Plasma synthesis of carbon nanotube-gold nanohybrids: efficient catalysts for green oxidation of silanes in water†
Ting
Liu‡
,
Fan
Yang‡
,
Yongfeng
Li
*,
Liang
Ren
,
Liqiang
Zhang
,
Kai
Xu
,
Xian
Wang
,
Chunming
Xu
and
Jinsen
Gao
State Key Laboratory of Heavy Oil Processing, China University of Petroleum, Changping 102249, Beijing, China. E-mail: yfli@cup.edu.cn; Fax: +86-010-89739028; Tel: +86-010-89739028
First published on 25th October 2013
Abstract
We report the green synthesis of silanols from hydrosilanes in high yields by using oleylamine (OA) stabilized gold nanoparticles (AuNPs) supported on oxidized multi-walled carbon nanotubes (o-CNTs) as catalysts in H2O. The Au catalyst can be easily synthesized by a one-pot gas–liquid interfacial plasma method, and the catalyst exhibited much more remarkable catalytic activity in the oxidation of various organosilanes by using water as the solvent compared with other organic solvents (for example THF, ethyl acetate, and acetone), which is very important for organic synthesis from both the standpoint of practical reasons and an economic perspective. The Au catalyst can be readily recovered and reused without any loss of catalytic activity. In addition, our findings indicate that o-CNTs and OA are the key components of the catalyst in which the o-CNT support makes the hybrid materials hydrophilic, and the OA stabilizer makes the hybrid materials lipophilic, resulting in the high activity of the catalyst in H2O.
Introduction
Catalysis using gold nanoparticles (AuNPs) has attracted increasing interest due to their potentially green and sustainable catalytic properties.1 It has been reported that AuNPs supported on various metal oxide surfaces, such as Au/TiO2, Au/Al2O3, Au/Fe2O3, Au/SiO2 and Au/SnO2 with controllable shape, exhibit unprecedented catalytic activities for transformation of organic substrates.1,2 Also, carbon nanotubes (CNTs) have been normally used as an ideal support for the dispersion and stabilization of AuNPs, due to their large chemically active surface, unique physical properties, inherent size, hollow geometry and stability at high temperatures.3 In order to control the size and morphology of AuNPs, various kinds of specific surfactant or ligand molecules such as sulfur, phosphine, phosphine oxide, amine and carboxylate ligands are used to prevent the nanoparticle aggregation.4 Many approaches have been employed in synthesizing AuNPs on supports, including thermal, photolysis, electroless deposition, electrodeposition, chemical decoration, π–π stacking, electrostatic interactions, sonochemical, microwave assisted, plasma, adsorption, deposition–precipitation, wet impregnation, or by other means.2,5–7 On the other hand, the gas–liquid interfacial plasma (GLIP) method has recently attracted much attention for the formation of AuNPs owing to its unique properties such as ultra-high density, high reactivity, high process rate, preparation of nanomaterials in large scale, avoiding use of toxic stabilizers and reducing agents, the continuous synthesis, reaction at room temperature, and no need to stir during the nanoparticle formation process.8Although some homogeneous catalytic systems are known for the selective oxidation of silanes to silanols,9 the development of a highly active and selective heterogeneous catalyst for this reaction is still highly desirable from the point of view of organic synthesis and industrial interest. Heterogeneous catalysis based on Au metal has been studied more extensively to oxidize silanes to silanols with water compared with Pt,10 Ru,11 Ag,12 and Pd13 metals due to the higher activity and perfect selectivity. For example, Kaneda et al. have reported that hydroxyapatite supported AuNPs were an effective catalyst for this transformation.14 In addition, Asao et al. reported that nanoporous gold was also a highly active and selective catalyst.15 Recently, the elegant examples of AuNP heterogeneous catalysts for silane oxidation including layer-by-layer assembly of AuNPs on a carbon nanotube were reported by Doris et al., which afforded the turnover frequency (TOF) as high as 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 in THF.16 For silica-supported AuNPs reported by Zhang and co-workers, the TOF of the Au/SiO2 reaches 59400 h−1 calculated based on the surface gold atoms,17 and the aluminium oxyhydroxide supported AuNP catalysts for silane oxidation were reported by Park et al.13a,18 However, the development of a green, selective, and efficient catalyst is still required, which will enlarge its practical applications.
000 h−1 in THF.16 For silica-supported AuNPs reported by Zhang and co-workers, the TOF of the Au/SiO2 reaches 59400 h−1 calculated based on the surface gold atoms,17 and the aluminium oxyhydroxide supported AuNP catalysts for silane oxidation were reported by Park et al.13a,18 However, the development of a green, selective, and efficient catalyst is still required, which will enlarge its practical applications.
Herein, we report a facile GLIP one-pot synthesis of oleylamine stabilized AuNPs supported on oxidized multi-walled carbon nanotubes (o-CNTs) as highly efficient catalysts for the selective oxidation of silanes in water, as seen in Fig. 1. Usually, the presence of ligands is detrimental to catalytic activity because the ligands coat the active surface of the AuNPs.5 In this work, however, we report for the first time that the oleylamine (OA) coated AuNPs supported on oxidized CNTs are extremely active for the selective oxidation of a variety of silanes in water. The presence of OA benefits the formation of AuNPs with small and narrow particle size distribution, and affects the carboxyl group formed ammonium salt, leading to a more hydrophilic hybrid. The long chain of OA surrounding the AuNPs also makes silanes easily connect with the AuNPs in water. The above characters make the hybrid material catalyze the silanes oxidation reaction in water with high reactivity, and chemical reactions using water as solvent is expected to receive more attention in organic synthesis from both a practical and an economic standpoint.
 | ||
| Fig. 1 Selective oxidation of silanes to silanols in water by an AuNP decorated o-CNT catalyst. | ||
Experimental
Materials
All chemicals were used as received without further purification: hydrogen tetrachloroaurate(III) tetrahydrate, 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM]BF4), phenyldimethylsilane, and triisopropylsilane (Aladdin industrial corporation). Oleylamine and polyvinylpyrrolidone-K30 (PVP-K30) (Tokyo Chemical Co., Ltd. >40%). Triethylsilane, triphenylsilane, diphenylsilane, chloroform-d, and 1-dodecanethiol (J&K Scientific Co., Ltd.). Ethanol, methylene chloride (CH2Cl2), acetone, acetic ether, tetrahydrofuran, toluene, and N,N-dimethylformamide (DMF) (Beijing chemical works).Fabrication of AuNPs decorated o-CNTs
The CNTs were prepared by electrically arcing graphite rods in argon (99.99%) in a stainless steel chamber with an inner diameter of 165 mm and a height of 410 mm. The anode was a graphite rod (6 × 6 mm2, 100–200 mm in length) and the cathode was a high-purity graphite electrode (20 mm in diameter, 38 mm in length). The argon (Ar) gas functioned as buffer gas and its pressure was varied in the range of 0.04–0.05 MPa in the experiment. The voltage and current for arcing were controlled at 20 V and 70–90 A, respectively. After the arc discharge was finished, the soft material in the inner core of the deposited rod was taken out and examined using a transmission electron microscope (TEM). The synthesized CNTs (1 g) were pre-treated by sonication in 14 M HNO3 for 1 h and then refluxed for 12 h in a mixture of HNO3 (50 mL, 14 M) and H2SO4 (98%, 50 mL). The mixture was then diluted with ice water, the suspension was centrifuged at 4000 rpm for 10 min and the resulting solution was discarded, washed with distilled water until the pH value reached 7, and washed with ethanol. The o-CNTs were dried at 120 °C for 12 h in the oven. The decoration of AuNPs on the o-CNT catalyst was carried out by a GLIP method with HAuCl4·4H2O, o-CNTs and OA at room temperature for 15 min. The glow plasma was generated between the top flat stainless steel (SUS) and the bottom ionic liquid electrode by using a DC power source (KIKUSUI PMC 500-0.1A). Ar gas was introduced and used as the plasma-forming gas, the chamber was a stainless steel with an inner diameter of 70 mm and four glass windows, and the gap between electrodes is 4 mm. A direct current (DC) power source negative bias prepared by VDC = 220–250 V is applied to a stainless steel electrode in the gas phase for the generation of an Ar plasma, where the discharge current I is fixed to 0.02 A and the Ar gas is introduced up to a pressure of 290 Pa. 50 mg o-CNTs were added to the reactor, HAuCl4·4H2O were dispersed in 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM]BF4) and OA, then the Au solution was added to the reactor, and the Au solution and o-CNT mixture was left for 10 min. For the formation of AuNPs, electrons were irradiated toward the ionic liquid, and after the electron irradiation for t = 15 min, the mixture was sonicated in ethanol to remove the excess impurities and extracted from the ionic liquid using a centrifuge. Several kinds of AuNP decorated o-CNT hybrid materials with different Au weight ratios (4.8%, 7.7%, 13.3%, 15.2%) were prepared, and the respective materials were called Au-n (n = 1, 2, 3, 4).Au-2 catalyzed oxidation of organosilanes in water
The organosilanes in water solution (0.25 M, 2 mL) and A-2 (0.1 mol%, 1.3 mg) were added to PhMe2SiH (78 μL, 0.5 mmol) at room temperature. The reaction mixture was stirred at room temperature for 40 min and monitored by thin layer chromatography (TLC) analysis. The mixture was filtered through a polyvinylidene fluoride (PVDF) membrane with 0.2 μm pore size in order to isolate the catalysts which were washed with water (10 mL) and acetone (10 mL), and dried at 110 °C. The filtrate was extracted with diethylether three times. The organic extracts were dried with anhydrous magnesium sulfate, filtered, and evaporated to dryness. After concentration, the residue was purified by silica gel chromatography to afford PhMe2SiOH (75.6 mg, 99%) as yellowish oil.Results and discussion
Optical emission spectrometry (OES) was used to monitor the chemical species generated in the gas–liquid interfacial plasma region during the catalyst formation process, as shown in Fig. S2.† In the OES spectrum, the OH (308 nm), NH (336 nm), CN (365 nm), CH (430–440 nm) and Ar (700–850 nm) peaks are observed, giving evidence that the reduction activities are caused by the high-energy ion irradiation.19 All kinds of Au-n are examined by TEM as shown in Fig. S3,† the distribution histograms of the diameter are summarized in Fig. S4,† and the represented TEM image of Au-2 is shown in Fig. 2a. The results demonstrate that all synthesized AuNPs decorated on o-CNTs exhibit uniform morphologies and the particle diameters of Au-1, Au-2, Au-3 and Au-4 are 1.0, 1.1, 1.2, and 1.5 nm, respectively, as summarized in Table S1.† It is found that the particle size of AuNPs becomes larger with increasing Au loading ratios. In order to check the crystallinity of AuNPs, a relatively large Au nanoparticle (Au-4) was examined by high-resolution TEM (HRTEM), as seen in Fig. 2b where the interfinger distance of 0.234 nm and 0.34 nm was found, corresponding to the (111) lattice plane of the face-centred-cubic (fcc) gold and shell separation of CNTs, respectively. In addition, XRD spectra of o-CNTs and Au-2 are indicated in Fig. 2c, showing the characteristic diffraction peaks at 25.9°, 42.8°, 54.3° and 77.7°, corresponding to (002), (100), (004), and (110) reflections of graphite, respectively. The peaks at 38.1°, 44.4°, 64.5° and 77.6° correspond to (111), (200), (220), and (311) reflections of crystalline Au (0), respectively. The Au (111) peak gives a full-width at half-maximum of 2.1° at a 2θ value of 38.1°, and the average crystallite size was calculated to be 3.9 nm according to the Scherrer equation, which is slightly larger than the average size estimated from the TEM images. Fig. 2d shows the selected area electron diffraction (SAED) image for the Au-2, and the diffraction rings of lattice planes are in agreement with XRD results. | ||
| Fig. 2 A TEM image of Au-2 (a), a HRTEM image of Au-4 (b), and XRD spectra (c) and a SAED image (d) of Au-2. | ||
To gain more insight into the origin of the catalytic properties, the chemical element states of the hybrid material were measured by EDX (Fig. S5†) and XPS (Fig. S6†). Both results indicate that the Au-2 catalyst is composed of Au, C, N, and O. Two peaks in XPS for Au 4f7/2 and Au 4f5/2 at 83.9 and 87.7 eV are split into two types of Au electronic states (Au0 and Au+), as shown in Fig. 3a, and the C1s XPS is split into four functional groups, including a C sp2 bond at 284.3 eV, a C–O bond at 284.83 eV, a C–N bond at 285.9 eV, and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond at 287.7 eV, as indicated in Fig. 3b. Also, the N1s XPS can be divided into three components, an N–H bond at 399.2 eV, a C–N bond at 400.1 eV, and a C–N+ bond at 401.5 eV (Fig. 3c). The O1s XPS can be divided into two components, a CO bond at 531.2 eV and a C–O bond at 532.39 eV (Fig. 3d). In addition, the element distribution is also analyzed by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), and the elemental mappings of C, N, O and AuNPs are shown in Fig. 4. High loading of uniformly dispersed O, N, and AuNPs on the o-CNTs was observed, which suggests that the hybrid materials contain Au active species, a hydrophobic alkyl chain, and a hydrophilic carboxylic acid part. The synergistic action of these parts may play an important role for highly active catalytic species.
O bond at 287.7 eV, as indicated in Fig. 3b. Also, the N1s XPS can be divided into three components, an N–H bond at 399.2 eV, a C–N bond at 400.1 eV, and a C–N+ bond at 401.5 eV (Fig. 3c). The O1s XPS can be divided into two components, a CO bond at 531.2 eV and a C–O bond at 532.39 eV (Fig. 3d). In addition, the element distribution is also analyzed by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), and the elemental mappings of C, N, O and AuNPs are shown in Fig. 4. High loading of uniformly dispersed O, N, and AuNPs on the o-CNTs was observed, which suggests that the hybrid materials contain Au active species, a hydrophobic alkyl chain, and a hydrophilic carboxylic acid part. The synergistic action of these parts may play an important role for highly active catalytic species.
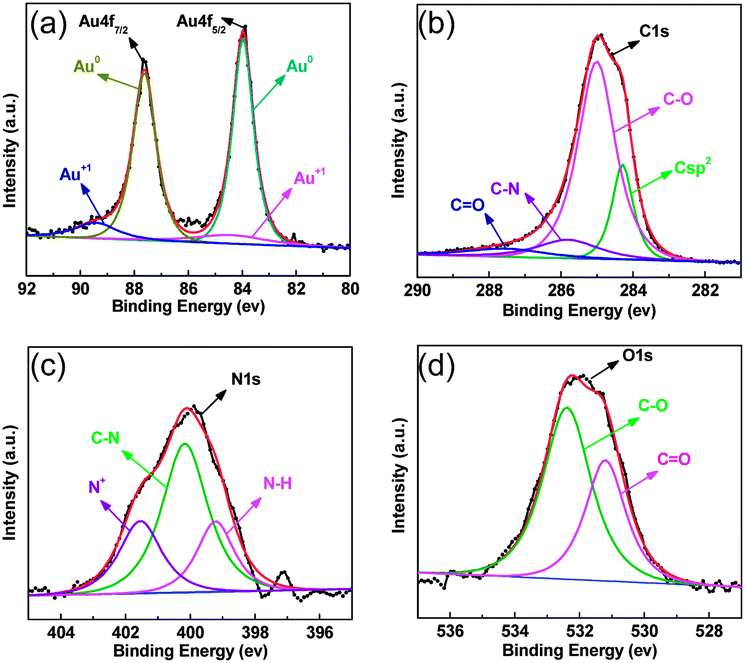 | ||
| Fig. 3 The high-resolution XPS spectra of Au-2, Au4f (a), C1s (b), N1s (c), and O1s (d). | ||
 | ||
| Fig. 4 HAADF-STEM images and elemental mappings of AuNPs decorated on o-CNTs. | ||
In the preliminary experiments, we performed the oxidation of dimethylphenylsilane 1a with water in acetone by using AuCl3 as a catalyst, the reaction was proceeded smoothly, and the product 2a:2a′ was 25:75 in 99% yield (Table 1, entry 1). During the reaction, it was found that the solid Au was produced on the surface of the vial, demonstrating that the Au3+ could be easily reduced to Au0 in the reaction system. After that, we tried to add some common ligands for the purpose of stabilizing the formed AuNPs as catalysts in order to further increase the reaction selectivity (Table 1, entries 1–4). We found that the oleylamine (OA) was a more efficient ligand to limit the formation of disiloxane 2a′. In our case, using the Au-2 catalyst in acetone leads to producing silanol 2a in 99% yield in 5 hours (entry 5). In the presence of various organic solvents, including THF, EtOAc and DMF afforded the corresponding silanols 2a in 99% yield in 55 min, 70 min and 85 min, respectively (entries 6–8). When using toluene as solvent, the reaction cannot proceed (entry 9). In comparison, the product 2a was surprisingly obtained in 99% yield as a single product in only 7 min in H2O. Usually, heterogeneous catalyzed oxidation of silanes to silanols was more effective by using the organic solvent compared with H2O, except for using H2O as solvent to suppress the disiloxane 2a′. Further experiments performed by different Au catalysts Au-1, Au-2, Au-3, and Au-4 using H2O as solvent show that the amount of o-CNTs exhibited a significant effect on the value of TOF (entries 11–14), for example, catalysts Au-1 and Au-2 were active, giving 2a in 99% yield in 40 min, whereas catalysts Au-3 and Au-4 gave 2a in 99% yield in 105 min and 150 min, respectively. It is worth mentioning that the catalyst Au-2 shows the highest activity in water reported to date. The control experiment with the presence of only o-CNTs as catalysts has been performed, and it is found that there is no reaction proceeded (entry 15). This finding indicates that the Au active species are necessary for the current silane oxidation reaction.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | t [min] | Yield (%) (2a:2a′)b |
| a Reaction conditions: dimethylphenylsilane (0.5 mmol), H2O (50 μL), and Au (1 mol%) in 1 mL of solvent at room temperature. b Isolated yield. c The OA, PVP, and C12H25SH were 20 mol%. d Au (0.1 mol%). e 13 mg of o-CNTs was used. | ||||
| 1 | AuCl3 | Acetone | 300 | 99 (25:75) |
| 2c | AuCl3 + OA | Acetone | 300 | 99 (89:11) |
| 3c | AuCl3 + PVP | Acetone | 300 | 99 (87:13) |
| 4c | AuCl3 + C12H25SH | Acetone | 300 | 99 (0:100) |
| 5 | Au-2 | Acetone | 300 | 99 (100:0) |
| 6 | Au-2 | THF | 55 | 99 (100:0) |
| 7 | Au-2 | EtOAc | 70 | 99 (100:0) |
| 8 | Au-2 | DMF | 85 | 99 (100:0) |
| 9 | Au-2 | Tol | 720 | Trace |
| 10 | Au-2 | H2O | 7 | 99 (100:0) |
| 11d | Au-2 | H2O | 40 | 99 (100:0) |
| 12d | Au-1 | H2O | 40 | 99 (100:0) |
| 13d | Au-3 | H2O | 105 | 98 (100:0) |
| 14d | Au-4 | H2O | 150 | 97 (100:0) |
| 15e | o-CNTs | Acetone | 300 | 0 |

Moreover, we have compared the Au-2 with other catalysts reported in the literature, as summarized in Table 2. It is obvious that the Au-2 catalyst prepared in this work is much more active than Au/AlO(OH),13a,18 nanoporous Au,15 and AuHAP14 under comparable reaction conditions (entries 1–4). Even compared with AuCNT catalyst reported in literature,16 the Au-2 catalyst still has comparable activity (entry 5). The oxidation of 1a was catalyzed with 0.001 mol% of Au-2 in H2O to reach a striking turnover number (TON) of 61000 and a TOF of 10167 h−1 (entry 6), better than the AuCNT catalyst. However, the preparation of AuCNTs involved a multi-step layer-by-layer assembly process, which will limit their practical applications. In addition, the oxidation of 1a was catalyzed with 0.4 mol% of the Au/SiO2 catalyst to reach a TON of 248 and a TOF of 14850 h−1 (entry 7),13a however, on using an extremely low Au/substrate ratio or H2O as solvent, the Au/SiO2 catalyst cannot perform well.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | t (min) | Yieldb (%) |
| a Reaction conditions: dimethylphenylsilane (0.5 mmol) and Au-2 (0.1 mol%) in 1 mL of H2O at room temperature. b Isolated yield. c Reaction temperature was 80 °C. | ||||
| 1a | Au-2 (0.1) | H2O | 40 | 99 |
| 2 | Au/AlO(OH) (0.1) | EtOAc | 120 | 98 |
| 3c | AuNPore (1) | Acetone | 60 | 100 |
| 4 | AuHAP (0.83) | H2O | 180 | 99 |
| 5 | AuCNT (0.1) | THF | 45 | 98 |
| 6 | Au-2 (0.001) | H2O | 720 | 61 |
| 7 | Au/SiO2 (0.4) | THF | 1 | 99 |

Reusability is the most important feature of a heterogeneous catalyst, which is superior to a homogenous one. First, to confirm that the reaction was indeed catalyzed by solid Au-2 rather than by homogenous gold species, we have carried out the following leaching experiments (eqn (1)). After the catalytic oxidation of 1a was carried out for 10 min under standard conditions, the Au catalyst was removed from the vessel by centrifugation with 2a produced in 81% yield at this time. When no further reaction took place after removing the catalyst, the Au catalyst was then put back into the mixture. As a result, the oxidation reaction was restarted, and the product 2a was obtained in 99% yield in 30 min. In addition, the leaching of the AuNPs in the reaction of 1a was also examined using an inductively coupled plasma optical emission spectrometer (ICP-OES), and no leaching of AuNPs was detected by the ICP analysis. To assess recyclability of Au-2, multiple dimethylphenylsilane 1a oxidation cycles were carried out, and the recovery of the heterogeneous catalyst is carried out by filtration for the separation of the catalyst from the reaction mixture. The catalyst was repeatedly used four times, but no significant loss of activity was observed. The product 2a was obtained nearly quantitatively every time (Table 3, entries 1–4). After the reaction, the catalyst was again examined by TEM, and the image indicated that the particle size was increased (Fig. S7†), however, the morphology and distribution of the AuNPs have no significant changes in comparison with observations in Fig. 1a.
 | (1) |

Furthermore, the catalytic oxidation reactions with a variety of organosilanes using the Au-2 catalyst were carried out (Table 4). First, triethylsilane (1b) exposed sterically was quantitatively oxidized to the corresponding silanol 2b in 15 minutes without the formation of disiloxane (entry 2). Next, the oxidation of two reputedly challenging substrates (1c and 1d) was examined; the corresponding silanols 2c and d were obtained in high yields by increasing the catalyst loading amount and the reaction time (entries 3–4). The Au-2 catalyst could also be used in the oxidation of diphenylsilane (1e), and the corresponding silanol (2e) was obtained in high yield (entry 5).
|
|
||||
|---|---|---|---|---|
| Entry | R1R2R3SiH (1) | t (min) | R1R2R3SiOH (2) | Yieldb (%) |
| a Reaction conditions: organosilane (0.5 mmol) and Au-2 (0.1 mol%) in 1 mL of H2O at room temperature. b Isolated yield. c Au-2 (1 mol%) was used. d 7/3 of H2O and THF was used. | ||||
| 1 | PhMe2SiH (1a) | 40 | PhMe2SiOH (2a) | 99 |
| 2 | Et3SiH (1b) | 15 | Et3SiOH (2b) | 99 |
| 3cd | iPr3SiH (1c) | 660 | iPr3SiOH (2c) | 97 |
| 4d | Ph3SiH (1d) | 170 | Ph3SiOH (2d) | 99 |
| 5 | Ph2SiH2 (1e) | 110 | Ph2Si(OH)2 (2e) | 97 |

Several control experiments were conducted to understand the high catalytic activity in H2O (eqn (2)): the reaction of dimethylphenylsilane 1a with H2O in the presence of AuNPs stabilized by OA as the catalyst and acetone as the solvent, the reaction cannot proceed, yielding the corresponding silanol 2ain 99% yield in 70 min, whereas on using H2O as the solvent, the reaction time was prolonged to 180 min. It is interesting to find that owing to the presence of o-CNTs in the reaction mixture, the reaction time was reduced to 60 min; these results indicate evidently that the o-CNTs were important for oxidation of 1a in H2O to improve the catalytic activity. Another control experiment was also performed, and due to the absence of oxygen, the current reaction cannot proceed at all. A similar phenomenon was reported in the previous work, and O2 activates water which, in turn, oxidizes the substrate at the surface of AuNPs, which has been proved by Davis and Liu.20
 | (2) |
Based on the above results, the reason for the high activity of Au-2 catalyzed organosilane oxidation is schematically shown in Scheme 1.
 | ||
| Scheme 1 A possible reaction pathway for the high activity of Au-2. | ||
Initially, Au-2 was dispersed in H2O which is absorbed via the –OH group on the surface of the CNTs and the –NH2, –COO–NH4+– groups on the surface of the AuNPs. Next, the organosilane was easily linked to the AuNPs because the OA surrounding the AuNPs makes the catalyst lipophilic. Both the above effects make the H2O and silane more easily gather onto the surface of the low-coordinated Au atoms in H2O, leading to the high activity of the current catalyst in H2O. Our scenario is similar to the related mechanistic studies on the hydrolytic oxidation of hydrosilanes by metal nanoparticles reported by Park et al.18
Conclusions
In summary, our results demonstrate that AuNPs supported on o-CNTs are highly efficient heterogeneous catalysts for the selective oxidation of silanes in H2O. The control experiments indicated clearly that o-CNTs and OA were key components of the current catalyst. The o-CNT support makes the hybrid materials hydrophilic, and the OA stabilizer makes the hybrid materials lipophilic, which improve the catalytic activity in H2O. A wide range of organosilanes can be oxidized to the corresponding organosilanols without the formation of the disiloxane by-product. Moreover, the Au-2 catalyst can be recovered several times without leaching and loss of activity. It is worth noting that the current catalyst could be easily prepared by using the one-pot GLIP method, which will expand its practical applications. Further work is in progress to extend this green synthesis concept to other chemical catalytic reactions using water as a solvent.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21106184 and 21202203), the Science Foundation Research Funds provided to New Recruitments of China University of Petroleum, Beijing (no. YJRC-2011-18 and YJRC-2012-31), the Foundation for the Author of National Excellent Doctoral Dissertation of PR China (no. 201252) and Thousand Talents Program.References
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- (a) V. Georgakilas, D. Gournis, V. Tzitzios, L. Pasquato, D. M. Guldi and M. Prato, J. Mater. Chem., 2007, 17, 2679–2694 RSC; (b) N. Karousis, N. Tagmatarchis and D. Tasis, Chem. Rev., 2010, 110, 5366–5397 CrossRef CAS PubMed; (c) R. Singh, T. Premkumar, J. Y. Shin and K. E. Geckeler, Chem.–Eur. J., 2010, 16, 1728–1743 CrossRef CAS PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- Z. Ma and S. Dai, ACS Catal., 2011, 1, 805–818 CrossRef CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS PubMed.
- (a) R. Zanella, E. V. Basiuk, P. Santiago, V. A. Basiuk, E. Mireles, I. Puente-Lee and J. M. Saniger, J. Phys. Chem. B, 2005, 109, 16290–16295 CrossRef CAS PubMed; (b) T. Nakamura, T. Ohana, M. Ishihara, M. Hasegawa and Y. Koga, Diamond Relat. Mater., 2007, 16, 1091–1094 CrossRef CAS PubMed; (c) L. Qu and L. Dai, J. Am. Chem. Soc., 2005, 127, 10806–10807 CrossRef CAS PubMed; (d) X. Ye, L. Chen, C. Wang, J. Aubuchon, I. Chen, A. Gapin, J. Talbot and S. Jin, J. Phys. Chem. B, 2006, 110, 12938–12942 CrossRef CAS PubMed; (e) Y. Wang, X. Xu, Z. Tian, Y. Zong, H. Cheng and C. Lin, Chem.–Eur. J., 2006, 12, 2542–2549 CrossRef CAS PubMed; (f) G. Wei, C. Pan, J. Reichert and K. D. Jandt, Carbon, 2010, 48, 645–653 CrossRef CAS PubMed; (g) X. Hu, T. Wang, X. Qu and S. Dong, J. Phys. Chem. B, 2006, 110, 853–857 CrossRef CAS PubMed; (h) Y. Xing, J. Phys. Chem. B, 2004, 108, 19255–19259 CrossRef CAS; (i) M. Cano, A. Benito, W. K. Maser and E. P. Urriolabeitia, Carbon, 2011, 49, 652–658 CrossRef CAS PubMed; (j) F. Yang, Y. Li, T. Liu, K. Xu, L. Zhang, C. Xu and J. Gao, Chem. Eng. J., 2013, 226, 52–58 CrossRef CAS PubMed.
- (a) T. Kaneko, K. Baba, T. Harada and R. Hatakeyama, Plasma Processes Polym., 2009, 6, 713–718 CrossRef CAS; (b) O. Höfft and F. Endres, Phys. Chem. Chem. Phys., 2011, 13, 13472–13478 RSC.
- (a) E. A. Ison, R. A. Corbin and M. M. Abu-Omar, J. Am. Chem. Soc., 2005, 127, 11938–11939 CrossRef CAS PubMed; (b) R. A. Corbin, E. A. Ison and M. M. Abu-Omar, Dalton Trans., 2009, 2850–2855 RSC; (c) U. Schubert and C. Lorenz, Inorg. Chem., 1997, 36, 1258–1259 CrossRef CAS; (d) M. Lee, S. Ko and S. Chang, J. Am. Chem. Soc., 2000, 122, 12011–12012 CrossRef CAS; (e) Y. Lee, D. Seomoon, S. Kim, H. Han, S. Chang and P. H. Lee, J. Org. Chem., 2004, 69, 1741–1743 CrossRef CAS PubMed.
- B. P. S. Chauhan, A. Sarkar, M. Chauhan and A. Roka, Appl. Organomet. Chem., 2009, 23, 385–390 CrossRef CAS.
- (a) E. Choi, C. Lee, Y. Na and S. Chang, Org. Lett., 2002, 4, 2369–2371 CrossRef CAS PubMed; (b) K. Mori, M. Tano, T. Mizugaki, K. Ebitani and K. Kaneda, New J. Chem., 2002, 26, 1536–1538 RSC.
- T. Mitsudome, S. Arita, H. Mori, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2008, 47, 7938–7940 CrossRef CAS PubMed.
- (a) M. Jeon, J. Han and J. Park, ChemCatChem, 2012, 4, 521–524 CrossRef CAS; (b) K. Shimizu, T. Kubo and A. Satsuma, Chem.–Eur. J., 2012, 18, 2226–2229 CrossRef CAS PubMed.
- T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2009, 5302–5304 RSC.
- N. Asao, Y. Ishikawa, N. Hatakeyama, Menggenbateer, Y. Yamamoto, M. W. Chen, W. Zhang and A. Inoue, Angew. Chem., Int. Ed., 2010, 49, 10093–10095 CrossRef CAS PubMed.
- J. John, E. Gravel, A. Hagege, H. Y. Li, T. Gacoin and E. Doris, Angew. Chem., Int. Ed., 2011, 50, 7533–7536 CrossRef CAS PubMed.
- W. J. Li, A. Q. Wang, X. F. Yang, Y. Q. Huang and T. Zhang, Chem. Commun., 2012, 48, 9183–9185 RSC.
- M. Jeon, J. Han and J. Park, ACS Catal., 2012, 2, 1539–1549 CrossRef CAS.
- (a) H. H. Chen, C. C. Weng, J. D. Liao, L. M. Whang and W. H. Kang, J. Hazard. Mater., 2012, 201, 185–192 CrossRef PubMed; (b) T. Kaneko, Q. Chen, T. Harada and R. Hatakeyama, Plasma Sources Sci. Technol., 2011, 20, 034014 CrossRef.
- (a) B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed; (b) C. Shang and Z. P. Liu, J. Am. Chem. Soc., 2011, 133, 9938–9947 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, experimental setup, OES, XPS, EDX, Au loading, AuNP size, and other TEM images of carbon nanotube–gold nanohybrids. See DOI: 10.1039/c3ta13693k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |