 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synthesis of metallic and semiconducting nanoparticles from reactive melts of precursors
Edward
Lewis
a,
Sarah
Haigh
a and
Paul
O'Brien
*b
aSchool of Materials, University of Manchester, Manchester M13 9PL, UK. E-mail: Paul.O'Brien@manchester.ac.uk
bSchools of Materials and of Chemistry, University of Manchester, Manchester M13 9PL, UK
First published on 9th October 2013
Abstract
The decomposition of melts of single-source precursors has yielded a range of interesting nanocrystals. The metal and semiconductor nanomaterials produced often display very exotic morphologies. These solventless, self-capping reactions are exceedingly simple and potentially scalable. In this review we survey all examples known to us of this approach to nanoparticle synthesis.
 Edward Lewis | Edward Lewis received his MChem degree from the University of Oxford in 2011. He is working towards a PhD in nanoscience at the University of Manchester under the supervision of Sarah Haigh and Paul O'Brien. His research involves the development of techniques for in situ observation of nanoparticle growth. |
 Sarah Haigh | Sarah Haigh is a Lecturer in Material Science at the University of Manchester. She received her MEng degree in Material Science from the University of Oxford in 2004 and her D.Phil degree in Advanced Microscopy in 2008 in the same department working under the supervision of Prof. Angus Kirkland and Prof. John Hutchison. She was a postdoctoral research fellow working with JEOL UK until moving to the University of Manchester in 2010. Her current research focuses on using high resolution imaging and elemental analysis to study a range of novel materials systems, including graphene based high mobility transistors, mixed-metal nanoparticle catalysts and high performance alloys. In 2013 she was awarded the Institute of Materials, Minerals and Mining Silver Medal for outstanding contribution to the broad field of materials science. |
 Paul O'Brien | Paul O'Brien is Chair of Inorganic Materials Chemistry in the Schools of Chemistry and of Materials at the University of Manchester; he was Research Dean 2000–2003, Head of the School of Chemistry 2002–09 and is now Head of the School of Materials 2011. He held academic positions at Chelsea, Queen Mary and Imperial Colleges in the University of London. External appointments have included: Vice President of the RSC 2009–11, visiting Professor at Georgia Tech 1996–99, visiting Fellow Magdalen College Oxford 2009 and Distinguished Fellow at the Institute for Advanced Studies Durham 2011. He has received distinguished alumni awards: the Potts Medal (Liverpool) and the A. G. Evans Memorial Medal Lecture (Cardiff). He was also awarded the Kroll and Sir Colin Humphreys Medals of the IOMMM and the first Peter Day Award from the RSC. His research centres on developing new chemical processes for thin films and nanoparticles; especially of chalcogenides. He holds honorary D.Scs. from both the Universities of Zululand and Liverpool. In 2002 he founded Nanoco, now listed on AIM/LSE. In May 2013 he was elected a Fellow of the Royal Society London. |
1. Introduction
This review aims to highlight a promising class of reactions that potentially provide a simple and scalable route to nanomaterials. These processes, which we shall refer to as melt reactions, are solventless reactions carried out by heating a precursor, often under an inert atmosphere, to bring about its decomposition. The melt approach has been successful in preparing a broad range of nanomaterials including metals, metal oxides, and metal chalcogenides. A wide range of morphologies including spheres,1,2 nanorods,3,4 nanowires,5–7 nanofabrics,5 and nanodisks3,8,9 have been prepared using this process.The dominant chemical approach to nanoparticle synthesis is currently hot-injection.10 There is also considerable interest in heating-up reactions, which have attracted attention because of their scalability.11 In comparison, melt reactions are simpler and potentially easier to scale up than most other routes. Melt reactions are solventless: this gives the melt approach a potential economic and environmental advantage over both hot-injection and heating-up approaches which use high boiling point solvents and precursors.
An interesting feature of these reactions is that they are usually “self capping”. Capping ligands play an important role in nanoparticle synthesis: passivating the particle's surface, controlling the rate of growth and, in some cases, influencing the shape of particles formed. In a typical hot-injection synthesis the solvent into which the precursor is injected acts as the capping ligand. In melt reactions, ligands produced during the precursor's decomposition can cap the nanoparticles. Removing the need for solvents has both economic and environmental benefits.
Another interesting feature of melt reactions is the proposed relationship between precursor structure and nanoproduct shape, most convincingly observed in the thermolysis of layered thiolate precursors.8,12,13 This approach is a relatively new, and largely unexplored, way to control nanocrystal shape. Some researchers also claim that solventless methods reduce interparticle collisions, ensuring growth occurs solely by monomer addition.3,6 Consequently, this approach has the potential to give high quality monodisperse products.3,6
A wide range of single-source precursors have already been developed for use in metal–organic chemical vapour deposition (MOCVD) and hot-injection syntheses.14,15 While some of these precursors have been used in melt reactions, a huge number of promising molecules have yet to be investigated. Fig. 1 shows the structures of some of the ligands that have been used as precursors in melt reactions. We hope that this review will stimulate further investigations into this class of reactions. There is great potential to explore a more diverse range of precursor chemistry and gain a better understanding of how the reaction conditions and precursor structure influence the size, shape, and composition of the nanoproducts. Ultimately this approach could prove to be commercially important, providing a simple, cheap, and scalable way to mass produce nanoparticles.
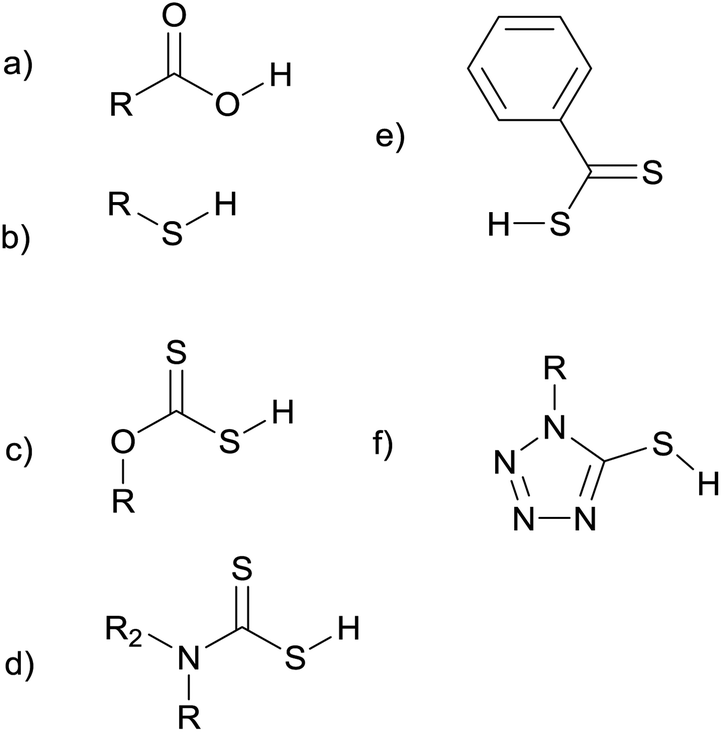 | ||
| Fig. 1 Some of the ligands used as precursors in melt reactions: carboxylic acids (a), thiols (b) xanthates (c), dithiocarbamates (d), dithiocarboxylates (e), and thiotetrazoles (f). | ||
This review aims to summarise all the examples to date of solventless nanoparticle synthesis using single-source self-capping precursors. The review is divided into two parts; the first concerning elemental (metals) and the second compound materials such as sulfides or oxides.
2. Metals
2.1 Metal nanocrystals from the salts of carboxylic acids
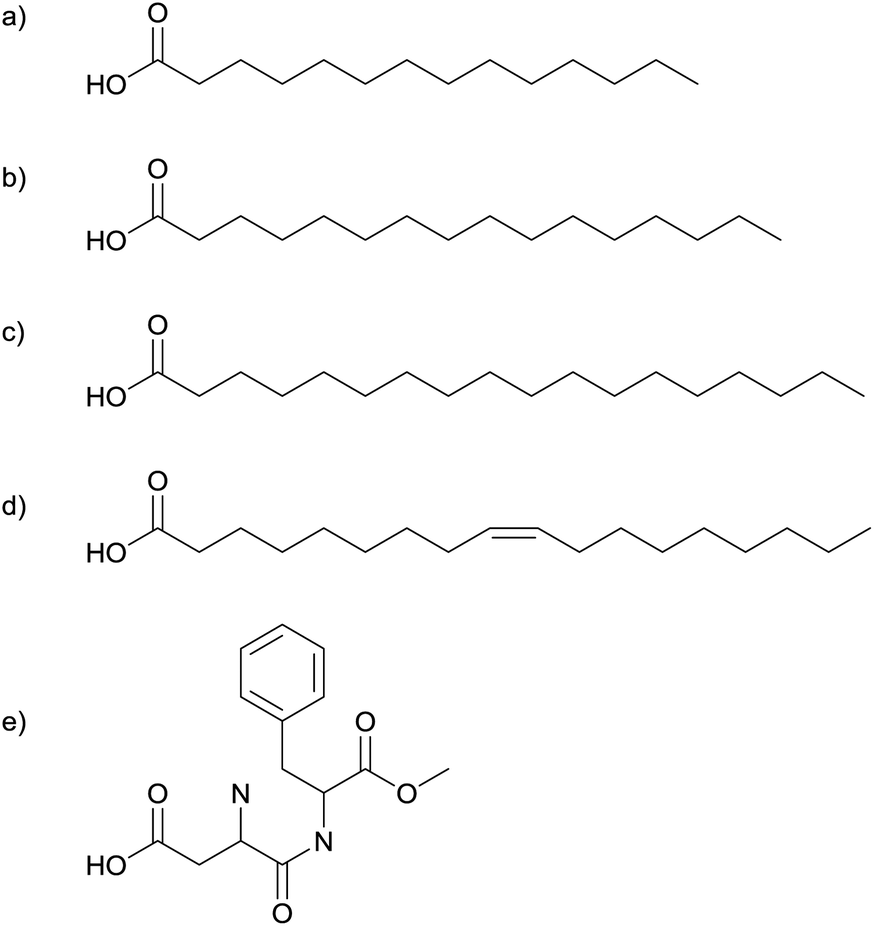 | ||
| Fig. 2 Myristic- (a), palmitic- (b), stearic- (c) and oleic-acid (d), and aspartame (e) have been used to synthesise silver nanoparticles. | ||
The spherical silver nanoparticles were assembled into hexagonally close packed two-dimensional arrays by spreading a toluene suspension of the nanoparticles on a water surface.16 The silver core size was found to be independent of the organic acid used. The separation between nanoparticles was dependent on the alkyl chain length; longer chains resulted in larger gaps between particles. This correlation supports the particles being self-capped with the precursor fatty acids.1,16
Further investigation of the silver nanoparticles formed by heating the silver salts of myristic-, stearic-, and oleic-acid under nitrogen at 250 °C for 1–2 hours showed that the products from all three precursors were silver nanoparticles with a mean diameter of 4.7 ± 0.6 nm.17 High resolution TEM imaging revealed that only ca. 2% of the particles were single crystals and the majority contained multiple twin defects.17 The fatty acid used in the precursor caps the nanoparticles, preventing aggregation. It was found that these capping ligands could be removed by heating at 210 °C in air. Thin films of silver can be produced by coating a surface with these nanoparticles and heating it to 250 °C.17
In a similar reaction, Lee et al. heated the silver salt of oleic acid (Fig. 2d), to produce silver nanoparticles.18,19 However, their experiments were carried out at low pressures: they transferred the dry silver complex to a Pyrex tube, flushed it with nitrogen and then sealed it at 0.3 Torr. The temperature was gradually increased at 2 °C min−1 and then maintained at 290 °C for an hour before cooling back to room temperature.18 Thermogravimetric analysis (TGA) shows that the silver–oleate decomposes at 287 °C.18 Decomposition is believed to occur by homolytic cleavage of the metal–carboxylate bond, forming free radicals.19 The oleate molecules act as capping ligands, hindering addition to the surface, causing slow steady growth. TEM images show a very narrow size distribution and interparticle separations of 2 nm.18
This separation is significantly less than twice the approximate length of the oleate ligand (1.75 nm) suggesting interdigitation. Energy dispersive X-ray spectroscopy (EDX) confirmed that the nanocrystals were pure silver.18
Yang et al. produced a series of fatty acid stabilized silver nanoparticles with different length alkyl chains.20 The particles were synthesised by heating the dry silver salts of fatty acids at 250 °C in a nitrogen atmosphere for 2 hours. A series of six acids were used: –OOC–(CH2)m−2–CH3 (m = 8, 10, 12, 14, 16, and 18). In all cases the spherical silver nanoparticles produced were found to be 4.7 ± 0.6 nm in diameter.20
The silver salt of aspartame (Fig. 2e) has also been used in the solventless synthesis of silver nanoparticles.21 Heating in a furnace under a nitrogen atmosphere produces spherical silver nanoparticles. Particle size depends on the temperature used; 185 °C for 10 minutes gave 5 ± 2 nm particles whilst 200 °C for 10 minutes gave 45 ± 10 nm particles. TGA reveals a mass loss starting at 185 °C and ending at 200 °C, infra red spectroscopy (IR) reveals that this process corresponds to the loss of the ester group. It is believed that the formaldehyde molecule released forms H2 and CO, two strongly reducing molecules, and these species are key in reducing Ag(I) to Ag(0). The larger particle size at 200 °C is explained as a consequence of the higher concentration of these reducing molecules.21
Silver–carboxylate decomposition in the presence of an amine has also been investigated.22,23 Silver–myristate (Fig. 2a) in triethylamine (NEt3) decomposes at the very low temperature of 80 °C to produce highly monodisperse silver nanoparticles, 4.4 ± 0.2 nm in diameter. The precursor is insoluble in the tertiary amine. It is proposed that an intermediate silver–myristate–amine adduct forms and this intermediate undergoes rapid thermal decomposition, forming myristate capped silver nanoparticles dispersed in the amine. Gas chromatography and mass spectrometry confirm that the silver nanoparticles formed are capped only with myristate and not the amine, the interparticle separations seen in TEM images also lend support to the view that the particles are myristate capped.22 All other reactions described in this review involve a solid precursor being heated in a furnace to produce nanoparticles. At first glance this reaction appears to be different as it was performed in a liquid. However, the liquid amine acts neither as a solvent for the precursor, nor as a capping agent for the nanoparticles formed. The myristate is a solid that undergoes solventless thermolysis producing nanoparticles capped by groups originating solely from the precursor. The amine's only role is that of a catalyst, making a reaction that was previously performed at 250 °C take place at 80 °C.22
The approach to silver nanoparticle synthesis described in this section may find applications in the electronics industry. The European Union's waste electrical and electronic equipment (WEEE) legislation banned the use of lead in consumer electronics.24 This legislation prompted a shift in the electronics industry from the use of tin/lead solders towards lead-free alternatives.24 Electrically conductive adhesives (ECAs), consisting of a polymer resin combined with a metal filler, are one alternative to lead based solders.25 Silver nanoparticles have been incorporated into such ECAs, and the low temperature sintering of Ag nanoparticles can lead to highly conductive adhesives.26,27
The silver salts of fatty acids are known to form silver nanoparticles in melt reactions.1 These reactions have been used in the development of new lead-free solders.28,29 When using conductive epoxy based adhesives in printed electronics, high conductivity and short sintering times are both desirable properties. Wong et al. describe the sintering of a composite containing silver flakes.28 During sintering silver carboxylates on the surface of the flakes decomposes to form silver nanoparticles. These nanoparticles sinter at temperatures well below the bulk melting point of silver, rapidly forming metallic bridges between the flakes, improving conductivity.28 The lowered melting point of silver nanoparticles has also been exploited in the related area of screen printed thick films.29 The silver salt of palmitic acid (Fig. 2b) was heated under nitrogen at 250–270 °C for 90–120 minutes, producing silver nanoparticles ranging in size from 5–50 nm. This silver nanopowder was combined with an organic vehicle to form a conductive paste. This paste can be screen printed onto a substrate and then sintered. It is possible to sinter the paste at very low temperatures: 250–300 °C, compared to 650–800 °C required for pastes containing silver micropowders. The resulting films show impressive resilience to high temperatures and high currents.29
The low sintering temperatures of silver nanoparticles mean that melt reactions that yield silver nanoparticles may find applications in the electronics industry. However, some concerns remain regarding the toxicity and environmental impact of silver nanoparticles.30
2.2 Metal nanocrystals from the salts of thiols
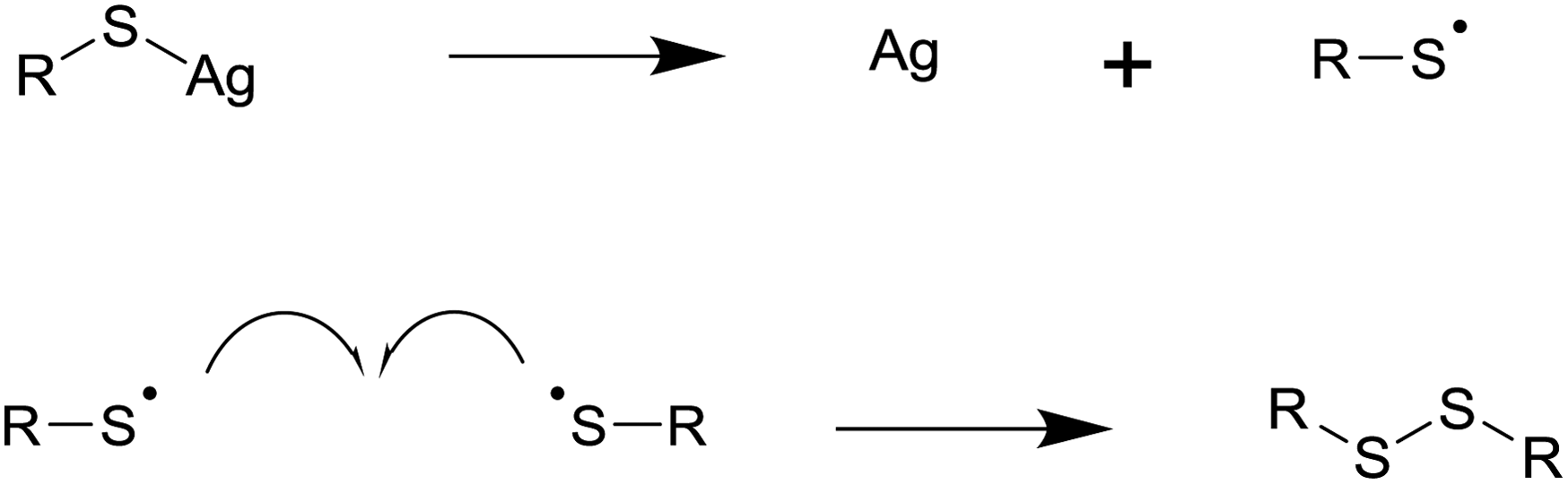 | ||
| Fig. 3 The decomposition of silver thiolates to form silver metal and a disulfide by-product. | ||
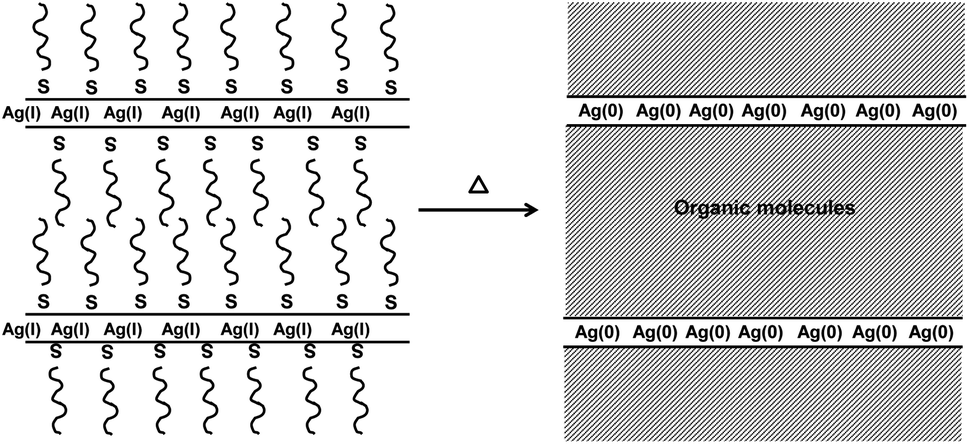 | ||
| Fig. 4 Decomposition of layered thiolate precursors is believed to lead to thin layers of metal atoms separated by thick layers of organic molecules. In plane diffusion of metal atoms is encouraged while interplane diffusion is limited, leading to asymmetric growth. This has been seen for both silver and bismuth thiolates.8,12 | ||
A further study of this system focused on the role of the ammonium ion; a series of precursors [RN(CH3)3][Au(SC12H12)2], where R = C8H17, C12H25, and C14H29 were investigated.36 Again, conditions of 180 °C for 5 hours under N2 were used to synthesise spherical gold nanoparticles stabilised by alkyl groups.36 It was found that ammonium cations with one long alkyl chain are better for nanoparticle synthesis than precursors with two long alkyl chains on the ammonium. An ammonium ion with a single long alkyl chain can easily eliminate trimethylamine and supply an alkyl group to cap the gold core.36 It was found that the addition of equimolar quantities of amine to the precursor improved size regulation and allow the reaction to proceed at lower temperatures.36 The exact nature of the amine used has an effect on particle size and polydispersity. The amine, a mild reductant, reduces Au(I) to Au(0) inhibiting the formation of disulfide molecules. This means that thiolate ligands are present to act as capping ligands.36
2.3 Metals summary
The melt approach has already found application in the synthesis of a range of metal nanoparticles. The metal salts of fatty acids can undergo solventless thermolysis to form self-capped silver, gold, and nickel nanoparticles. In all cases spherical particles with a fairly narrow size distribution were formed; interparticle separations, IR, and NMR support the idea that the particles are self-capped and that the capping ligand has the same chain length as the precursor.1,16,31 The reaction temperature appears to play an important role in controlling particle size, with higher temperatures generally yielding larger particles. In the case of nickel, temperature also influences the crystal phase formed.21,32 Amines can catalyse these reactions, making them viable at reduced temperatures.24 The decomposition of silver carboxylates to form silver nanoparticles may find applications in printable polymer nanocomposites where the reaction leads to short sintering times and high conductivities.28The metal salts of thiols have been used to synthesis silver, gold, palladium, and bismuth nanoparticles by solventless thermolysis. The decomposition mechanism involves the oxidation of the thiol to a radical and the reduction of a metal ion to its element. The radicals combine to form disulfide by-products, which make poor capping agents explaining the polydisperse products often seen with simple metal thiolate precursors. In some cases thermolysis of the same precursor at higher temperatures yields the metal sulfide product.12,37 Precursors containing an ammonium ion with one long alkyl chain have been used in gold nanoparticle synthesis, facilitating the capping of particles with alkyl groups. Thiolate precursors often adopt layered structures which can lead to anisotropic nanoparticle growth.12,13
3. Compound materials
3.1 Metal sulfide nanocrystals from dithiocarbamate precursors
The second published account of a self-capping solventless nanoparticle synthesis came from the O'Brien group.2 An asymmetric cadmium–dithiocarbamate precursor (Fig. 5) was heated under vacuum at temperatures ranging from 150 °C to 300 °C. The particles synthesised at 250 °C were approximately 5–7 nm in size. IR and NMR confirm that the particles were capped with the amine HNMe(C18H37). The crystalline phase of the CdS particles was shown to be dependent on the thermolysis temperature: at 150–250 °C XRD showed a cubic phase but at 300 °C the largest particles adopted the thermodynamically more stable hexagonal phase. The amount of capping agent was also shown to be temperature dependent. The particles prepared at 300 °C had significantly less capping agent that those prepared at 200 °C, as reflected in XPS C/Cd ratios and the particles' solubility in organic solvents.2 The same group later produced the bismuth analogue of this precursor, Bi(S2CN(C18H37)(CH3))3, and heated it under vacuum for an hour at a range of temperatures from 150–300 °C.38 Spherical particles of crystalline Bi2S3 were produced. The particles synthesised at 250 °C were approximately 8 nm in diameter and showed a fairly broad size distribution. Like the cadmium analogue these particles are believed to be self-capped with HNMe(C18H37).2,38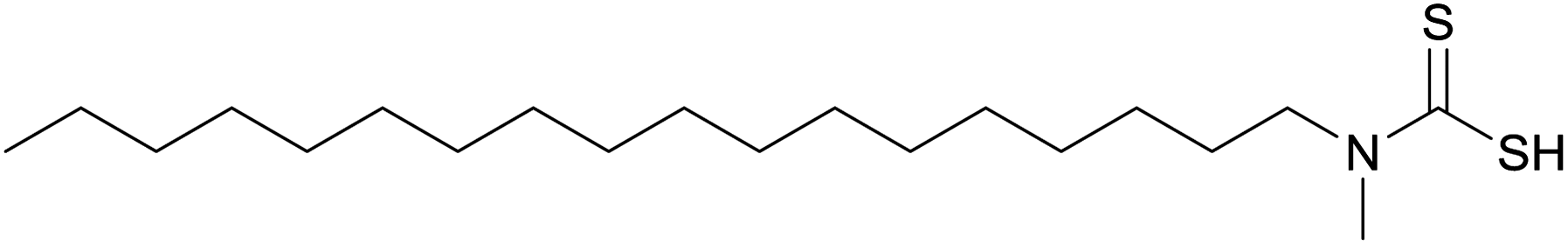 | ||
| Fig. 5 The Cd and Bi complexes of this asymmetric dithiocarbamate have been used in melt reactions, yielding CdS and Bi2S3 nanoparticles respectively.2,38 | ||
Somsook et al. synthesised the cadmium–dithiocarbamate complexes shown in Fig. 6. These precursors were heated at 700 °C under an N2 atmosphere.39 The thermolysis residue appeared to contain both CdO and CdS products. Particle sizes, estimated by the Scherrer equation, were 42 nm for the phenyl precursor and 59 nm for the ferrocenyl precursor. Electron microscopy showed nanofibres were produced by the ferrocenyl precursor but not by its phenyl analogue.39
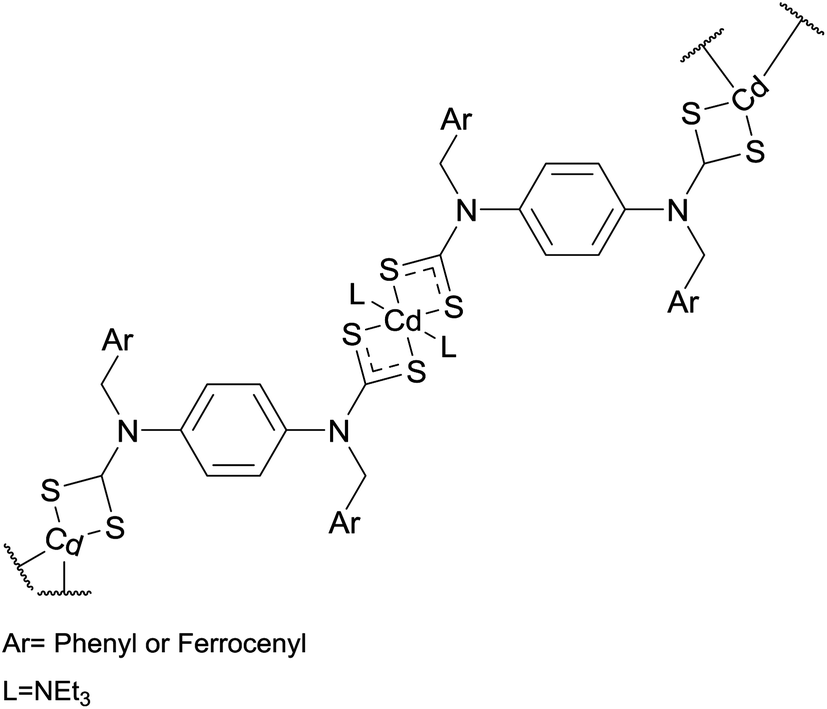 | ||
| Fig. 6 Precursors used by Somsook et al. in the melt synthesis of CdS nanoparticles and nanofibres.39 | ||
3.2 Metal sulfide nanocrystals from the salts of thiols
Later work looked at the decomposition of the same precursor under different reaction conditions; the precursor was heated in air at temperatures from 140–200 °C and with reaction times from 10–180 minutes. At 155 °C increasing reaction time brought about a shape evolution from small spherical particles to larger flat platelets.9 It was demonstrated that octanoate ligand plays an important role as a capping ligand: controlling particle growth by reducing the rate of monomer addition. In reactions where the octanoate was not present far larger particle sizes were seen but the shapes remained similar. The platelet shape is probably the result of face sensitive surface reactivity: with thiol adsorption and cleavage occurring preferentially on {100} facets. Differential scanning calorimetric (DSC) reveals the waxy solid precursor melting to a viscous fluid between 43 °C and 78 °C and C–S bond cleavage occurring at 113.5 °C.9 Cu2S nanodisks synthesised using the solventless method described above3,9 form self-assembled columnar structures upon evaporation. The disks stack face-to-face forming extended linear ribbon-like structures.40 Further work demonstrated that it is relatively easy to control the extent of polymerisation in the copper thiolate precursor and that the extent of polymerisation can dramatically alter product morphologies.6 The copper thiolate precursor is synthesised in the solution phase. Viscosity can be controlled by the speed and time of stirring during copper-thiolate coordination. Increased stirring speed and stirring time decreases viscosity.6 By reducing the stirring speed it is possible to increase the extent of polymerisation in the Cu(C12H25S)2 precursor. The more viscous, highly polymerised, precursor produces nanowires (Fig. 7c), while a less viscous, less extensively polymerised, analogue produces nanorods (Fig. 7b), and even lower viscosities yield spherical nanoparticles (Fig. 7a).3,6 The Cu2S nanowires produced by thermolysis at 155 °C for 120 minutes are 2–6 nm in diameter and 0.1 µm to several µm in length. Increased annealing temperatures appear to increase the diameter of the wires.6
 | ||
| Fig. 7 Micrographs demonstrating how the extent of polymerisation in the Cu(C12H25S)2 precursor influences Cu2S nanoproduct morphology. All are these images are from reactions carried out by heating the precursor in air at 155 °C for 120 minutes. Precursor viscosities were <10 mPa s−1, 13.2 mPa s−1, and 93.5 mPa s−1 for A, B, and C respectively.6 Adapted with permission from L. Chen et al., J. Am. Chem. Soc., 2004, 126, 16334–16335. Copyright 2004 American Chemical Society. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6 not the expected 1:2 which gives uneven wires and particles. This optimum precursor is found (from the lattice spacing of the layered solid) to contain two lead salts: lead dodecanethiol and lead octanoate. The lead thiolate is the source of PbS, while the lead octanoate appears to play a role in controlling morphology.7
:1.6 not the expected 1:2 which gives uneven wires and particles. This optimum precursor is found (from the lattice spacing of the layered solid) to contain two lead salts: lead dodecanethiol and lead octanoate. The lead thiolate is the source of PbS, while the lead octanoate appears to play a role in controlling morphology.7
3.3 Metal sulfide nanoparticles from dithiocarboxylate precursors
The metal dithiocarboxylates M(S2CAr)2(tmeda) (Ar = Ph, Tol) (M = Cd, Zn, Hg) (tmeda = (CH3)2NCH2CH2N(CH3)2) have been used to synthesise metal sulfide nanoparticles by solventless thermolysis. The precursors were heated at 350 °C for 2 hours under nitrogen. The CdS nanoproducts adopted the hexagonal crystalline phase, while the HgS and ZnS nanoparticles were shown to adopt the cubic phase.41 Increased reaction temperature increased particle size; the pyrolysis of the M = Cd, Ar = Ph precursor at 300, 400, and 500 °C gave CdS particles with particle sizes (estimated from the Scherrer equation) of 18, 36 and 43 nm respectively.413.4 Metal sulfide nanocrystals from xanthate precursors
Singhal et al. used an allyl-palladium(II)–xanthate complex and a palladium(II)–xanthate complex (Fig. 8) as single-source precursors for PdS nanoparticle synthesis.42 The precursors were heated in a furnace at 300 °C under an argon atmosphere. XRD showed the products to be crystalline tetragonal PdS. The two complexes shown in Fig. 8a and b gave particle sizes, estimated using the Scherrer equation,43 of 32 nm and 16 nm respectively.42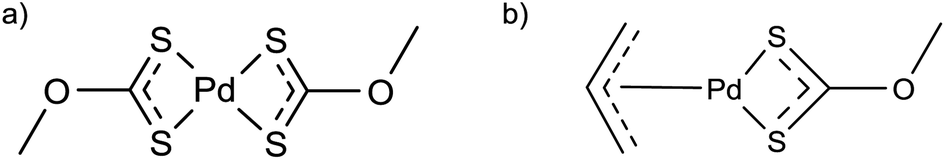 | ||
| Fig. 8 Palladium–xanthate precursors used in the solventless synthesis of PdS nanoparticles.42 | ||
3.5 Metal sulfides from the salts of thiotetrazoles
The cadmium salts of 1-substituted-5-thiotetrazoles (Fig. 1f) can be used as a single-source precursors to form CdS nanoparticles capped with thiotetrazoles.44 Two precursor were heated at a rate of 5 °C min−1 under a flow of nitrogen gas. The ethyl precursor was held at 250 °C for 1 hour whilst the adamantyl precursor was held at 250 °C for 2 hours. These reactions produced spherical CdS nanoparticles 2.5–3 nm in diameter. IR spectroscopy confirms the presence of thiotetrazolate anions on the nanoparticles' surfaces. The self-capping nature of the reaction is reflected in the solubility of the products: the ethyl precursor gives nanoparticles that are soluble in acetonitrile but not hexane, whilst the adamantyl gave particles soluble in hexane but not acetonitrile.44 Thiotetrazolates are potentially useful capping ligands because they decompose to give gaseous products. While capping ligands are vital in achieving monodispersity, preventing aggregation, and making nanoparticles dispersible in organic solvents, they are usually difficult to remove completely. Heating tetrazolate capped nanoparticles at 250–270 °C for 1.5–2 hours gives a solid product that is insoluble in organic solvent. IR spectroscopy shows no tetrazolate remains associated with the solid and it is believed that the tetrazolate ligand decomposes to give gaseous products (RN3, CS2, (CN)2 and N2) leaving uncapped nanoparticles behind.443.6 Metal chalcogenides from silyl precursors
DeGroot et al. investigated the solventless thermal decomposition of four single-source precursors for zinc chalcogenide synthesis: (1) (N,N′-tmeda)Zn(SSiMe3)2, (2) (N,N′-tmeda)Zn(SeSiMe3)2, (3) (3,5-Me2-C5H3N)2Zn(SSiMe3)2, and (4) (3,5-Me2-C5H3N)2Zn(SeSiMe3)2.45 TGA reveals that complete thermal decomposition of these complexes occurs at <300 °C. Unsurprisingly the sulfur complexes are more stable than their selenium counterparts. Heating up to 700 °C achieved complete thermolysis, producing bulk ZnE (E = S, Se). ZnS was shown to adopt the hexagonal wurtzite phase while ZnSe adopted the cubic sphaleritic phase. Heating to 200 °C avoided the loss of the nitrogen donor ligands, which act as capping ligands, and lead to the formation of nanoparticles. XRD of the 200 °C thermolysis product shows that ZnE is produced and peak broadening suggested that the material formed nanoparticles. This suggestion is supported by UV spectra of the thermolysis products of 1 and 2, which show the band gaps shifted to higher energies relative to the bulk. However, the decomposition products of 3 and 4 at 200 °C show no such quantum confinement effects, implying larger particles.453.7 Metal oxide nanocrystals from the salts of carboxylic acids
Iron(II)–oleate can be used as a precursor for solventless synthesis of Fe3O4 nanoparticles.19 The solid precursor was placed in a Pyrex tube which was flushed with nitrogen and sealed under low pressure. The tube was then heated to 320 °C and held at this temperature for two hours. The resulting Fe3O4 nanoparticles were spherical in shape, highly crystalline and showed a narrow size distribution, with an average diameter of 10.6 ± 1.2 nm. The nanoparticles were found to be capped with oleate ligands. The spherical nanoparticles can assemble into close-packed two-dimensional arrays, the spacing between nanoparticles was observed to be 2 nm, significantly less than twice the length of the oleate chain, suggesting interdigitation.19 Particle size and morphology was found to depend on temperature, annealing time, and pressure. Particle size increases with reaction time and particles annealed for longer than 3 hours become irregularly shaped. TGA reveals that the precursor decomposition temperature is pressure dependent. The decomposition temperatures increase under higher pressures. Whilst the reaction at 300 mTorr gave uniform spherical particles, the reaction at 500 mTorr give a mixture of morphologies with spheres, trigonal prisms, rods, and ‘diamond’ shaped particles all present.19 The authors suggest that the higher decomposition temperature at 500 mTorr leads to slower nucleation and consequently more morphological variety.19The above reaction has also been used as the first step in a two stage synthesis of magnetic iron nanoparticles. The Fe(II) salt of oleic acid is heated at 400 °C under a reduced pressure of 240 mTorr for 2 hours forming oleate capped Fe3O4 nanoparticles. TEM images of these particles show them to be roughly 18 to 24 nm in diameter. The iron oxide nanoparticles were then mixed with NaCl in a ratio of 1:40, ball milled to produce a fine powder and heated at 700 °C under an Ar–H2 gas mixture. The hydrogen gas reduced Fe3O4 to Fe, forming ferrite nanoparticles.46
PbCl–oleate has been used as a precursor for the solventless synthesis of Pb3O2Cl2 nanobelts.47 The solid precursor is heated in air for an hour at 190 °C; decomposition is believed to occur via the cleavage of the C–O bond in the oleate. The nanobelts produced have the mendipite crystal structure. In 90% of cases the nanobelt is elongated in the [010] direction. This elongation reflects the underlying crystal structure: accommodating the continuation of O2Pb3 chains along the length of the nanobelt. The belts are 29–170 nm wide, an average of 23.3 nm thick, and >4 µm long.47 The nanobelts are dispersible in non-polar organic solvents and it seems most likely that they are capped by alkyl groups formed in the decomposition process. The addition of ethylenediamine to the precursor improves the yield of nanobelts. Without this addition nanobelts elongated in the [010] direction are still produced, but nanocrystals roughly 5 nm in diameter are also formed.47
Thermolysis of the molecular cluster Mn12O12(O2C–R)16(H2O)4 gives MnO nano- and micro-products. Product size and morphology is found to depend on the temperature and time of thermolysis as well as the carboxylic acid employed.48 Heating of the precursor (R = C6H5) at 400 °C for 10 hours gives crystalline MnO nanoparticles 70 nm in diameter and spherical in shape. Higher temperatures give larger structures, for example heating at 500 °C for 10 hours produces pyramid shaped MnO particles 0.12 µm in diameter.48
3.8 Compounds materials summary
Nanocrystals of ZnS, CdS, HgS, Bi2S3, Cu2S, NiS, PbS, PdS, Fe3O4, MnO, and Pb3O2Cl2 have been synthesised by melt methods. A wider range of precursor chemistry has been applied to the synthesis of compound materials than to metals. The metal salts of fatty acids and thiols have been successfully used as precursors to both metal and compound nanoparticles. For some thiol salts the same precursor can yield either the metal or its sulfide, the compound forming at a higher reaction temperature.5,11,37 Ligands like dithiocarbamates and xanthates are already well established as single-source precursors in hot-injection syntheses and MOCVD.15,49 The successful application of these precursors to the melt approach suggests that the literature on MOCVD and hot-injection synthesis may contain a rich vein of promising single-source precursors as yet unused in melt reactions.14,15The complexes used as precursors can be divided into two classes: those where a single ligand acts as both the source of the anion and the capping ligand; and those where the metal ion is complexed to more than one type of ligand, one having the role of anion source the other acting solely as a capping agent. An example of the former would be the synthesis of CdS nanoparticles from a cadmium–dithiocarbamate precursors; whilst an example of the latter case would be the thermolysis of (N,N′-tmeda)Zn(SSiMe3)2, where SSiMe3 provides the sulfide but the amine is believed to cap the ZnS nanoparticles.2,45 In addition to conventional coordination complexes, molecular clusters have also been used as precursors in melt reaction.37,48
4. Concluding remarks
Abe and co-workers were apparently the first to report the potential practical importance of the melt approach to nanoparticle synthesis.1 The method attracted some attention, due mainly to its possible commercial applications as a route to novel silver solder,28,29 but the huge potential of this approach is yet to be more generally recognised. In the intervening years, the range of metal, metal chalcogenide and metal oxide nanocrystals synthesised in melt reactions has expanded, as has the precursor chemistry used. However, in many respects this is still a nascent field, the level of mechanistic understanding that has been developed for hot injection and heating up approaches is much greater.50 When compared to more established areas such as MOCVD and the hot-injection approach, relatively few single-source precursors have been investigated. There are also whole classes of compounds that have yet to be synthesised by this approach e.g. the III–V semiconductors.Melt reactions potentially offer the chemist precise control over a number of reaction parameters. These can be used to influence nanoparticle size, shape and chemistry. Higher temperatures typically lead to larger particles.4,9,19,32,41,48 Different temperatures can also yield different phases or materials5,11,32,37 and different particle shapes.3,5 Thiolate precursors react via M–S bond cleavage at lower temperatures to give metal nanoparticles whilst at higher temperatures S–C bond cleavage occurs, giving the sulfide.5,11,37 Extended reaction time can also have a pronounced effect on morphology, for example the evolution of nanofilms into nanorhombi12 or spheres into platelets.9 In some cases changes in the atmosphere under which the reaction is carried out can apparently change product morphology.31
The melt approach also offers some novel ways in which the size and shape of nanoproducts can be controlled. The layered structure of silver and bismuth thiolates appear to encourage anisotropic nanocrystal growth.8,12,13 In the synthesis of Cu2S nanostructures, it has been shown that the extent of polymerisation in copper-thiolate precursor dictates the product morphology.6 Work on the iron salts of fatty acids has demonstrated that the pressure under which thermolysis occurs can alter the morphology of the Fe3O4 nanoparticles formed.19
As well as offering new ways to control shape, solventless synthesis has a number of practical advantages: the precursors are easy to handle, the need for harsh reactants such as reducing agents or sulfonating agents is eliminated, and the reaction parameters (temperature and time of thermolysis) are easily controlled. The process of heating a precursor in a furnace should be relatively easy and cheap to scale up. The fact that melt reactions are solventless and, often, self-capping, means the solvents and capping agents employed in traditional nanoparticle synthesis are not needed and we note that the yields of these reactions are often very high. For these reasons the melt approach has the potential to provide an economical means of mass producing nanocrystals.
Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (UK) and the North West Nanoscience Doctoral Training Centre (NOWNano DTC) at the University of Manchester.Notes and references
- K. Abe, T. Hanada, Y. Yoshida, N. Tanigaki, H. Takiguchi, H. Nagasawa, M. Nakamoto, T. Yamaguchi and K. Yase, Thin Solid Films, 1998, 327–329, 524 CrossRef CAS.
- M. Lazell and P. O'Brien, Chem. Commun., 1999, 2041 RSC.
- T. H. Larsen, M. Sigman, A. Ghezelbash, R. C. Doty and B. A. Korgel, J. Am. Chem. Soc., 2003, 125, 5638 CrossRef CAS PubMed.
- A. Ghezelbash, M. B. Sigman and B. A. Korgel, Nano Lett., 2004, 4, 537 CrossRef CAS.
- M. B. Sigman and B. A. Korgel, Chem. Mater., 2005, 17, 1655 CrossRef CAS.
- L. Chen, Y. B. Chen and L. M. Wu, J. Am. Chem. Soc., 2004, 126, 16334 CrossRef CAS PubMed.
- J. Chen, L. Chen and L.-M. Wu, Inorg. Chem., 2007, 46, 8038 CrossRef CAS PubMed.
- Y.-B. Chen, L. Chen and L.-M. Wu, Inorg. Chem., 2005, 44, 9817 CrossRef CAS PubMed.
- M. B. Sigman, A. Ghezelbash, T. Hanrath, A. E. Saunders, F. Lee and B. A. Korgel, J. Am. Chem. Soc., 2003, 125, 16050 CrossRef CAS PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS.
- J. Park, K. J. An, Y. S. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891 CrossRef CAS PubMed; M.-Y. Chiang, S.-H. Chang, C.-Y. Chen, F.-W. Yuan and H.-Y. Tuan, J. Phys. Chem. C, 2011, 115, 1592 Search PubMed.
- J. Chen, L.-M. Wu and L. Chen, Inorg. Chem., 2007, 46, 586 CrossRef CAS PubMed.
- L. Chen and L.-M. Wu, in Inorganic Chemistry in Focus III, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 295–303 Search PubMed.
- M. A. Malik, M. Afzaal and P. O'Brien, Chem. Rev., 2010, 110, 4417 CrossRef CAS PubMed; D. Fan, M. Afzaal, M. A. Mallik, C. Q. Nguyen, P. O'Brien and P. J. Thomas, Coord. Chem. Rev., 2007, 251, 1878 CrossRef PubMed; M. A. Malik, N. Revaprasadu and P. O'Brien, Chem. Mater., 2001, 13, 913 CrossRef; N. L. Pickett and P. O'Brien, Chem. Rec., 2001, 1, 467 CrossRef PubMed.
- P. O'Brien, J. R. Walsh, I. M. Watson, M. Motevalli and L. Henriksen, J. Chem. Soc., Dalton Trans., 1996, 2491–2496 RSC.
- K. Abe, Y. Ichino, N. Takada, T. Hanada, Y. Yoshida, N. Tanigaki, T. Yamaguchi, H. Nagasawa and K. Yase, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1999, 337, 31 CrossRef CAS.
- H. Nagasawa, M. Maruyama, T. Komatsu, S. Isoda and T. Kobayashi, Phys. Status Solidi A, 2002, 191, 67 CrossRef CAS.
- D. K. Lee and Y. S. Kang, ETRI Journal, 2004, 26, 252 CrossRef.
- H. G. Cha, D. K. Lee, Y. H. Kim, C. W. Kim, C. S. Lee and Y. S. Kang, Inorg. Chem., 2008, 47, 121 CrossRef CAS PubMed.
- N. Yang and K. Aoki, J. Phys. Chem. B, 2005, 109, 23911 CrossRef CAS PubMed.
- M. Cavicchioli, L. C. Varanda, A. C. Massabni and P. Melnikov, Mater. Lett., 2005, 59, 3585 CrossRef CAS PubMed.
- M. Yamamoto and M. Nakamoto, J. Mater. Chem., 2003, 13, 2064 RSC.
- Y. Kashiwagi, M. Yamamoto and M. Nakamoto, J. Colloid Interface Sci., 2006, 300, 169 CrossRef CAS PubMed.
- M. Pecht, Y. Fukuda and S. Rajagopal, IEEE Trans. Electron. Packag. Manuf., 2004, 27, 221 CrossRef.
- Y. Li, K. S. Moon and C. P. Wong, Science, 2005, 308, 1419 CrossRef CAS PubMed.
- H. J. Jiang, K. S. Moon, J. X. Lu and C. P. Wong, J. Electron. Mater., 2005, 34, 1432 CrossRef CAS PubMed.
- Y. Li and C. P. Wong, Mater. Sci. Eng., 2006, 51, 1 CrossRef CAS PubMed; H. J. Jiang, K. S. Moon, Y. Li and C. P. Wong, Chem. Mater., 2006, 18, 2969 CrossRef.
- R. W. Zhang, W. Lin, K. S. Moon and C. P. Wong, ACS Appl. Mater. Interfaces, 2010, 2, 2637 CAS.
- M. Jakubowska, M. Jarosz, K. Kielbasinski and A. Mlozniak, Microelectron. Reliab., 2011, 51, 1235 CrossRef CAS PubMed.
- P. V. Asharani, Y. L. Wu, Z. Y. Gong and S. Valiyaveettil, Nanotechnology, 2008, 19, 255102 CrossRef CAS PubMed; E. Navarro, F. Piccapietra, B. Wagner, F. Marconi, R. Kaegi, N. Odzak, L. Sigg and R. Behra, Environ. Sci. Technol., 2008, 42, 8959 CrossRef; J. Y. Roh, S. J. Sim, J. Yi, K. Park, K. H. Chung, D. Y. Ryu and J. Choi, Environ. Sci. Technol., 2009, 43, 3933 CrossRef; T. M. Tolaymat, A. M. El Badawy, A. Genaidy, K. G. Scheckel, T. P. Luxton and M. Suidan, Sci. Total Environ., 2010, 408, 999 CrossRef PubMed.
- M. Yamamoto and M. Nakamoto, Chem. Lett., 2003, 32, 452 CrossRef CAS.
- S. G. Kim, Y. Terashi, A. Purwanto and K. Okuyama, Colloids Surf., A, 2009, 337, 96 CrossRef CAS PubMed.
- Y. Wang, J. Chen, L. Chen, Y.-B. Chen and L.-M. Wu, Cryst. Growth Des., 2010, 10, 1578 CAS.
- G. Carotenuto, C. L. Hison, F. Capezzuto, M. Palomba, P. Perlo and P. Conte, J. Nanopart. Res., 2009, 11, 1729 CrossRef CAS.
- M. Nakamoto, M. Yamamoto and M. Fukusumi, Chem. Commun., 2002, 1622 RSC.
- M. Nakamoto, Y. Kashiwagi and M. Yamamoto, Inorg. Chim. Acta, 2005, 358, 4229 CrossRef CAS PubMed.
- D. Jose and B. R. Jagirdar, J. Solid State Chem., 2010, 183, 2059 CrossRef CAS PubMed.
- M. Lazell, S. J. Norager, P. O'Brien and N. Revaprasadu, Mater. Sci. Eng., C, 2001, 16, 129 CrossRef.
- N. Thammakan and E. Somsook, Mater. Lett., 2006, 60, 1161 CrossRef CAS PubMed.
- A. E. Saunders, A. Ghezelbash, D.-M. Smilgies, M. B. Sigman, Jr and B. A. Korgel, Nano Lett., 2006, 6, 2959 CrossRef CAS PubMed.
- G. Kedarnath, V. K. Jain, S. Ghoshal, G. K. Dey, C. A. Ellis and E. R. T. Tiekink, Eur. J. Inorg. Chem., 2007, 1566 CrossRef CAS.
- A. Singhal, D. P. Dutta, A. K. Tyagi, S. M. Mobin, P. Mathur and I. Lieberwirth, J. Organomet. Chem., 2007, 692, 5285 CrossRef CAS PubMed.
- A. L. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- S. V. Voitekhovich, D. V. Talapin, C. Klinke, A. Kornowski and H. Weller, Chem. Mater., 2008, 20, 4545 CrossRef CAS.
- M. W. DeGroot, C. Khadka, H. Rosner and J. F. Corrigan, J. Cluster Sci., 2006, 17, 97 CrossRef CAS PubMed.
- Y. C. Han, H. G. Cha, C. W. Kim, Y. H. Kim and Y. S. Kang, J. Phys. Chem. C, 2007, 111, 6275 CAS.
- M. B. Sigman and B. A. Korgel, J. Am. Chem. Soc., 2005, 127, 10089 CrossRef CAS PubMed.
- L. Y. Chen, H. Xing, Y. M. Shen, J. F. Bai and G. Q. Jiang, J. Solid State Chem., 2009, 182, 1387 CrossRef CAS PubMed.
- P. O'Brien, J. R. Walsh, I. M. Watson, L. Hart and S. R. P. Silva, J. Cryst. Growth, 1996, 167, 133 CrossRef CAS; J. Akhtar, M. Afzaal, M. A. Vincent, N. A. Burton, I. H. Hillier and P. O'Brien, Chem. Commun., 2011, 47, 1991 RSC; B. Ludolph, M. A. Malik, P. O'Brien and N. Revaprasadu, Chem. Commun., 1998, 1849 RSC.
- C. de Mello Donegá, P. Liljeroth and D. Vanmaekelbergh, Small, 2005, 1, 1152 CrossRef CAS PubMed; S. G. Kwon, Y. Piao, J. Park, S. Angappane, Y. Jo, N.-M. Hwang, J.-G. Park and T. Hyeon, J. Am. Chem. Soc., 2007, 129, 12571 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |