New conjugated alternating benzodithiophene-containing copolymers with different acceptor units: synthesis and photovoltaic application
M. L.
Keshtov
a,
D. V.
Marochkin
a,
V. S.
Kochurov
b,
A. R.
Khokhlov
a,
E. N.
Koukaras
cd and
G. D.
Sharma
*e
aInstitute of Organoelement Compounds of the Russian Academy of Sciences, Vavilova st., 28, 119991 Moscow, Russian Federation
bLomonosov Moscow State University, Faculty of Physics, 1-2 Leninskiye Gory, Moscow, 119991, Russian Federation
cInstitute of Chemical Engineering Sciences, Foundation for Research & Technology, Hellas, Stadiou Str. Platani, Patras, 26504, Greece
dMolecular Engineering Laboratory, Department of Physics, University of Patras, Patras, 26500, GR, Greece
eR & D Center for Engineering and Science, JEC group of Colleges, Jaipur Engineering College, Kukas, Jaipur 303101, India. E-mail: sharamgd_in@yahoo.com; gdsharma273@gmail.com; Fax: +91-1426-511240; Tel: +91-1426-227345
First published on 3rd October 2013
Abstract
Two new alternating low band gap D–A copolymers with different acceptor structures of 4,8-bis-(5-bromothiophene-2-yl)-benzo[1,2,5]thiadiazole (P1) and 4,8-dithiophene-2-yl-benzo[1,2-c;4,5-c′]-bis-[1,2,5]thiadiazole (P2) and a common BDT donor segment have been synthesized under Stille reaction conditions and characterized. The polymers showed good solubility, broad absorption bands and optical band gaps of 1.62 eV and 1.16 eV for P1 and P2, respectively. Bulk heterojunction (BHJ) polymer solar cells based on P1 and P2 as electron donors and fullerene derivatives (PC60BM and PC70BM) as acceptor were fabricated and their photovoltaic response was investigated. The overall power conversion efficieny (PCE) achieved for BHJ solar cells based on P1:PC60BM, P2:PC60BM, P1:PC70BM and P2:PC70BM blends cast from THF solvent is about 2.17%, 0.80%, 3.45% and 1.19%, respectively. The higher PCE for the device based on P1 has been attributed to the high value of hole mobility for P1 as compared to P2 and a larger driving force i.e. LUMO–LUMO offset, for photo-induced charge transfer for P1:PCBM BHJ active layer. The PCE has been further increased up to 5.30% and 1.58% for P1:PC70BM and P2:PC70BM blends cast from DIO/THF solvent, which is attributed to the improved crystallinity and a more balanced charge transport in the device.
1. Introduction
Polymer solar cells (PSCs) have attracted considerable attention as renewable energy sources due to their unique advantages such as low cost, low weight and the possibility of flexible large-area device fabrication.1 So far the most successful PSCs are based on bulk heterojunction (BHJ) active layers which consist of a phase separated blend of organic electron donor (polymers or small molecules) and acceptor (mostly fullerene derivatives), sandwiched between two electrodes with different work functions.2To date, poly(3-hexylthiophene) (P3HT) and [6,6]-phenyl-C61-butyric acid methyl ester (PCBM) are the most representative donor and acceptor materials, respectively. The power conversion efficiency (PCE) of the PSC based on P3HT/PCBM has reached more than 4%.3 However, P3HT has a relatively large band gap (∼2.0 eV) and absorbs photons with wavelengths shorter than 650 nm, in addition, the relatively high location of the HOMO (−4.80 to −4.90 eV) leads to a lower open circuit voltage (∼0.6 V).4 Thus, the design and synthesis of new donor conjugated polymers with broad absorption bands and low HOMO energy level is one of the most important issues.5 In order to achieve high efficiency polymer solar cells, it is necessary to prepare conjugated polymers with the following desired features: good optical and mechanical properties, long term stability and processability and efficient charge transfer. For a BHJ active layer consisting of fullerene as electron acceptor and polymer as electron donor, the ideal polymer should have a band gap between 1.2 and 1.9 eV with a broad absorption spectrum to absorb as much sunlight as possible and to generate the high short circuit current, and the LUMO level should be in the range of 3.5–3.8 eV to create enough driving force to promote an efficient exciton dissociation at the D–A interface.
As is well known, the PCE of solar cells is proportional to the open circuit voltage (Voc), short-circuit current (Jsc) and the fill factor (FF) of the PSC. Thus, the key issues that must be taken into account in the molecular design of conjugated polymers are: broad absorption spectra to improve the collection of sunlight with a high Jsc, suitable energy levels i.e. highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) matching with the fullerene acceptor, low lying HOMO with maximum Voc, high hole mobility, and appropriate compatibility with fullerene acceptor to form nanoscale bi-continuous interpenetrating networks.
Among the many possible directions, one feasible approach towards broadening the absorption spectrum and tuning the energy levels is to design alternating donor–acceptor copolymers, in which the electron rich (donor) monomers are copolymerized with electron deficient (acceptor) monomers. The incorporation of alternating electron donating units (push) and electron withdrawing units (pull) within the polymer backbone results in intra-molecular charge transfer (ICT) which lowers the band gap. Moreover, the HOMO and LUMO energy levels of D–A copolymers can also be independently controlled by their donor and acceptor components, respectively, which provides a means for narrowing the band gap of the copolymer with appropriate energy level alignments with fullerene derivative acceptors.6 To date, the efficiency of PSCs based on donor–acceptor (D–A) copolymers has reached ∼5–8%.7 Recently, PCEs more than 10% in single junction and tandem solar cells have been achieved by development of new materials and device architectures.8 Benzo[1,2-b:3,4-b′]dithiophene (BDT) is one of the most successful donor units in the construction of the D–A copolymers for high performance photovoltaic materials.9 The advantages of BDT-coupled planar structure are easy to purify BDT monomers, and a high hole mobility (for example, the hole mobility of the copolymer of BDT and thiophene reaches 0.25 cm2 V−1 s−1).10
For these reasons, we opted to synthesize copolymers P1 and P2 derivatives of benzo[1,2-b:4,3-b′]dithiophene (BDT) as the donor and two new acceptor structures for the copolymerization with 4,8-bis-(5-bromothiophene-2-yl)-benzo[1,2,5]thiadiazole and 4,8-dithiophene-2-yl-benzo[1,2-c;4,5-c′]-bis-[1,2,5]thiadiazole, respectively. Acceptor structures are easily synthesized and have a relatively simple and flat structure, which may be useful for the delocalization of the electrons when they are included in the D–A copolymers. In addition, their relatively strong electron acceptor ability will lead to a decrease in the HOMO and LUMO levels of D–A copolymers, which will lead to a high open-circuit voltage (Voc) in the PSC. Due to the presence of alkyl substituents the copolymers exhibit good solubility in common organic solvents. We have used these copolymers as electron donors along with fullerene derivatives, i.e. PC60BM and PC70BM, as electron acceptors for the fabrication of BHJ polymer solar cells. The PCE of the devices based on P1:PC60BM, P2:PC60BM, P1:PC70BM and P2:PC70BM is about 2.17%, 0.80%, 3.45% and 1.19%, respectively. The higher value of PCEs for solar cells based on P1 as electron donors in the BHJ active layer was attributed to the relatively higher value of hole mobility and larger driving force for photoinduced charge transfer at the D–A interface present in the BHJ active layer. The PCE has been further improved up to 5.30% and 1.58%, for P1:PC70BM and P2:PC70BM blends processed from DIO/THF solvent.
2. Experimental details
2.1 Instruments and characterization methods
1H and 13C NMR spectra of the starting compounds and polymers were recorded on the spectrometer “Bruker Avance-400” with a working frequency of 400.13 and 100.62 MHz, respectively. IR spectra were recorded by a FT-IR spectrometer “Perkin-Elmer 1720-X”, TGA and DSC analysis was performed on “Perkin-Elmer TGA-7” and “Perkin-Elmer DSC-7” devices with heating rate of 20 deg min−1.The absorption spectra in the range 190–1100 nm were recorded on the spectrophotometer “Varian Cary 50.” The source of the exciting light was a xenon lamp L8253, which is part of the block radiator with optical fiber output radiation “Hamamatsu LC-4.” Cyclic voltammetry measurements were performed on a potentiostat-galvanostat μAUTOLAB Type III equipped with standard three-electrode scheme in an acetonitrile solution of 0.1 mol l−1 tributylammonium hexafluorophosphate (n-Bu4NPF6) at a potential scan rate of 50 mV s−1. Films of investigated polymers were deposited on a glass surface coated with ITO and then dried and used as working electrode. Ag/Ag+ and platinum were used as reference and counter electrodes, respectively.
2.2 Synthesis of copolymers P1 and P2
2.3 Methodology of the theoretical calculations
In addition to the experimental measurements, we have studied the structures theoretically within the framework of density functional theory (DFT) and time-dependent density functional theory (TD-DFT). The P1 and P2 monomers exhibit structural flexibility which led us to design several stereoisomers as initial geometries for our calculations. The initial stereoisomers also included cis- and trans-configurations of the alkane units.The calculations have been performed employing the gradient corrected functional PBE17 and the hybrid exchange–correlation functional PBE018a (without adjustable parameters) of Perdew, Burke and Ernzerhof, as well as the hybrid functional of Becke, Lee, Yang and Parr, B3LYP.18b,c Jacquemin et al. have reported an extensive comparative study19 on the performance of hybrid functionals with and without long-range corrections for the calculation of the visible absorption spectra of a wide range of organic dyes. They find that among the functionals under study, while also taking into account solvent effects, the PBE0 functional provides un-calibrated excitation energies with the smallest average absolute deviation of 0.14 eV. The long-range corrected functionals may outperform PBE0 on average only when the excitation energies are calibrated with (per case) linear corrections. Compared to our experimental results we find that the widely used B3LYP functional performs very well, and can also serve as a reference to facilitate comparison with the literature.
For each structure we have used several stereoisomers as initial geometries for the geometry optimizations. This first round of calculations was performed employing the PBE functional. The resolution of the identity method20 was used for the treatment of the two-electron integrals. The TZVP basis set21 which is of triple-ζ quality was used throughout. The resulting structures were further optimized using both the hybrid PBE0 and B3LYP functionals, using the same basis set and also taking into account solvent effects. The polarization continuum model (PCM)22 was employed to account for effects from solute–solvent interactions. The importance of accounting for solvation effects when calculating the absorption spectrum has been extensively shown for a wide range of systems, including, but not limited to organic dyes, dye-sensitized TiO2 nanoparticles and Ru(II)–polypyridyl complexes.23–27 Tight convergence criteria were in place for the SCF energy (up to 10−7 Eh) and the one-electron density (rms of the density matrix up to 10−8) as well as for the norm of the Cartesian gradient (residual forces both average and maximum smaller than 1.5 × 10−5 a.u.) and residual displacements (both average and maximum smaller than 6 × 10−5 a.u.).
The TD-DFT excited state calculations have been performed, using the same functionals, on the corresponding ground state structures. In each case we have calculated the optical gap in the presence of solvents. For solvents we have used tetrahydrofuran (THF) and ortho-dichlorobenzene (o-DCB or 1,2-DCB) using the self-consistent reaction field solvent parameters as defined in the Gaussian 09 electronic structure package.28 The first round of geometry optimization was performed using the Turbomole package.29 All of the follow up calculations were performed using the Gaussian package.28
2.4 Device fabrication and characterization
Indium tin oxide (ITO) coated glass substrates were ultrasonically cleaned sequentially in detergent, deionized water and isopropyl alcohol and dried. A layer of poly(3,4-ethylenedioxythiophene):poly(styrene sulphonate) (PEDOT:PSS) (thickness about 65 nm) was deposited on the ITO coated glass substrates by spin coating from an aqueous solution and then annealed on a hot plate at 90 °C for 20 min in air and subsequently cooled to room temperature. The blends of P1 or P2:PC60BM or PC70BM of different weight ratios were prepared by dissolving the copolymers and fullerene derivatives in THF solution and stirring for about 2 h. For the solvent additive, i.e. DIO/THF, a small amount of DIO (3% by volume) is added to the THF solution. The photoactive layer of P1 or P2:PC60BM or PC70BM (in different ratios with and without solvent additive DIO) was deposited onto the PEDOT:PSS layer by spin coating and then dried in ambient atmosphere. Finally, an aluminum (Al) metal top electrode was deposited in vacuum on the active layer at a pressure of ca. 5 × 10−5 Pa. The active area of the device was ca. 0.14 cm2. The current density–voltage (J–V) characteristics were measured on a computer-controlled Keithley 236 source meter unit. A xenon lamp coupled with AM 1.5 solar spectrum filter was used as the light source, and the optical power at the sample was 100 mW cm−2.3. Results and discussion
3.1 Synthesis and characterization of the copolymers
Benzo[1,2,5]thiadiazole is one of the most attractive electron-withdrawing fragments. Its functionalization by different substituents makes it possible to obtain a wide range of heteroaromatic compounds that are promising as “building blocks” for new electroactive polymers. In this regard, on the basis of key compound – 4,7-dibromobenzo[1,2,5]thiadiazole 1 – we have developed a number of thiophene-containing derivatives presented in Scheme 1. Herewith, compound 6 has been previously described in the literature. | ||
| Scheme 1 | ||
By the interaction of 4,7-dibromobenzo[1,2,5]thiadiazole 1 with 2-tributylstannylthiophene in Stille coupling reaction using Pd(PPh3)2Cl2 as catalyst, followed by bromination of product 2 by N-bromosuccinimide, we obtained 4,7-bis-(5-bromothiophene-2-yl)-benzo[1,2,5]thiadiazole 3 in 66% yield,12 the purity, composition and structure of which are confirmed by melting point, NMR spectroscopy and elemental analysis. Nitration of 4,7-dibromobenzo[1,2,5]thiadiazole 1 by fuming nitric acid in 100% sulfuric acid yielded dinitroderivative 4, which is then converted to the 5,6-dinitro-4,7-dithiophene-2-yl-benzo[1,2,5]thiadiazole 5 by Stille cross-coupling in THF.13 Reduction of 5 by iron powder in acetic acid resulted in 4,7-dithiophene-2-yl-benzo[1,2,5]thiadiazole-5,6-diamine 6.13 By condensation of compound 6 with N-thionylaniline in the presence of (CH3)3SiCl, 4,8-dithiophene-2-yl-benzo[1,2-c;4,5-c′]-bis-[1,2,5]thiadiazole 7 was obtained,14 which proved to have a quite high melting point (Mp = 332–334 °C) and is a barely soluble product.
Further bromination in the α-positions of the thiophene fragments was performed using N-bromosuccinimide in a mixture of chloroform–acetic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature. The desired product 8 was purified by recrystallization from DMF.15
:1) at room temperature. The desired product 8 was purified by recrystallization from DMF.15
On the basis of the synthesized heteroaromatic thiophene-containing monomers, a series of new low band gap copolymers with the quinoid π-conjugation nature and a strict alternation of donor–acceptor units has been developed, obtained by the Stille cross-coupling reaction. Polycondensation was carried out under argon in a mixture of toluene and DMF at 115 °C for 48 h, using tetrakis(triphenylphosphine) palladium(0) as a catalyst. The role of electron-donor component in the structures of polymers P1 and P2 is served by 2,6-bis-trimetylstannyl-4,8-didodecyloxybenzo[1,2-b;4,5-b′]dithiophene 9, and the electron acceptor fragments are monomers 3 and 8 (Scheme 2).
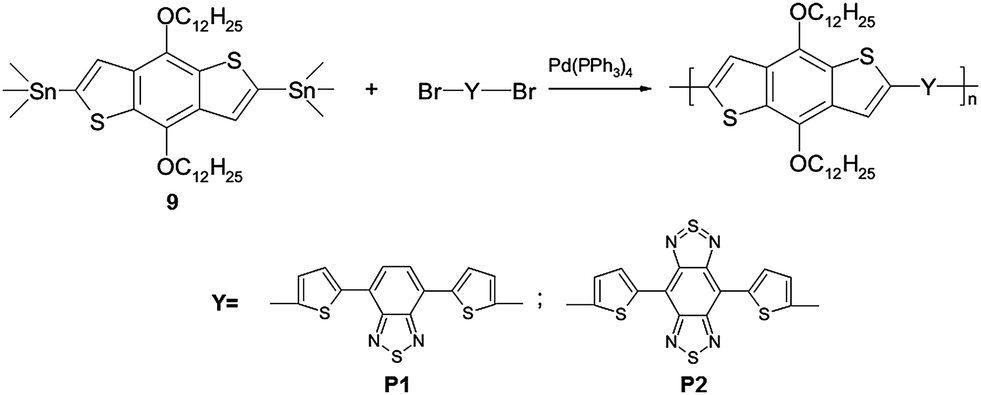 | ||
| Scheme 2 | ||
The resulting polymers were purified from the residual metal catalyst, organotin and low molecular weight impurities by re-precipitation twice from solution in methanol and subsequent extraction with methanol, hexane and acetone. The yield of the desired high molecular mass products was 60–76%.
The composition and structure of the low band gap polymers P1 and P2 were confirmed by 13C NMR spectroscopy as shown in Fig. 1a and b for P1 and P2, respectively.
 | ||
| Fig. 1 13C NMR spectra of copolymer (a) P1 and (b) P2. | ||
For P113C NMR (100 MHz, CdCl3, δ) 14.10 (82, 100); 22.72 (81, 99); 26.48 (73, 91); 28.24 (74, 92), 29.36 (79, 97); 29.54 (78, 96); 29.58 (77, 95); 30.09 (75, 93); 30.22 (76, 94); 30.50 (72, 90); 31.98 (80, 98); 69.47 (71, 89) (Aliph); 118.00 (63); 118.38 (69); 118.85 (29, 57); 121.52 (60); 121.92 (66); 122.01 (30, 56); 123.66 (51, 52); 131.26 (68); 131.66 (62); 132.98 (44, 50); 134.71 (26, 55); 137.06 (61, 67); 137.48 (64); 139.21 (58); 144.92 (28, 53); 151.08 (45, 49) (Ar).
For P213C NMR (100 MHz, CdCl3, δ) 14.10 (85, 103); 22.72 (84, 102); 26.48 (76, 94); 28.24 (77, 95), 29.36 (82, 100); 29.54 (81, 99); 29.58 (80, 98); 30.09 (78, 96); 30.22 (79, 97); 30.50 (75, 93); 31.98 (83, 101); 69.47 (74, 92) (Aliph); 111.65 (44, 50); 113.75 (29, 60); 118.00 (66); 118.38 (72); 121.52 (63); 121.92 (69); 122.28 (30, 59); 131.26 (71); 131.66 (65); 134.98 (26, 58); 137.06 (64, 70); 137.48 (67); 140.71 (61); 141.32 (28, 56); 150.79 (45, 49); 153.19 (51, 55) (Ar).
Average molecular weights and polydispersity of the polymers were determined by GPC (eluent – THF, standard – polystyrene) and varied in the range of (1.22–1.43) × 104 and 1.48–1.72, respectively. Both the copolymers P1 and P2 are soluble in common organic solvents such as DMF, N-methylpyrrolidone, chloroform, THF, and o-dichlorobenzene. The glass transition temperature of polymers determined by TMA is in the range of 344–348 °C (Table 1).
| Polymer | –Y– | M n (×10−4) | M w (×10−4) | M w/Mn | T g (°C) | T 10% (°C) |
|---|---|---|---|---|---|---|
| a In the numerator – the air, in the denominator – the atmosphere of argon. | ||||||
| P1 |

|
1.22 | 1.81 | 1.48 | 196 | 341, 344 |
| P2 |

|
1.43 | 2.46 | 1.72 | 333 | 338, 348 |
As can be seen from Table 1, the presence of long alkyl substituents in the structure of macromolecules generally leads to lower glass transition temperature of the polymers. However, a larger number of annelation of heterocyclic fragments into monomeric units, which reduces the flexibility of individual structural fragments, tends to increase Tg.
T 10% values were determined by TGA both in air and argon in the range 295–341 °C and 316–348 °C, respectively. Fig. 2 shows the typical TGA curves of polymers P1 and P2 in the air. Both the synthesized copolymers exhibit relatively high thermal stability.
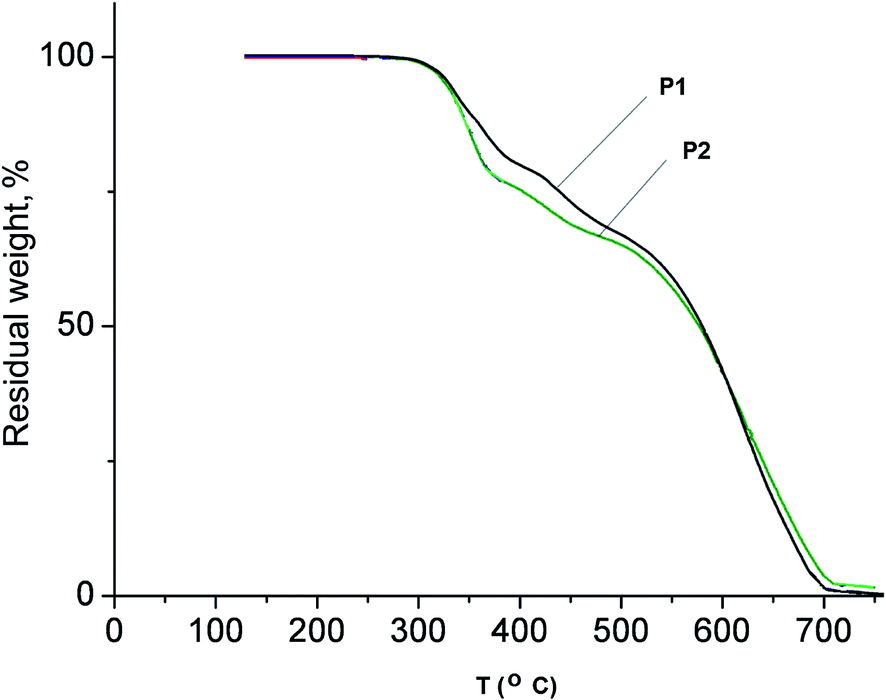 | ||
| Fig. 2 TGA curves of copolymers P1 and P2 in the air. | ||
3.2 Optical and electrochemical properties
One of the main properties of the copolymers resulting in their perspectives for application as photoactive materials in solar cells is their absorption coefficient in the visible and near IR ranges.30 The electronic absorption properties of the synthesized copolymers were measured in both THF solution and as thin films spin coated onto quartz substrates and are shown in Fig. 3a (P1) and b (P2). The results of the spectral data are presented in Table 2. | ||
| Fig. 3 The normalized absorption spectra of copolymers (a) P1 and (b) P2 in solution and thin film form cast from THF solution. | ||
| Polymer | λ absmax solution (nm) (molar extinction coefficient) | λ absmax film (nm) (molar extinction coefficient) | λ absonset film (nm) | E optg (eV) |
|---|---|---|---|---|
| P1 | 398 (3.20 × 104 cm−1); 539 (2.34 × 104 cm−1) | 419; 588 | 786 | 1.58 |
| P2 | 479 (3.14 × 104 cm−1); 866 (1.56 × 104 cm−1) | 481; 896 | 1042 | 1.18 |
The UV-visible spectrum of the copolymers is similar to that of the D–A copolymers31 with two characteristics absorption bands observed near 410–481 and 588–1050 nm (Table 2, Fig. 3). We assign the high energy absorption bands to π–π* transitions from the BT unit and the longer wavelength absorption bands to the intramolecular charge transfer (ICT) between the BT donor and different acceptor units in the copolymers. An interesting observation is the impressive bathochromic shift (415 nm) of the absorption band of polymer P2 with respect to polymer P1 (Fig. 3a and b), which differs from the first fragment by the absence of a second thiadiazole ring in co-monomer 3. This band of polymer P2 is almost completely located in the near-infrared area and determines the ability of this material to absorb photons of the solar spectrum with the lowest energy.32 These results also indicate that the conjugated side chains can easily tune the absorption behavior of BT based D–A copolymers, which is beneficial for the design and preparation of efficient photovoltaic materials with enhanced absorption and suitable energy levels. The peaks of the long-wavelength absorption bands of copolymers P1 and P2 (which are based on benzo[1,2,5]thiadiazole derivatives) are bathochromically shifted in the order λabsmax (P1) < λabsmax (P2). This observed shift is consistent with the general ideas on the nature of electron-withdrawing condensed heterocyclic systems as the number of “pyridine type” nitrogen atoms increases. As shown in Fig. 3a and b, the absorption peaks in the longer wavelength region are red shifted relative to their counterparts in the solution. The absorption bands become broader than that in solution presumably due to the better planarity of the polymer backbone, the formation of π-stacked structures as well as a strong electronic interaction between the energy values of the optical band gaps of P1 and P2 that were calculated from the absorption spectra of the polymers in the thin films, specifically from the onset absorption edge in the longer wavelength region. The values have been complied in Table 2. The optical band gap of polymer P2 (1.18 eV) is lower than P1 (1.58 eV) which indicates an increase in the ability of the monomer to lower the HOMO–LUMO energy gap of polymers. The small band gap observed in P2 can be attributed to the high electron affinity and large quinoid contribution of the BDT unit.33
The electrochemical properties of polymers P1 and P2 were examined by cyclic voltammetry. It was observed that both copolymers exhibit reversible or partially reversible redox properties due to the high electro-activity. As shown in Fig. 4, both copolymers show a predominant oxidation peak (p doping) due to the electron donating benzothiophene and aryl-vinylene segment and a minor reduction peak (n doping) related to the electron withdrawing benzothiadiazole unit. The onset oxidation potential (Eoxdonset) and onset reduction potential (Eredonset) observed in the cyclic voltammogram for P1 and P2 are summarized in Table 3. The values of HOMO and LUMO energy levels for copolymers were estimated from the following expressions
| HOMO = −q(Eoxonset + 4.71) eV |
| LOMO = −q(Eredonset + 4.71) eV |
 | ||
| Fig. 4 Cyclic voltammograms of P1 andP2 in 0.1 M solution of lithium perchlorate in acetonitrile at a scan rate 50 mV s−1. | ||
| Polymer | E redonset (V) | E oxonset (V) | E HOMO (eV) | E LUMO (eV) | E ecg (eV) |
|---|---|---|---|---|---|
| P1 | −1.14 | 0.60 | −5.24 | −3.48 | 1.76 |
| P2 | −0.92 | 0.34 | −5.10 | −3.82 | 1.28 |
From the onset potentials of the p-doping process, the HOMO energy levels were estimated to be −5.24 eV and −5.10 eV for P1 and P2, respectively. On the other hand, the LUMO energy levels were estimated from the onset reduction potentials of n doping, to be −3.48 eV and −3.82 eV for P1 and P2, respectively. The values found for the energy band gap (Eecg) of the polymers using cyclic voltammetry according to the equation Eecg = (HOMO − LUMO) are 1.76 eV and 1.28 eV for P1 and P2, respectively. The values and trends are consistent with those estimated by the optical method described above. The higher electrochemical band gap is a common phenomenon for the conjugated polymers because of the energy barrier of charge transfer at the electrodes during electrochemical measurements.
3.3 Theoretical calculations
The stereoisomers for each of the monomers (P1 and P2), optimized at the PBE/TZVP level of theory, are found to be nearly isoenergetic. This is to be expected for rotamers and cis/trans isomers. The maximum energy difference for each case does not exceed 65 meV (1.5 kcal mol−1), with the trans isomers energetically lower. This energy difference can be considered as the largest vertical rotational barrier in the gas phase. Corresponding adiabatic values would of course be larger and would account for possible steric effects.The PBE functional belongs to the generalized gradient approximation (GGA) family of functionals which are known to underestimate the highest occupied molecular orbital (HOMO) − lowest unoccupied molecular orbital (LUMO) gap (HL gap). The calculated HOMO and LUMO energy levels, HL gap, optical band gap, oscillator strength and dipole moment computed at different levels of theory in the gas phase as well as THF and o-DCB solvent phases are shown in Tables 4 and 5. From Tables 4 and 5 we can see that the PBE functional systematically underestimates the HL gap, compared to the experimental values, on average by about 0.25 eV.
| Gas phase | |||||
|---|---|---|---|---|---|
| HOMO (eV) | LUMO (eV) | HL gap (eV) | |Dipole moment| (D) | ||
| P1 | PBE | −4.73 | −3.52 | 1.21 | 0.50 |
| PBE0 | −5.50 | −2.85 | 2.65 | 0.26 | |
| B3LYP | −5.23 | −2.93 | 2.30 | 0.21 | |
| P2 | PBE | −4.65 | −4.13 | 0.52 | 1.72 |
| PBE0 | −5.30 | −3.73 | 1.58 | 0.80 | |
| B3LYP | −5.04 | −3.75 | 1.30 | 1.09 | |
| HOMO (eV) | LUMO (eV) | HL gap (eV) | Optical gap (eV) | f, osc. strength | |Dipole moment| (D) | ||
|---|---|---|---|---|---|---|---|
| Solvent THF | |||||||
| P1 | PBE | −4.84 | −3.55 | 1.28 | 1.43 | 0.23 | 0.75 |
| PBE0 | −5.63 | −2.89 | 2.75 | 2.22 | 0.80 | 0.51 | |
| B3LYP | −5.44 | −3.02 | 2.42 | 2.06 | 0.68 | 0.57 | |
| P2 | PBE | −4.72 | −4.15 | 0.57 | 0.87 | 0.28 | 1.76 |
| PBE0 | −5.44 | −3.76 | 1.68 | 1.29 | 0.57 | 0.78 | |
| B3LYP | −5.25 | −3.86 | 1.38 | 1.16 | 0.52 | 0.94 | |
| Solvent o-DCB | |||||||
| P1 | PBE | −4.85 | −3.56 | 1.29 | 1.43 | 0.26 | 0.77 |
| PBE0 | −5.64 | −2.89 | 2.75 | 2.21 | 0.83 | 0.54 | |
| B3LYP | −5.45 | −3.02 | 2.43 | 2.05 | 0.72 | 0.60 | |
| P2 | PBE | −4.73 | −4.15 | 0.57 | 0.86 | 0.31 | 1.74 |
| PBE0 | −5.45 | −3.76 | 1.69 | 1.28 | 0.60 | 0.79 | |
| B3LYP | −5.26 | −3.87 | 1.39 | 1.15 | 0.55 | 0.94 | |
The energetically lowest isomers of each case were further optimized, initially in the gas phase, employing the hybrid functionals PBE0 and B3LYP. No significant structural differences arise from these optimizations which suggests that the PBE functional produces high quality structures. For the structure P2 the calculated values for the HL gaps are in excellent agreement with the experimental values, as is shown in Tables 2–5. A discrepancy is noted for the P1 structure for which the calculated HL gaps are overestimated by both hybrid functionals with regards to the experiment. More so, the overestimation is not the same for the two functionals but differs by about 0.16 eV.
The HOMO and LUMO of the P1 and P2 monomers are shown in Fig. 5, plotted using an isovalue of 0.02. In both cases the HOMO is delocalized over the dithiophene, thiophene and thiadiazole units of the structures as is shown in Fig. 5a and b. Contribution of alkane groups to the HOMO is negligible. In both cases the LUMO is highly localized on the thiadiazole units extending slightly over the linking thiophene units. The localization of the LUMO on the thiadiazole is more pronounced in the case of P2, compared to P1.
 | ||
| Fig. 5 HOMO and LUMO isosurfaces of the P1 and P2 monomers. | ||
The UV/visible absorption spectra, calculated at the DFT/B3LYP level of theory and using THF as a solvent, are shown in Fig. 6 for both the P1 and P2 monomers. The spectra have been produced by convoluting Gaussian functions with HWHM = 0.1 eV centered at the excitation energies. The P2 spectrum exhibits three clear bands with high intensities, centered at 1.2 (1033 nm), 2.8 (443 nm) and 3.6 eV (344 nm) respectively. All remaining peaks have significantly lower intensities. The most prominent difference between the two spectra is the existence of a gap between the first and second significant peaks in the spectrum of P2 which is about 1.6 eV (775 nm). The spectrum of the P1 monomer exhibits two well defined main bands centered at 2.0 (620 nm) and 3.0 eV (413 nm). A third band is also present which is much broader and has a fine structure of two lower intensity peaks centered at 3.8 eV (326 nm) and 4.2 (295 nm). Secondary peaks of low intensities are also found between the bands. However, the intensities are larger that the corresponding secondary peaks of the P2 spectrum.
 | ||
| Fig. 6 Theoretical UV/vis absorption spectra of the (a) P1 and (b) P2 monomers estimated from DFT. | ||
3.4 Electrical properties of the copolymers
We have investigated the current–voltage characteristics of the copolymers P1 and P2 to investigate their semiconductor nature, in the dark employing the ITO/PEDOT:PSS/P1 or P2/Al device structure and illustrated in Fig. 7. The J–V characteristics of the devices in the dark showed a rectification effect when positive potential was applied to the ITO/PEDOT:PSS electrode with respect to the Al electrode. Since the HOMO level of both copolymers (in the range of −5.02 to −5.24 eV) lies very close to the work function of PEDOT:PSS (−5.1 eV), it forms a nearly ohmic contact for hole injection from the ITO/PEDOT:PSS into the HOMO level of copolymer. However the LUMO level of the copolymers (in the range of −3.56 to −3.80 eV) is very far from the work function of the Al electrode (−4.2 eV) and they form a Schottky barrier for electron injection from Al into the LUMO level. Therefore the rectification effect observed in the devices is due to the formation of the Schottky barrier at the Al–copolymer interface which indicates that the copolymer behaves as a p-type organic semiconductor, and thus can be used as electron donors for the BHJ organic solar cells. We have also estimated the hole mobility of the pristine P1 and P2 from the current–voltage characteristics in the dark using the SCLC method and found them to be 6.62 × 10−5 cm2 V−1 s−1 and 2.14 × 10−6 cm2 V−1 s−1 for P1 and P2, respectively. We assume that the polymer solar cell based on P1 as electron donor in the BHJ active layer may achieve higher power conversion efficiency compared to its P2 counterpart. | ||
| Fig. 7 J–V characteristics of ITO/PEDOT:PSS/P1 or P2/Al devices in the dark at room temperature. | ||
3.5 Photovoltaic properties
The energy band diagrams of the copolymers along with fullerene derivatives are shown in Fig. 8. As can be seen from this figure, the LUMO offset for the P1:PCBM BHJ is higher than that for the P2:PCBM BHJ active layer, therefore we assume that the photo-induced charge transfer in the P1 based device is more efficient that for the P2 based device. | ||
| Fig. 8 Energy band diagrams of the P1, P2 and PCBMs. | ||
The bulk heterojunction PSCs were fabricated with a device structure ITO/PEDOT:PSS/P1 or P2:PC60BM (1:1 by wt)/Al to investigate the photovoltaic properties of the copolymers. Fig. 9 shows the typical current–voltage (J–V) characteristics of the devices under illumination of AM 1.5, 100 mW cm−2. The corresponding photovoltaic parameters such as short circuit current (Jsc), open circuit voltage (Voc), fill factor (FF) and overall power conversion efficiency (PCE) are summarized in Table 6. The BHJ photovoltaic device based on P1:PC60BM showed a Voc of 0.86 V, a Jsc of 5.26 mA cm−2 resulting in a PCE of 2.17% and FF of 0.48. Under the same conditions, the P2:PC60BM device exhibits a lower PCE of 0.80% with a Voc of 0.61 V, a Jsc of 3.28 mA cm−2, and FF of 0.40. The higher value of Voc for the device based on P1 is due to its low lying HOMO energy level, since Voc is directly related to the difference between HOMO energy level of donor and LUMO energy level of the acceptor.
 | ||
| Fig. 9
J–V characteristics of BHJ photovoltaic devices based on P1:PC60BM (1:1 wt ratio) and P2:PC60BM (1:1 wt ratio) blends cast from THF solvent, under illumination (100 mW cm−2). | ||
:1 w/w)
It was anticipated that copolymer P2 should show a high Jsc value, since it absorbed strongly in both visible and NIR regions, as confirmed by the absorption spectra. However, results gave a lower Jsc value than P1. One of the reasons for the low Jsc value was attributed to the strong electron accepting ability of BDT. The high electron withdrawing capability of the BDT would result in a low lying LUMO level, and thus excitons generated after the photo-excitation might not have enough driving force to allow the generated exciton to dissociate.34 Another possibility can be ascribed to the small LUMO–LUMO offset between P2 and PCBMs which would not provide enough driving force for the electron injection process to occur.
The solar cell based on polymer P2:PC60BM showed a low value of efficiency (0.80%). Despite the low band gap, the short-circuit current of the device based on this polymer was significantly lower than that for polymer P1. Jsc is defined as the product of the density of photoinduced charge carriers on their mobility in organic semiconductors. The mobility of charge carriers are related to the parameters of the device, and not to the photoactive material and in a more significant extent depends on film nano-morphology. The morphology, in turn, is affected by the conditions of solar cell manufacturing: solvent evaporation time, the temperature of the solution and application method. Probably, the smaller solubility of macromolecules P2 in THF (effective solvent for the formation of bulk heterojunction) was the reason which led to lower values of the characteristics of photovoltaic devices based on this polymer.
The solar cells’ morphology of the active layer plays an important role in improving the photovoltaic characteristics. We have measured the images of the surface morphology of the active layer P1:PC60BM and P2:PC60BM obtained by AFM. The rms roughness of the P1:PC60BM and P2:PC60BM films are 0.76 nm and 2.71 nm, respectively. The P1:PC60BM blend film has well-distributed small-sized domains with small roughness of 0.76 nm, suggesting that the polymer is suitably compatible with the PC60BM. Film P2:PC60BM shows relatively large domains and phase separation. The rough morphology and large size of the phase separation of the P2:PC60BM film does not promote effective separation of the excitons, leading to a lower short-circuit current. Nano-sized aggregated domains and the improved phase separation of the P1:PC60BM film contributes to a better separation of the charge and increase in PCE.
The PCE of the BHJ solar cells which are based on a blend of polymer P1 or P2 (donor) and PC60BM (acceptor) is quite low. To further improve the PCE, we have used PC70BM as the electron acceptor instead of PC60BM. PC70BM has a similar electronic structure and energy levels, but has considerably stronger absorption in the visible region.35 Therefore, the copolymer based BHJ polymer solar cells can be expected to exhibit improved performance when PC70BM is used in place of PC60BM as the acceptor due to the compensation of the poor absorption of the copolymer:PC60BM blend in the 400–550 nm range. We have fabricated the BHJ devices with the P1:PC70BM and P2:PC70BM blends and also investigated their photovoltaic properties. The J–V characteristics of these devices are shown in Fig. 10a and photovoltaic parameters are listed in Table 6. As shown in Fig. 10a and Table 6, both devices exhibit improvement in the PCE compared to the corresponding PC60BM based devices. The P1 and P2 devices show Voc values of 0.84 V and 0.62 V, Jsc values of 7.94 mA cm−2 and 4.48 mA cm−2, and FFs of 0.52 and 0.43, which results in PCEs of 3.45% and 1.19%, respectively. The enhancement in the PCE of the devices based on the PC70BM acceptor is mainly due to the increased value of Jsc. This increase is attributed to the strong absorption of PC70BM in the range of 400–550 nm, which is missing in the case of PC60BM. The increased absorption of PC70BM enhances the light harvesting efficiency when PC70BM is used instead of PC60BM, which results in further improved PCE. The Voc values in these device change slightly, since both PC60BM and PC70BM have almost the same LUMO energy level.
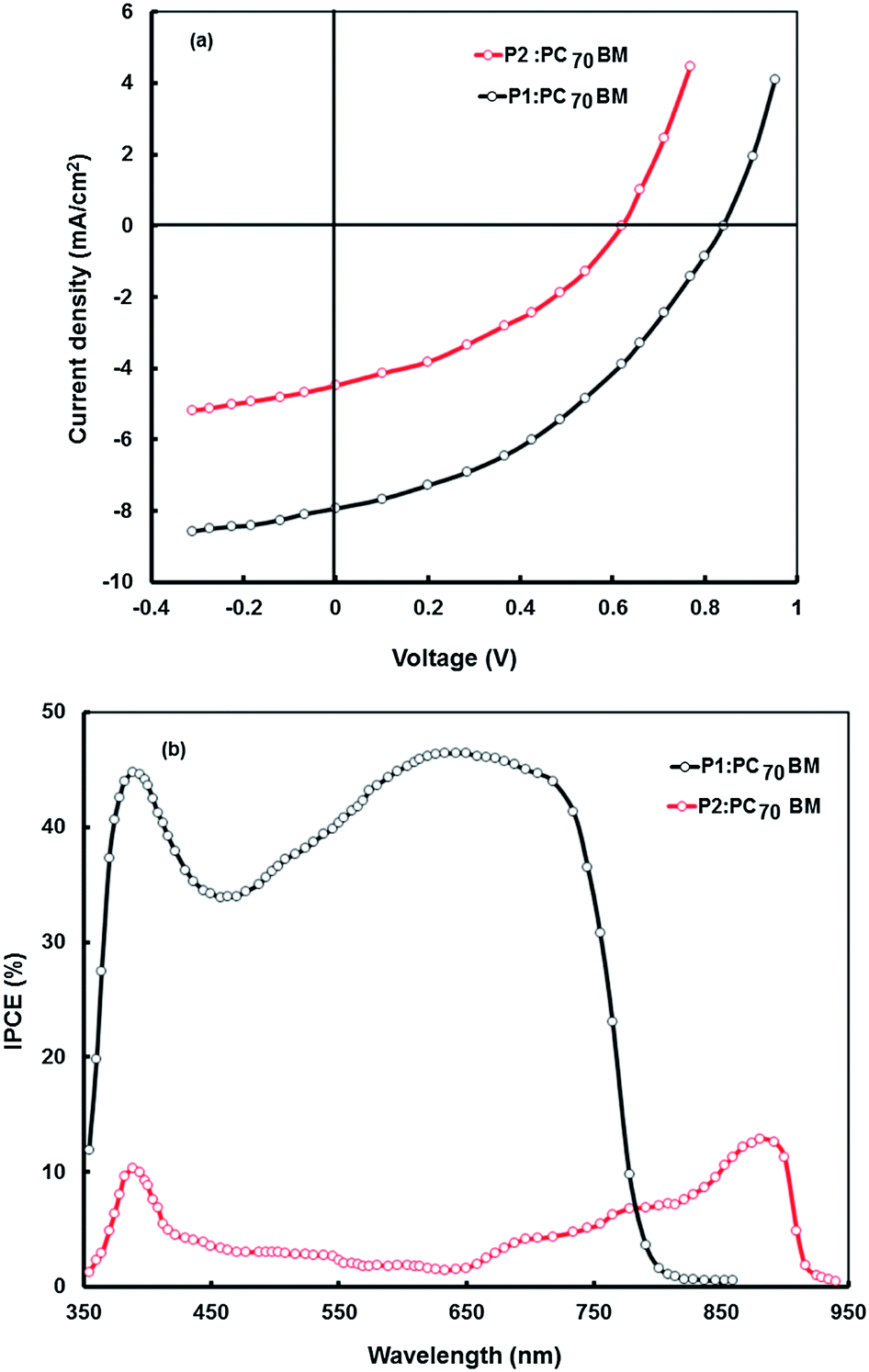 | ||
| Fig. 10 (a) J–V characteristics under illumination intensity of 100 mW cm−2 and (b) IPCE spectra of BHJ photovoltaic devices based on P1:PC70BM (1:2 wt ratio) and P2:PC70BM (1:2 wt ratio) blends cast from THF solvent. | ||
The IPCE values for the P1:PC70BM based solar are significantly higher than that for the P2:PC70BM counter part (Fig. 10b) indicating a more efficient generation and transport charge carrier in the P1:PC70BM BHJ active layer. Similar results were also observed for the BHJ solar cells with PC60BM as electron acceptor. The significant increase in the Jsc in the device based on P1 as electron donor in the BHJ active layer either with PC60BM or PC70BM as electron acceptor, is due to the high value of IPCE. The IPCE values for the P2:PC70BM device are relatively very small as compared to P1:PC70BM, although both P1 and P2 have a broad absorption spectral response, even P2 has a broader and low band gap. The difference in the values of IPCE can be ascribed to the higher value of LUMO level of P1, leading to a large LUMO–LUMO offset towards the PCBM and an increased driving force for exciton dissociation.
3.6 Effect of solvent additive on the photovoltaic response
The values of the PCE for these copolymers as donor and fullerene derivatives are still low. To improve the PCE of BHJ polymer solar cells, it is necessary to properly form continuous pathways in the active layer to transfer the electrons and holes towards the collection electrodes without leading to recombination. In addition, the morphology of the BHJ active layer plays an important role in determining the performance of the BHJ polymer solar cells, because charge carrier generation, recombination and transport are influenced by the spontaneously formed micro/nanostructure of the active layer during solution processing.36 The morphology of the active layer can be significantly improved through different processing methods to prepare the active layer i.e. thermal annealing36b or solvent annealing.36c For the P3HT based BHJ solar cells, the thermal annealing of the BHJ active layer has led to a remarkable enhancement in the PCE compared to that of its pristine film because P3HT consists of an ordered crystalline phase.36b,37 On the other hand, D–π–A type copolymer does not usually exhibit higher crystallinity than P3HT even after thermal annealing,38 therefore the thermal annealing method does not always produce the optimal intermolecular self organization in amorphous morphologies.39 As an alternative, Heeger et al. introduced a solvent mixture method in which the BHJ active layer was prepared through a mixture of two different solvents having different boiling points.40 The addition of small amounts of high boiling point additive into the host solvent for the preparation of BHJ active layer also improves the morphology of the active layer.37,41 In recent years, remarkable progress has been achieved for the BHJ polymer solar cells through the combination of D–A copolymers as electron donor and solvent additive42 and recently a PCE of 8.62% has been reported for BHJ polymer solar cells using this processing method.43 We have also prepared the P1:PC70BM and P2:PC70BM BHJ active layers by adding (1%, 3% and 5% by volume) 1,8-diiodooctane (DIO) into the host solvent. The optimized concentration was found to be 3% by volume. The current–voltage characteristics of P1:PC70BM and P2:PC70BM solar cells processed from the DIO/THF solvent are shown in Fig. 11a and the photovoltaic parameters are summarized in Table 6. The Jsc was increased from 4.48 to 6.05 mA cm−2 and from 7.94 to 10.52 mA cm−2 for the P2:PC70BM and P1:PC70BM based solar cells, respectively. Similarly the FF has been increased from 0.43 to 0.45 and from 0.52 to 0.60 for the P2:PC70BM and P1:PC70BM based solar cells, respectively. However, the Voc was slightly lowered for the devices processed with DIO/THF solvent. The increase in Jsc and FF leads to enhancement of the PCE from 1.19 to 1.58 and from 3.45 to 5.30% for the P2:PC70BM and P1:PC70BM based solar cells, respectively. To investigate the reason for the increase in Jsc with the addition of DIO in the host THF solvent for the processing of the BHJ film, we have measured the series resistance (Rs) of the device from the slope of the J–V characteristics under illumination near the open circuit voltage. We found that the value of Rs has been reduced from 6.4 Ω cm2 (THF processed) to 4.7 Ω cm2 (DIO/THF processed). The decrease in the Rs for the device processed from DIO/THF solvent as compared to that of THF indicates that the mobility of charge carriers increases. The higher IPCE value (as shown in Fig. 11b) of the device processed from DIO/THF than that processed from THF indicates an increased charge carrier generation and transportation and also supports the enhancement in the value of Jsc.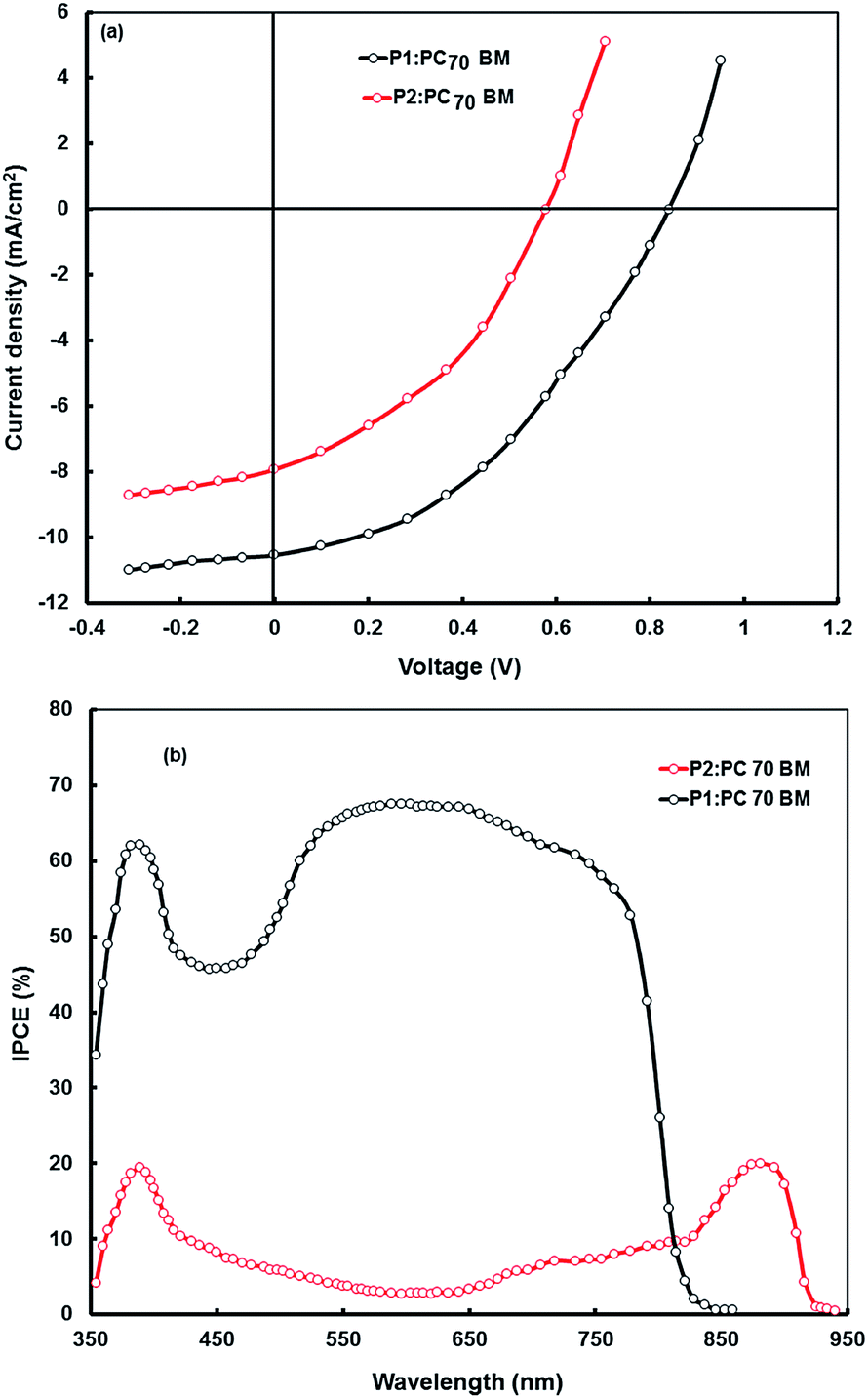 | ||
| Fig. 11 (a) J–V characteristics under illumination intensity of 100 mW cm−2 and (b) IPCE spectra of BHJ photovoltaic devices based on P1:PC70BM (1:2 wt ratio) and P2:PC70BM (1:2 wt ratio) blends cast from DIO/THF solvent. | ||
To acquire information about the increase in the PCE of the devices by adding the DIO as solvent additive, the effect of the additive on the absorption and morphology of the active layers was investigated. The UV-visible absorption spectra of the P1:PC70BM films with and without DIO are shown in Fig. 12. As shown in Fig. 12, the absorption of the blend film with DIO was increased, which could enhance the efficiency of charge carrier generation due to a better photon harvesting. It was already reported earlier that the additives were able to influence the polymer absorption profiles.37a,44 When 3% DIO was added, an increase in the absorbance over the wavelength region 500–750 nm was observed, because of the π–π transition among the copolymer chains. It contributes to the increase in the PCE compared to the device without processing additive. We assume that the crystallinity of P2:PC70BM blend cast from DIO/THF may be improved as compared to the blend cast from THF without DIO.
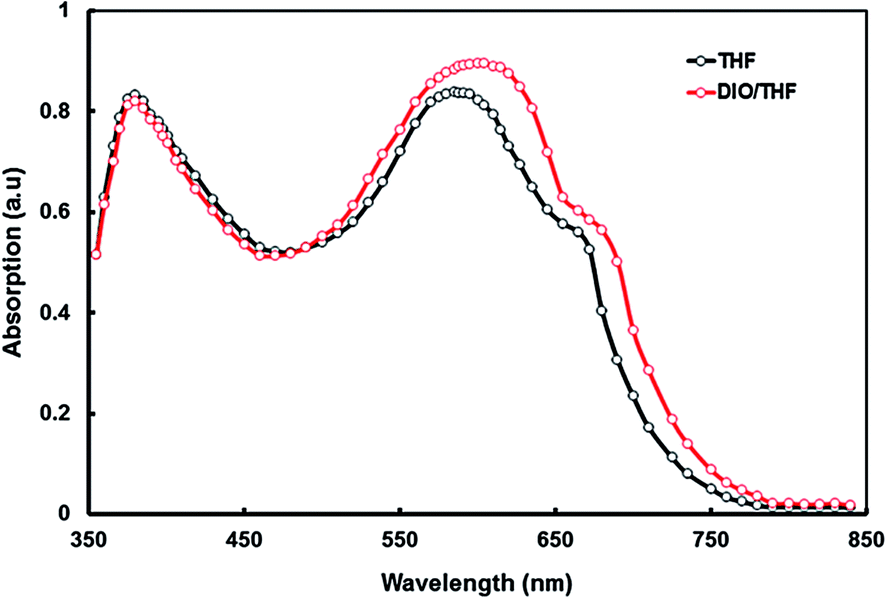 | ||
| Fig. 12 Normalized absorption spectra of P1:PC70BM thin films processed from THF and DIO/THF solvents. | ||
In order to examine the ordering of the copolymer chain by addition of DIO additive, XRD patterns were recorded and are shown in Fig. 13. The increase in the intensity of (100) diffraction peak was observed around 2θ = 7.58° in the P1:PC70BM blend film processed from DIO/THF compared to the blend processed from THF. The increase in the intensity means increase in the crystallinity in copolymer. Therefore addition of DIO as additive has an impact on the phase separation of copolymer and PC70BM, because PCBM selectively dissolved in the DIO. In addition, the DIO has a higher boiling point than the host THF solvent, PC70BM tends to remain in the solution mixture (during drying) longer than copolymer, thereby enabling control of the phase separation and morphology of the BHJ active layer. We have estimated the crystalline size (d) of copolymer (100) using the Scherrer's equation (d = kλ/Bcosθ), where B is the full width at half maximum (FWHM) of the (100) peak, λ is the wavelength of incident X-ray (0.154 nm), θ is the angle of refraction and k is Scherrer's constant (0.9). The crystalline size of the copolymer P1 was estimated at 11.2 nm and 13.4 nm for the film processed from THF and DIO/THF solvents, respectively. Increase of crystalline size in P1 leads to an increase in the hole mobility created in the BHJ active layer, as a result the Jsc and PCE of the device improves significantly.
 | ||
| Fig. 13 XRD patterns of P1:PC70BM active layers processed from THF and DIO/THF. | ||
Besides the absorption, charge carrier mobility and balance between the hole and electron mobilities are also crucial factors for achieving high PCE with BHJ organic solar cells. Both hole and electron mobility were calculated using the space charge limited current model45 using hole only and electron only devices, respectively. The J–V characteristics in the dark under forward bias were fitted with the model (Fig. 14a and b for hole and electron only devices using P1:PC70BM, respectively), described by the Mott–Gurney Law. We found about over an order of magnitude increase in the hole mobility (μh) for the device processed with DIO/THF as compared to that processed from THF, with a mobility increased from 1.7 × 10−5 cm2 V−1 s−1 to 3.6 × 10−4 cm2 V−1 s−1. However, the electron mobility (μe) increases from 4.8 × 10−4 to 5.3 × 10−4 cm2 V−1 s−1. Moreover, the device processed from DIO/THF shows relatively well balanced mobility (μe/μh = 1.47) compared to the device processed from THF (μe/μh = 28.23). The more balanced mobility contributes to higher Jsc and FF as observed in the device processed from DIO/THF solvent because the accumulated space charge limited current (SCLC) charges and hence recombination processes are reduced by the increase in the hole mobility and enhanced charge collection efficiency.46
 | ||
| Fig. 14 Current–voltage (J–V) characteristics of (a) hole only and (b) electron only devices, in the dark, Vapp is applied voltage and Vbi is built in potential. | ||
The nanoscale morphology plays an important role in the PCE of the BHJ organic solar cells. Proper morphology is essential not only for exciton dissociation but also for the charge transport to respective electrodes for their efficient collection.35b,47 An ideal domain size of 10–20 nm of copolymer and PCBM with an interpenetrating bi-continuous network is needed for achieving high PCE of BHJ polymer solar cells. However, both larger (>10–20 nm) and smaller (<10–20 nm) domain sizes of BHJ active layers may limit both charge transfer and separation. The morphology of the BHJ active layer processed from THF and DIO/THF was investigated through atomic force microscopy (AFM) images as shown in Fig. 15. From the height images we found that the root mean square (rms) of P1:PC70BM cast from THF (0.95 nm) was lower than that processed from DIO/THF (1.6 nm). The surface roughness indicates the self-organization ordering of the polymer. Therefore, the increase in surface roughness and highly ordered crystalline domain may effectively provide separate pathways for electrons and holes, and increase the Jsc. With the addition of DIO, the nanoscale domain can provide a pathway through which electrons and holes are separated after forming an interpenetrating network.
 | ||
| Fig. 15 AFM phase images (image area 3 μm × 3 μm) of P1:PC70BM blended thin films cast from (a) THF and (b) DIO/THF solvents. | ||
4. Conclusion
We have synthesized two novel D–A copolymers with different acceptor structures of 4,8-bis-(5-bromothiophene-2-yl)-benzo[1,2,5]thiadiazole (P1) and 4,8-dithiophene-2-yl-benzo[1,2-c;4,5-c′]-bis-[1,2,5]thiadiazole (P2) with a common BDT donor segment and investigated their optical and electrochemical properties. Both donor–acceptor copolymers exhibit good solubility in common organic solvents and a broad absorption from 350 nm to the near infrared region. The optical bands of P1 and P2 estimated from the onset absorption edge are 1.58 eV and 1.16 eV, respectively. These copolymers were used as electron donors along with the fullerene derivatives (PC60BM and PC70BM) as electron acceptors for the fabrication of BHJ polymer solar cells. Preliminary results show that the solar cells based on P1: PC60BM, P2:PC60BM, P1:PC70BM and P2:PC70BM exhibit PCEs of 2.17%, 0.80%, 3.45%, 1.19%, respectively. The higher value of PCE for P1 as electron donor has been attributed to its relatively high mobility and high open circuit voltage due to the deep lying HOMO level and high Jsc due to the large driving force for exciton dissociation at the D–A interface in the BHJ active layer. The PCE has been further improved up to 5.30% and 1.58% for P1:PC70BM and P2:PC70BM blends cast from DIO/THF solvent. This increase in the PCE is attributed to the improved crystallinity of copolymer phase, more balanced charge transport in the device and larger D–A interface in the active layer for exciton dissociation, when the blend was processed from the DIO/THF solvent. These results show that the new copolymers are useful materials for organic photovoltaic applications.Acknowledgements
This work was supported by the Presidium of the Russian Academy of Sciences under Program P-8: Polyfunctional Materials for Molecular Electronics; by the Ministry of Education and Science of the Russian Federation under State Contract agreement no. 8848; and by the Division of Chemistry and Materials Sciences, Russian Academy of Sciences, under programs of fundamental research OKh-3 (Creation and Study of Macromolecules and Macromolecular Structures of New Generations) and OKh-2 (Creation of New Metal, Ceramic, Glass, Polymer, and Composite Materials).References
- (a) S. Gunes, H. Neugebauer and N. S. Sariciftci, Conjugated polymer based organic solar cells, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed; (b) G. Dennler, M. C. Scharber and C. J. Brabes, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS; (c) A. Facchetti, π-conjugated polymers for organic electronics and photovoltaic cell applications, Chem. Mater., 2011, 23, 733–758 CrossRef CAS; (d) G. Li, R. Zhu and Y. Yang, Polymer solar cells, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS; (e) J. Y. Kim, K. Lee, N. E. Coates, D. Moses, T. Q. Nguyen, M. Dante and A. J. Heeger, Efficient tandem solar cells fabricated by all solution processing, Science, 2007, 317, 222–225 CrossRef CAS PubMed; (f) S. Sista, M. H. Park, Z. Hong, Y. Wu, J. Hou, W. L. Kwan, G. Li and Y. Yang, Highly efficient tandem polymer solar cells, Adv. Mater., 2010, 22, 380–383 CrossRef CAS PubMed; (g) D. J. Lipomi, B. C. K. Tee, M. Vosgueritchian and Z. Bao, Stretchable organic solar cells, Adv. Mater., 2011, 23, 1771–1775 CrossRef CAS PubMed; (h) R. Zhu, A. Kumar and Y. Yang, Polarizing organic photovoltaic, Adv. Mater., 2011, 23, 4193–4198 CrossRef CAS PubMed; (i) C. H. Peters, I. T. Sachs-Quintana, J. P. Kastrop, S. Beaupre, M. Leclerc and M. D. McGehee, High efficiency polymer solar cells with long operating times, Adv. Energy Mater., 2011, 1, 491–494 CrossRef CAS; (j) L. Dou, J. Jou, J. Yang, C. C. Chen, Y. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Tandem polymer solar cells featuring a spectrally matched low band gap polymer, Nat. Photonics, 2012, 6, 180–185 CrossRef CAS.
- (a) T. Chu, J. Lu, S. Beaupre, Y. Zhang, J. R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, Bulk heterojunction solar cells using thieno[3,4-c]pyrrole-4,6-dioneand dithieno[3,2-b:2′,3′-d] silole copolymer with a power conversion efficiency of 7.3%, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS PubMed; (b) J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Efficiency enhancement in low band gap polymer solar cells by processing with alkane dithiols, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed; (c) G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Polymer photovoltaic cells: enhanced efficiencies via a network of internal donor–acceptor heterojunctions, Science, 1995, 270, 1789–1791 CAS; (d) Y. F. Li and Y. P. Zou, Conjugated polymer photovoltaic materials with broad absorption band and high charge carrier mobility, Adv. Mater., 2008, 20, 2952–2958 CrossRef CAS; (e) J. Chen and Y. Cao, Development of novel conjugated donor polymers for high efficiency bulk heterojunction photovoltaic devices, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS PubMed.
- G. Li, V. Shrotriva, J. S. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, High efficiency solution processable polymer photovoltaic cells by self organization of polymer blends, Nat. Mater., 2005, 4, 864–868 CrossRef CAS.
- J. H. Hou, Z. A. Tan, Y. Yan, Y. J. He, C. H. Yang and Y. F. Li, Synthesis and photovoltaic properties of two dimensional conjugated polythiophene with bi(thienylenevinylene) side chains, J. Am. Chem. Soc., 2006, 128, 4911–4916 CrossRef CAS PubMed.
- H. Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Polymer solar cells with enhanced open circuit voltage and efficiency, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS.
- (a) R. Kroon, M. Lenes, J. C. Hummelen, P. W. M. Blom and B. Boer, Small band gap polymers for organic solar cells (polymer materials development in the last five years), Polym. Rev., 2008, 48, 531–582 CrossRef CAS; (b) Z. G. Zhang, K. L. Zhang, G. Liu, C. X. Zhu, K. G. Neoh and E. T. Kang, Triphenylamine-fluorene alternating conjugated copolymers with pedant acceptors groups: synthesis, structure–property relationship and photovoltaic application, Macromolecules, 2009, 42, 3104–3111 CrossRef CAS; (c) H. Zhou, L. Yang and W. You, Rational design of high performance conjugated polymers for organic solar cells, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- (a) A. Najari, S. Beaupre, P. Berrouard, Y. Zou, J. R. Pouliot, C. Lepage-Perusse and M. Leclerc, Synthesis and characterization of new thieno[3,4-c]pyrrole-4,6-dione derivatives for photovoltaic applications, Adv. Funct. Mater., 2011, 21, 718–728 CrossRef CAS; (b) C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Frechet, Synthetic control of structural order in N-alkylthieno[3,4-c]pyrrole-4,6 dione based polymers for efficient solar cells, J. Am. Chem. Soc., 2010, 132, 7595–7597 CrossRef CAS PubMed; (c) Y. Liang, Z. Xu, J. Xia, S. T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, For the bright future – bulk heterojunction polymer solar cells with power conversion efficiencies of 7.4%, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed; (d) J. M. Jiang, M. C. Yuan, K. Dinakaran, A. Hariharan and K.-H. Wei, Crystalline donor–acceptor conjugated polymers for bulk heterojunction photovoltaics, J. Mater. Chem. A, 2013, 1, 4415–4422 RSC.
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta and E. D. Dunlop, Solar cells efficiency tables (version 39), Prog. Photovoltaics, 2012, 20, 12–20 Search PubMed.
- (a) L. J. Huo, X. Guo, S. Zhang, Y. F. Li and J. Hou, PBDTTTZ: A broad band gap conjugated polymer with high photovoltaic performance in polymer solar cells, Macromolecules, 2011, 44, 4035–4037 CrossRef CAS; (b) N. Allard, S. Beaupre, B. R. Aich, A. Najari, Y. Tao and M. Leclerc, Synthesis and characterization of new poly(thio[3,4-d]thiazole) derivatives for photovoltaic applications, Macromolecules, 2011, 44, 7184–7187 CrossRef CAS; (c) Y. Liang and L. Yu, A new class of semiconducting polymers for bulk heterojunction solar cells with exceptionally high performance, Acc. Chem. Res., 2010, 43, 1227–1236 CrossRef CAS PubMed.
- H. Pan, Y. Li, Y. Wu, P. Liu, B. S. Ong, S. Zhu and G. Xu, Low temperature, solution processed, high mobility polymer semiconductors for thin films, J. Am. Chem. Soc., 2007, 129, 4112–4113 CrossRef CAS PubMed.
- C. Kitamura, S. Tanaka and Y. Yamashita, Chem. Mater., 1996, 8, 570–578 CrossRef CAS.
- Q. Hou, Y. Xu, W. Yang, M. Yuan, J. Peng and Y. Cao, Novel red emitting fluorene based copolymers, J. Mater. Chem., 2002, 12, 2887–2892 RSC.
- E. Perzon, X. Wang, S. Admassie, O. Inganäs and M. R. Andersson, An alternative low band gap polyfluorene for optoelectronic devices, Polymer, 2006, 47, 4261–4268 CrossRef CAS PubMed.
- M. Karikomi, C. Kitamura, S. Tanaka and Y. Yamashita, New narrow band gap polymer composed of benzobis(1,2,5-thiazole) and thiophenes, J. Am. Chem. Soc., 1995, 117, 6791–6792 CrossRef CAS.
- E. Bundgaard and F. C. Krebs, Low band gap conjugated polymers based on thiophene, benzothiadiazole, and benzobis(thiadiazole), Macromolecules, 2006, 39, 2823–2831 CrossRef CAS.
- Y. Liang, D. Feng, Y. Wu, S. T. Tsai, G. Li, C. Ray and L. Yu, High efficient solar cell polymer developed via fine tuning of structural and electronic properties, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- (a) C. Adamo and V. Barone, Quantum Monte Carlo calculations for a large numbers of Bosons in a harmonic trap, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef; (b) A. D. Becke, Density functional thermochemistry III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, On certain integrals useful in molecular orbital calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–89 CrossRef CAS.
- D. Jacquemin, E. A. Perpète, G. E. Scuseria, I. Ciofini and C. Adamo, TD-DFT performance for the visible absorption spectra of organic dye: convention versus long range hybrids, J. Chem. Theory Comput., 2008, 4, 123–135 CrossRef CAS.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Auxiliary basis sets to appropriate coulomb potentials, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, Fully optimized contracted Gaussian basis sets for triple zeta valence quality for atoms Li to Kr, J. Chem. Phys., 1994, 100, 5829 CrossRef.
- G. Scalmani and M. J. Frisch, Continuous surface charge polarizable continuum models of salvation. I General formalism, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, Combined experimental and DFT-TDDFT computational study of photoelectrochemical cell ruthenium sensitizers, J. Am. Chem. Soc., 2005, 127, 16835–16847 CrossRef CAS PubMed.
- S. Fantacci, F. De Angelis and A. Selloni, Absorption spectrum and solvatochromism of [Ru(4,4′-COOH-2,2′bpy)2(NCS)2] molecular dye by time dependent density functional theory, J. Am. Chem. Soc., 2003, 125, 4381–4387 CrossRef CAS PubMed.
- F. De Angelis, S. Fantacci and A. Selloni, Time dependent density functional theory study of the absorption spectrum of [Ru(4,4′-COOH-2,2′bpy)2(NCS)2] in water solution: Influence of pH, Chem. Phys. Lett., 2004, 389, 204–208 CrossRef CAS PubMed.
- S. Fantacci, F. De Angelis, J. Wang, S. Bernhard and A. Selloni, A combined computational and experimental study of polynuclear Ru-TPPZ complexes: Insight into the electronic and optical properties of coordination polymers, J. Am. Chem. Soc., 2004, 126, 9715–9723 CrossRef CAS PubMed.
- F. De Angelis, A. Tilocca and A. Selloni, Time dependent DFT study of [Fe(CN)6]4+ sensitization of TiO2 nanoparticles, J. Am. Chem. Soc., 2004, 126, 15024–15025 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 03, revision C.01, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- TURBOMOLE (version 5.6), Universitat Karlsruhe, 2000 Search PubMed.
- (a) J. Roncalli, Synthetic principles for low bandgap control in linear π-conjugated systems, Chem. Rev., 1997, 97, 173–205 CrossRef PubMed; (b) R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Streamlined microwave assisted preparation of narrow bandgap conjugated polymers for high performance bulk heterojunction solar cells, Nat. Chem., 2009, 1, 657–661 CrossRef CAS PubMed; (c) H. Y. Chen, J. H. Hou, A. E. Hayden, H. Yang, K. N. Houk and Y. Yang, Silicon atom substitution enhances interchain packing in a thiophene based polymer system, Adv. Mater., 2010, 22, 371–375 CrossRef CAS PubMed; (d) Q. Peng, X. J. Liu, Y. C. Qin, M. J. Li, J. Xu, G. W. Fu and L. M. Dai, Pyrazino[2,3-g] quinoxaline based conjugated copolymers with indolocarbazole coplanar moieties designed for efficient photovoltaic applications, J. Mater. Chem., 2011, 21, 7714–7722 RSC; (e) X. H. Zhou, L. Q. Yang and W. You, Rational design for high performance conjugated polymers for organic solar cells, Macromolecules, 2012, 45, 607–632 CrossRef.
- Q. Peng, K. Park, T. Lin, M. Durstock and L. M. Dai, Donor–π–acceptor conjugated copolymers for photovoltaic applications: tuning the open circuit voltage by adjusting the donor/acceptor ratio, J. Phys. Chem. B, 2008, 112, 2801–2808 CrossRef CAS PubMed.
- (a) Y. Yang, R. T. Farley, T. T. Steckler, S. H. Eom, J. R. Reynolds, K. S. Schanze and J. Xue, Efficient near infrared organic light emitting devices based on low gap fluorescent oligomers, J. Appl. Phys., 2009, 106, 044509–044515 CrossRef; (b) T. T. Steckler, K. A. Abboud, M. Craps, A. G. Rinzler and J. R. Reynolds, Low bandgap EDOT-benzobis(thiadiazole) hybrid polymer characterized on near IT transmissive single walled carbon nanotube electrodes, Chem. Commun., 2007, 4904–4906 RSC; (c) M. Karikomi, C. Kitamura, S. Tanaka and Y. Yamashita, New narrow bandgap polymer composed of benzobis(1,2,5-thiadiazole) and thiophenes, J. Am. Chem. Soc., 1995, 117, 6791–6792 CrossRef CAS.
- (a) P. Peuman, V. Bulovic and S. R. Forrest, Efficient photon harvesting at high optical intensities in ultra-thin organic double hetero-structure photovoltaic diodes, Appl. Phys. Lett., 2000, 76, 2650 CrossRef; (b) H. Ohkita, S. Cook, Y. Asututi, W. Duffy, S. Tierney, W. Zhang, M. Heeny, I. McCulloch, J. Nelson, D. D. C. Bradley and J. R. Durrant, Charge carrier formation in polythiophene/fullerene blend films studied by transient absorption spectroscopy, J. Am. Chem. Soc., 2008, 130, 3030–3042 CrossRef CAS PubMed.
- M. M. Wienk, J. M. Kroon, W. J. H. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. J. Janssen, Efficient methano[70] fullerene/MDMo-PPV bulk heterojunction photovoltaic cells, Angew. Chem., 2003, 115, 3493–3497 CrossRef.
- (a) W. W. Li, Y. Zhou, B. V. Andersson, L. M. Andersson, Y. Thomann, C. Veit, K. Tvingstedt, R. P. Qin, Z. S. Bo, O. Inganäs, U. Würfel and F. L. Zhang, The effect of additive on performance and self assembly of HSX-1/PCBM photovoltaic devices, Org. Electron., 2011, 12, 1544–1551 CrossRef CAS PubMed; (b) W. L. Ma, C. Y. Yang, X. Gong, K. Lee and A. J. Heeger, Thermally stable, efficient polymer solar cells with nanoscale control of the interpenetrating network morphology, Adv. Funct. Mater., 2012, 15, 1617–1622 CrossRef; (c) S. S. Bavel, M. Barenklau, G. With, H. Hoppe and J. Loos, P3HT/PCBM bulk heterojunction solar cells: impact of blend composition and 3D morphology on device performance, Adv. Funct. Mater., 2010, 20, 1458–1463 CrossRef; (d) J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Efficiency enhancement in low bandgap polymer solar cells by processing with alkane dithiols, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed; (e) M. S. Su, C. Y. Kuo, M. C. Yuan, U. S. Jeng, C. J. Su and K. H. Wei, Improving device efficiency of polymer/fullerene bulk heterojunction solar cells through enhanced crystallinity and reduced grain boundaries induced by solvent effect, Adv. Mater., 2011, 23, 3315–3319 CrossRef CAS PubMed.
- (a) J. Y. Kim, S. H. Kim, H. H. Lee, K. Lee, W. Ma, X. Gong and A. J. Heeger, New architecture for high efficiency polymer photovoltaic cells using solution based titanium oxide as an optical spacer, Adv. Mater., 2006, 18, 572–576 CrossRef CAS; (b) F. Padinger, R. S. Rittberger and N. S. Sariciftci, Effect of postproduction treatment on plastic solar cells, Adv. Funct. Mater., 2003, 13, 85–88 CrossRef CAS; (c) C. R. McNeill, J. J. M. Halls, R. Wilson, G. L. Whiting, S. Berkebile, M. G. Ramsey, R. H. Friend and N. C. Greenham, Efficient polythiophene/polyfluorene copolymer bulk heterojunction photovoltaic devices: device physics and annealing effects, Adv. Funct. Mater., 2008, 18, 2309–2321 CrossRef CAS.
- (a) F. Liu, Y. Gu, J. W. Jung, W. H. Jo and T. P. Russell, On the morphology of polymer based photovoltaics, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1018–1044 CrossRef CAS; (b) C. Shim, M. Kim, S. G. Ihn, Y. S. Choi, Y. Kim and K. Cho, Controlled nanomorphology of PCDTBT-fullerene blends via polymer end group functionalization for high efficiency organic solar cells, Chem. Commun., 2012, 48, 7206–7208 RSC; (c) T. Wang, A. J. Pearson, A. D. F. Dunbar, P. A. Staniec, D. C. Watters, H. Yi, A. J. Ryan, R. A. L. Jones, A. Iraqi and D. G. Lidzey, Correlating structure with function in thermally annealed PCDTBT:PC70BM photovoltaic blends, Adv. Funct. Mater., 2012, 22, 1399–1408 CrossRef CAS.
- (a) H. Cha, H. Kong, D. S. Chung, W. M. Yun, T. K. An, J. Hwang, Y. H. Kim, H. K. Shim and C. E. Park, Thermally stable amorphous polymeric semiconductors containing fluorene and thiophene for use in organic solar cells, Org. Electron., 2010, 11, 1534–1542 CrossRef CAS PubMed; (b) H. Cha, J. W. Park, D. S. Chung, T. K. An, Y. H. Kim, S. K. Kwon and C. E. Park, A side chain modified quarterthiophene derivative for enhancing the performance of organic solar cells, J. Mater. Chem., 2012, 22, 15141–15145 RSC; (c) B. S. Ong, Y. Wu, P. Liu and S. Gardner, High performance semiconducting polythiophenes for organic thin films, J. Am. Chem. Soc., 2004, 126, 3378–3379 CrossRef CAS PubMed; (d) N. Blouin, A. Michaud and M. Leclerc, A low band gap poly(2,7-carbazole) derivative for use in high performance solar cells, Adv. Mater., 2007, 19, 2295–2300 CrossRef CAS.
- F. Zhang, K. G. Jespersen, C. Bjorstrom, M. Svensson, M. R. Andersson, V. Sundstrom, K. Magnusson, E. Moons, A. Yartsev and O. Inganas, Influence of solvent mixing on the morphology and performance of solar cells based on polyfluorene copolymer/fullerene blends, Adv. Funct. Mater., 2006, 16, 667–674 CrossRef CAS.
- J. K. Lee, W. L. Ma, C. J. Brabec, J. Yuen, J. S. Moon, J. Y. Kim, K. Lee, G. C. Bazan and A. J. Heeger, Processing additives for improved efficiency from bulk heterojunction solar cells, J. Am. Chem. Soc., 2008, 130, 3619 CrossRef CAS PubMed.
- (a) F. Huang, K. S. Chen, H. L. Yip, S. K. Hau, O. Acton, Y. Zhang, J. D. Luo and A. K. Y. Jen, Development of new conjugated polymers with donor–π–acceptor side chains for high performance solar cells, J. Am. Chem. Soc., 2009, 131, 13886–13887 CrossRef CAS PubMed; (b) J. H. Hou, H. Y. Chen, S. Q. Zhang, R. I. Chen, Y. Yang, Y. Wu and G. Li, Synthesis of a low band gap polymer and its application in highly efficient polymer solar cells, J. Am. Chem. Soc., 2009, 131, 15586–15587 CrossRef CAS PubMed; (c) S. H. Chan, Y. S. Hsiao, L. I. Hung, G. W. Hwang, H. L. Chen, C. Ting and C. P. Chen, Morphology evolution of spin coated films of poly (thiophene–phenylene–thiophene) and [6,6]-phenyl-C71-butyric acid methyl ester by solvent effect, Macromolecules, 2010, 43, 3399–3405 CrossRef CAS.
- N. Ni, B. Qu, D. Tian, Z. Cong, W. Wang, C. Gao, L. Xiao, Z. Chen, Q. Gong and W. Wei, The effect of additive on the performance and phase separation of benzo[1,2-b:3,4-b′]dithiophene based polymer heterojunction photovoltaic devices, Macromol. Chem. Phys., 2013, 214, 985–993 CrossRef CAS.
- L. Dou, J. B. You, J. Yang, C. C. Chen, Y. J. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Tandem polymer solar cell featuring a spectrally matched low bandgap polymer, Nat. Photonics, 2012, 6, 180–185 CrossRef CAS.
- (a) V. D. Mihailetchi, L. J. A. Koster, P. W. M. Blom, C. Melzer, B. De Boer, J. K. J. vanDuren and R. A. J. Janssen, Compositional dependance of the performance of poly(p-phenylene vinylene):methanofullerene bulk heterojunction solar cells, Adv. Funct. Mater., 2005, 15, 795–801 CrossRef CAS; (b) V. D. Mihailetchi, H. Xie, B. deBoer, L. J. A. Koster and P. W. M. Blom, Charge transport and photocurrent generation in poly(3-hexylthiophene):methanofullerne bulk heterojunction solar cells, Adv. Funct. Mater., 2006, 16, 699–708 CrossRef CAS.
- (a) L. J. A. Koster, S. E. Shaheen and J. C. Hummelen, Pathways to a new regime for organic solar cells, Adv. Energy Mater., 2012, 2, 1246–1253 CrossRef CAS; (b) R. A. J. Janssen and J. Nelson, Factors Limiting device efficiency in organic photovoltaic devices, Adv. Mater., 2013, 25, 1847–1858 CrossRef CAS PubMed.
- W. L. Leong, S. R. Cowan and A. J. Heeger, Differential Resistance Analysis of Charge Carrier Losses in Organic Bulk Heterojunction Solar Cells: Observing the Transition from Bimolecular to Trap-Assisted Recombination and Quantifying the Order of Recombination, Adv. Energy Mater., 2011, 1, 517–522 CrossRef CAS.
- G. Li, V. Shrotriya, J. S. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, High-efficiency solution processable polymer photovoltaic cells by self-organization of polymer blends, Nat. Mater., 2005, 4, 864–868 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |