIF-7. They also reveal the importance of gas loading pressure on the guest–host relationships in the ZIF-7 (CO2) system.
Deuterated ZIF-7 (D-ZIF-7) powder (0.14 g) was prepared based on the procedure described previously, but employing deuterated benzimidazole as reagent.12 The sample was placed into an aluminium can with gas loading system. Neutron powder diffraction measurements were performed under pCO2 = 0, 50, 100, 200 kPa at 300 K using high-resolution neutron powder diffractometer D2B at the Institut Laue-Langevin. Before data collection, in order to remove all possible residing guest molecules, the sample was heated from 300 to 393 K at a rate of 1 K min−1 under vacuum; the temperature was then kept at 393 K for 2 hours before cooling to 300 K (1 K min−1) for neutron diffraction measurements. The crystal structure of guest-free D-ZIF-7 was refined by the Rietveld method15,16 using Topas 4.1,17 using the hydrogenated ZIF-7 structure given by Yaghi et al. as the starting model.1 The determination of the location of CO2 molecules in the D-ZIF-7 (CO2) structure under pCO2 = 50 kPa was carried out using Fourier difference maps generated by GSAS.18,19 Further input from DFT calculation indicated that two CO2 adsorption sites are most reasonable for Rietveld refinement of the internal structure of D-ZIF-7 (CO2) under pCO2 = 50, 100, 200 kPa. In particular, the modelling of D-ZIF-7 (CO2) structure (pCO2 = 100 kPa) was carried out by combined-Rietveld refinement on two neutron diffraction datasets obtained using different incident beam wavelengths λ = 1.5946 and 2.3909 Å. Further experimental details are provided in the ESI.†
Both CO2 adsorption sites reside in the small cavities formed by the benzimidazolate ligands in the six-membered rings of zinc atoms (Fig. 1). They are designated A and B according to their host cavity features. Cavity A has the largest void in ZIF-7 and has previously been proposed as the primary gas adsorption site.11 Its zinc ring plane is perpendicular to the 3-fold rotoinversion axis and the benzimidazolates point towards the axis to form a rhombohedral cage. Cavity B, on the other hand, has lower point symmetry and is open. Comparing it with Cavity A, one notes that the two opposing benzimidazolates in Cavity B point away from the cavity axis and the six-membered ring formed by the zinc atoms is distorted. The Wycoff multiplicities of cavities A and B in ZIF-7 structure are 6 and 18, respectively. There is no significant CO2 adsorption either around zinc four-membered rings or in the topological beta-cage of ZIF-7. The CO2 adsorption sites in ZIF-7 share certain similarities with those in ZIF-8: the dominant gas adsorption sites in ZIF-8 are in a Cavity A type pores formed by methylimidazolates.20–23 In ZIF-7 the benzimidazolate ligand the controlling factor on CO2 adsorption.
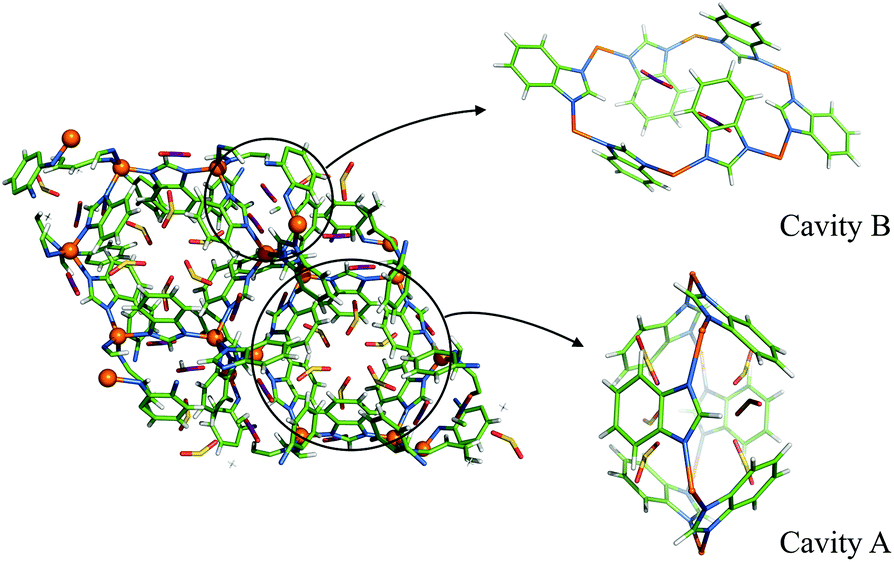 | ||
| Fig. 1 D-ZIF-7 (CO2) structure, pCO2 = 50 kPa. Zn: orange, C: cyan, N: blue, H/D: silver. CO2: C in Cavity A yellow, in Cavity B purple; O, red. | ||
CO2 adsorption site preferences revealed by our experimental data are in excellent agreement with recent simulation results from Morris et al.14 Simulated binding energies indicate Cavity B is more energetically preferable than Cavity A for CO2 adsorption. Experimental CO2 occupancies (Table 1) confirm this adsorption preference. This is due to the geometry differences between the two cavities. Since Cavity B is relatively open, it accommodates guest incorporation and transportation more readily. While one might anticipate small differences between the behaviour of hydrogenous ZIF-7 and deuterated ZIF-7, due to mass differences, the correspondence of our results with those of Morris et al.14 indicates that such differences are not significant within the framework of this study.
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) )
)
| pCO2 [kPa] | 0 | 50 | 100 | 200 | |
| CO2 occupancy | A | 0 | 0.35 | 0.26 | 0.25 |
| B | 0 | 0.53 | 0.68 | 0.68 | |
| Calculated total CO2 uptake [mmol g−1] | 0 | 2.60 | 2.76 | 2.73 | |
| a = b [Å] | 22.94 | 23.00 | 22.88 | 22.95 | |
| c [Å] | 15.75 | 15.76 | 15.66 | 15.71 | |
| V [Å3] | 7178 | 7224 | 7098 | 7165 | |
In guest-free D-ZIF-7, the diameter of the largest guest-accessible window in Cavity A is ∼3 Å. Considering the Lennard-Jones collision diameter of CO2 (4.05 Å),24 it is very difficult for CO2 to enter into Cavity A via this window if the structure is static. The rotational freedom of the benzimidazolate ligands plays an important role in accommodating guest molecule transport and dynamics.11 There are a number of examples from the literature showing that the static crystal structure and dynamic radii of guest molecules do not always match.25−29 Thus, it is reasonable that Cavity A has some adsorption capacity. Cavity B, on the other hand, has much larger guest accessibility than Cavity A. The benzimidazolates with phenyl group pointing away from the cavity axis open up to form a large channel (diameter ∼ 5–6 Å) for CO2 adsorption. The topological beta-cage of ZIF-7 plays little part in CO2 incorporation from both theoretical and experimental aspects; we attribute this to the strong steric effect from phenyl rings. It is worth noting that along with the increase of pressure, CO2 molecules tend to aggregate in Cavity B while leaving Cavity A relatively empty. This phenomenon is of interest for further investigation on the CO2 transportation in ZIF-7; the direct CO2 transport route from Cavity A to B is via the beta-cage, however, CO2 transport barriers from Cavity A and B into beta-cage are very high, therefore the actual CO2 transport route is uncertain and expected to be influenced by the benzimidazolate ligands.
A CO2-induced gate-opening process in D-ZIF-7 is indicated by our experimental results. Compared with the previous experimental CO2 adsorption isotherms,13,14 the calculated total CO2 uptake in our D-ZIF-7 (CO2) structures (Table 1) suggests that at pCO2 = 50 kPa the D-ZIF-7 sample is nearly fully saturated with CO2. Examining our D-ZIF-7 structural results we notice that the unit cell expands when CO2 pressure increases from 0 to 50 kPa. The benzimidazolate ligands of both Cavity A and B rotate to open up those cavities for CO2 adsorption (Fig. 2). When the CO2 pressure increases from 50 to 100 kPa, the total amount of CO2 adsorbed increases. At this stage, although CO2 begins to flow into Cavity B, the benzimidazolates at Cavity A continue to rotate to increase the accommodation space for more CO2 molecules to enter. Meanwhile, at Cavity B, the zinc six-membered ring becomes more distorted due to the movement of the ligands for gate-opening. It seems that the ligand movement in Cavity B induces an electrostatic field change in the cavity itself which helps to increase the affinity of CO2. The distortion of the zinc six-membered ring may be responsible for the subtle shrinkage in the unit cell dimensions.
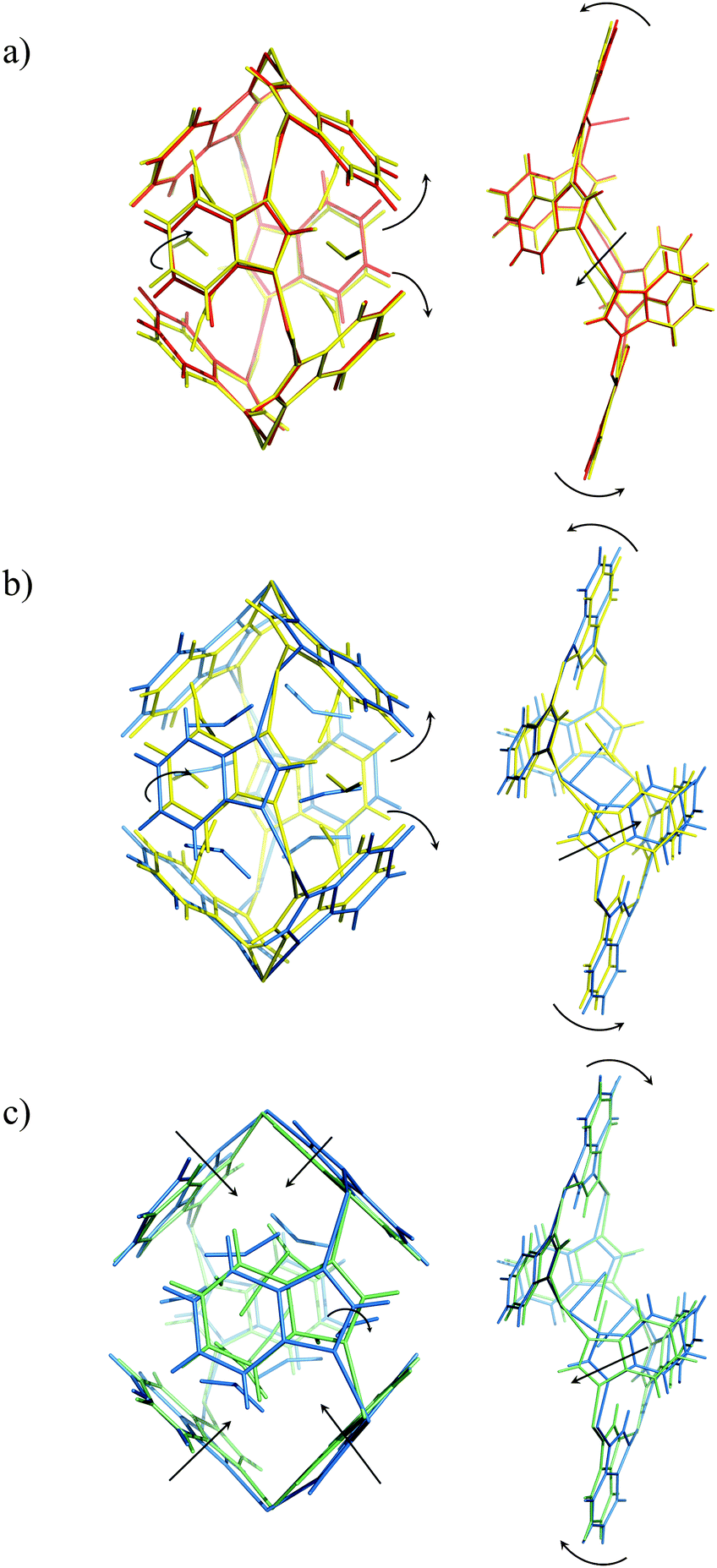 | ||
| Fig. 2 Dynamic structural behaviours of Cavity A (left) and B (right) at pCO2 = (a) 0 (red) to 50 (yellow) kPa; (b) 50 to 100 (blue) kPa; 100 to 200 (cyan) kPa. | ||
At higher external CO2 pressure, from pCO2 = 100 to 200 kPa, the influence of external pressure on the CO2 adsorption and structural behaviour of ZIF-7 becomes crucial. Fig. 2c clearly shows that Cavity A is squeezed by the external pressure instead of continuing to open up for internal CO2 adsorption in this pressure regime. At Cavity B, the zinc six-membered ring becomes less distorted and ligands move in the opposite sense compared to that seen upon pressure change from 50 to 100 kPa. This ligand movement induces anomalous unit cell expansion. For CO2 pressures from 100 to 200 kPa the total CO2 uptake and CO2 occupancy hardly increase any further. This suggests that the extrusion of Cavity A by increasing pressure inhibits further CO2 adsorption and transportation to Cavity B. The effect of external vs. internal pressure also influences the CO2 locations as a function of pCO2. This is illustrated in Fig. 3, where both CO2 adsorption sites are seen to move into the centre of Cavity A and B upon increasing pCO2.
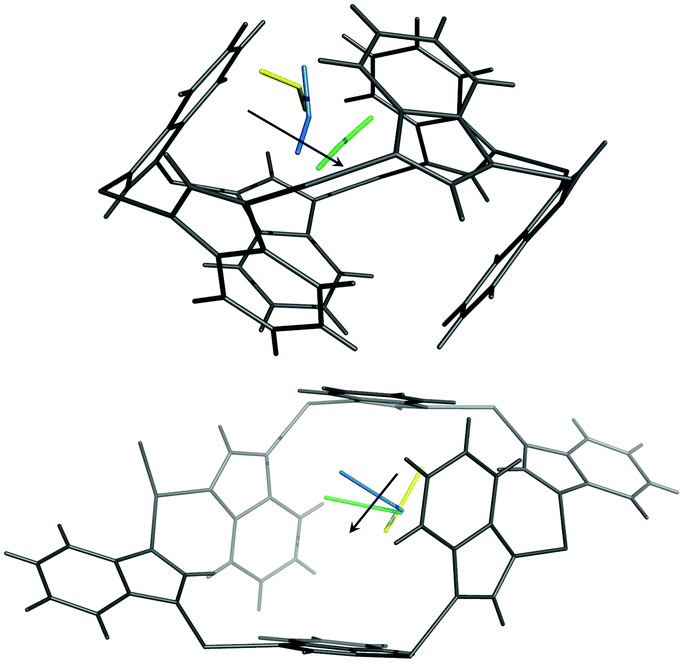 | ||
| Fig. 3 The CO2 adsorption site at Cavity A (upper figure) and B (lower figure) at pCO2 = 50 (yellow), 100 (blue), 200 (cyan) kPa. | ||
Conclusions
In summary, using high-resolution neutron powder diffraction, we have determined the CO2 adsorption geometry and site preference in ZIF-7. The results illustrate the importance of the benzimidazolate ligands in controlling the CO2 affinity and structural flexibility of ZIF-7. They also reveal the influence of pressure in the CO2 adsorption process in ZIF-7. This is of most importance in understanding the potential of these materials in gas storage and separation in industrial processes. We find that the gas adsorption properties of ZIF-7 depend upon a balance between the internal pressure of the guest molecule and the external gas pressure imposed at the higher range of pressures applied. We will explore the role of the latter in future studies of the structural response of ZIF-7 under different imposed pressures with a variety of guest molecules.Acknowledgements
This work was supported by the Cambridge Commonwealth, European and International Trust; China Scholarship Council; University of Cambridge; the Herchel Smith Fund (Cambridge); Heriot-Watt University; UK Science and Technology Facilities Council; Institut Laue-Langevin. We thank Mr Rajkrishna Dutta for his DFT calculation.Notes and references
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- X. Huang, J. Zhang and X. Chen, Chin. Sci. Bull., 2003, 48, 1531–1534 CAS.
- H. L. Jiang, B. Liu, T. Akita, M. Haruta, H. Sakurai and Q. Xu, J. Am. Chem. Soc., 2009, 131, 11302–11303 CrossRef CAS PubMed.
- S. A. Moggach, T. D. Bennett and A. K. Cheetham, Angew. Chem., Int. Ed., 2009, 48, 7087–7089 CrossRef CAS PubMed.
- G. Lu and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 7832–7833 CrossRef CAS PubMed.
- K. W. Chapman, D. F. Sava, G. J. Halder, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 18583–18585 CrossRef CAS PubMed.
- D. Peralta, G. Chaplais, A. Simon-Masseron, K. Barthelet, C. Chizallet, A. A. Quoineaud and G. D. Pirngruber, J. Am. Chem. Soc., 2012, 134, 8115–8126 CrossRef CAS PubMed.
- W. Chaikittisilp, M. Hu, H. Wang, H. S. Huang, T. Fujita, K. C. W. Wu, L. C. Chen, Y. Yamauchi and K. Ariga, Chem. Commun., 2012, 48, 7259–7261 RSC.
- S. Cao, T. D. Bennett, D. A. Keen, A. L. Goodwin and A. K. Cheetham, Chem. Commun., 2012, 48, 7805–7807 RSC.
- L. Zhang, Z. Hu and J. Jiang, J. Am. Chem. Soc., 2013, 135, 3722–3728 CrossRef CAS PubMed.
- J. van den Bergh, C. Gücüyener, E. A. Pidko, E. J. Hensen, J. Gascon and F. Kapteijn, Chem.–Eur. J., 2011, 17, 8832–8840 CrossRef CAS PubMed.
- C. Gücüyener, J. van den Bergh, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2010, 132, 17704–17706 CrossRef PubMed.
- S. Aguado, G. Bergeret, M. P. Titus, V. Moizan, C. Nieto-Draghi, N. Bats and D. Farrusseng, New J. Chem., 2011, 35, 546–550 RSC.
- W. Morris, N. He, K. G. Ray, P. Klonowski, H. Furukawa, I. N. Daniels, Y. A. Houndonougbo, M. Asta, O. M. Yaghi and B. B. Laird, J. Phys. Chem. C, 2012, 116, 24084–24090 CAS.
- L. B. McCusker, R. B. Von Dreele, D. E. Cox, D. Louër and P. Scardi, J. Appl. Crystallogr., 1999, 32, 36–50 CrossRef CAS.
- R. A. Young, The Rietveld Method, Oxford University Press, 1995 Search PubMed.
- A. A. Coelho, Coelho Software, Brisbane, 2007 Search PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, in Los Alamos National Laboratory Report LAUR, 1994, pp. 86–748 Search PubMed.
- J. Pérez-Pellitero, H. Amrouche, F. R. Siperstein, G. Pirngruber, C. Nieto-Draghi, G. Chaplais, A. Simon-Masseron, D. Bazer-Bachi, D. Peralta and N. Bats, Chem.–Eur. J., 2010, 16, 1560–1571 CrossRef PubMed.
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2007, 129, 5314–5315 CrossRef CAS PubMed.
- H. Wu, W. Zhou and T. Yildirim, J. Phys. Chem. C, 2009, 113, 3029–3035 CAS.
- B. Assfour, S. Leoni and G. Seifert, J. Phys. Chem. C, 2010, 114, 13381–13384 CAS.
- S. Matteucci, Y. Yampolskii, B. D. Freeman and I. Pinnau, in Materials Science of Membranes for Gas and Vapor Separation, John Wiley & Sons, Ltd, 2006, pp. 1–47 Search PubMed.
- L. Dobrzańska, G. O. Lloyd, H. G. Raubenheimer and L. J. Barbour, J. Am. Chem. Soc., 2006, 128, 698–699 CrossRef PubMed.
- P. K. Thallapally, G. O. Lloyd, J. L. Atwood and L. J. Barbour, Angew. Chem., Int. Ed., 2005, 44, 3848–3851 CrossRef CAS PubMed.
- T. Jacobs, G. O. Lloyd, J. A. Gertenbach, K. K. Müller-Nedebock, C. Esterhuysen and L. J. Barbour, Angew. Chem., Int. Ed., 2012, 51, 4913–4916 CrossRef CAS PubMed.
- J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000–1002 CrossRef CAS PubMed.
- X. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012–1015 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of D-ZIF-7 sample, neutron powder diffraction details, structure refinement methods and illustrations of D-ZIF-7 (CO2) structures. CCDC 973356–973359. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ta13981f |
| ‡ Current address: Institute of Chemical Sciences, School of Engineering and Physical Sciences, William Perkin Building, Heriot-Watt University, Edinburgh, EH14 4AS, UK. |
| This journal is © The Royal Society of Chemistry 2014 |