 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeveral different charge transfer and Ce3+ localization scenarios for Rh–CeO2(111)
Zhansheng
Lu
a,
Zongxian
Yang
a,
Kersti
Hermansson
*bc and
Christopher W. M.
Castleton
*d
aCollege of Physics and Electronic Engineering, Henan Normal University, Xinxiang, 453007, China
bMaterials Chemistry, The Ångström Laboratory, Uppsala University, Box 538, SE-75121 Uppsala, Sweden. E-mail: kersti@mkem.uu.se
cDepartment of Theoretical Chemistry, Royal Institute of Technology, Roslagstullsbacken 15, SE-106 91 Stockholm, Sweden
dSchool of Science and Technology, Nottingham Trent University, Nottingham, NG11 8NS, UK. E-mail: christopher.castleton@ntu.ac.uk
First published on 8th November 2013
Abstract
We present DFT+U based electronic structure calculations in a p(3 × 3) slab supercell, for low coverages of atomically dispersed Rh interacting with the CeO2(111) surface, comparing Rh as an adatom, and as a dopant substituted into the surface layer. We find that, energetically, a Rh atom approaching a ceria(111) surface with both sparse O and Ce vacancies present strongly prefers to heal the Ce vacancies, but next it prefers to adsorb on a stoichiometric region rather than healing an O vacancy. In the adatom system, Rh is oxidized by electron transfer to a 4f orbital on one Ce ion in the surface layer, which is then nominally converted from Ce4+ → Ce3+ (i.e. Rh adatoms are single donors). We show that there are a number of different local minima, with Ce3+ localization at 1st, 2nd or 3rd nearest neighbour Ce sites. The second neighbour is the most stable, but all are close in energy. In the Rh-doped system (Rh replaces Ce), Rh is oxidized by charge transfer to neighbouring O atoms, and Rh doping leads to deep acceptor and donor states. Rh is not stable in the O sublattice. Moreover, based on vacancy formation energies, we find that oxygen vacancy formation is strongly enhanced in the vicinity of Rh dopants, but slightly suppressed in the vicinity of Rh adatoms.
1. Introduction
Among the multiple roles of CeO2 in catalysis, one of the most important is to store and release oxygen, a process which is promoted by noble metals such as Pd, Pt, and Rh.1,2 Here, we focus on atomically dispersed Rh at low coverage on ceria (111), the most stable low index surface.3–5 In principle, Rh atoms can form adatom species, heal vacancies, or “dope” the surface by replacing individual cations or anions. Here we present a density functional theory (DFT)6 study comparing the energetics of these different possibilities, and the resulting geometric and electronic structures. We will use a p(3 × 3) surface supercell which enables us to compare alternative Ce3+ localization patterns, their accompanying structures, stabilities and charge transfer character.Rh–ceria systems have been studied intensively by experiments (see ref. 7 and references therein for earlier work), but a clear picture of the location and structure of low coverage Rh on ceria(111) has not emerged. The cerium ions in pure stoichiometric ceria are nominally in the Ce4+ state, with no 4f electrons, but it has long been known that the formation of oxygen vacancies leads to the appearance of Ce3+ ions.8,9 Pfau et al.10 showed that Ce3+ ions also appear after Rh adsorption. They deposited Rh from less than 0.1 monolayers (ML) up to 2.2 ML on ceria thin films, and found several regimes: for coverages below about 0.3 ML, they found that isolated Rh adatoms dominate; above 0.3 ML they found evidence of Rh aggregation, leading to the formation of metallic Rh layers from 2 ML. For all coverages, Ce3s XPS spectra showed the appearance of Ce3+ ions near the surface following Rh deposition. At low coverages (the focus of our paper), Pfau et al. attributed this to the formation of neutral Rh adatoms, which they suggested catalysed O desorption, which in turn led to Ce3+ creation. They acknowledged that charge transfer from Rh to Ce in the ground state was an alternative interpretation, but considered it unlikely due to the similarity of the work functions of bulk Rh and CeO2. However, they did find a new HREELS peak upon Rh adsorption, which they interpreted as (excited state) charge transfer from Rh4d to Ce4f, and suggested that this implies that Ce and Rh are close together, and hence that Rh must occupy a Ce-top adsorption site. Hosokawa et al.,11 on the other hand, used XANES to show that rather than remaining neutral, Rh is oxidized by the ceria surface, though not as much as in, say, Rh2O3. They also used XAFS measurements to show that Rh is “highly dispersed” on the surface, and forms Rh–O–Ce structures, indicating adsorption either above or neighbouring O, rather than on Ce as suggested by Pfau et al. Wang et al.12 also suggested that the Rh becomes charged, using XPS to identify both the Rh0/Rhδ+ and the Ce4+/Ce3+ redox couples, and temperature programmed reduction (TPR) to associate the redox couples with the higher catalytic activity of Rh–CeO2/Al2O3.
Further structural information comes from the work of Gayen et al.,13 who prepared Rh/ceria model catalysts containing 0.5–2 atom% Rh, which they characterised using a range of X-ray and chemical techniques. They again showed that Rh adsorption involves the creation of Ce3+ ions, and that Rh is dispersed over the ceria surface, with no indication of the presence of Rh metal, or of the oxides Rh2O3, RhO2 and CeRhO3, or of RhO-like species. They interpreted their H2 uptake data as indicating the presence of three different Rh sites on the surface. They also reported Rh–O and Rh–Ce distances of ∼2.0 Å and 3.2 Å, respectively, together with rough coordination numbers by fitting to EXAFS data. The data indicated that Rh is mostly associated with oxygen, and they interpreted this and their other data as indicating that Rh atoms can actually replace Ce atoms, principally in the surface layers, to form Ce1−xRhxO2−δ, leading to a higher catalytic activity for CO oxidation. It has been suggested14 that such substitution is a general method for modifying the reactivity of oxide-based catalysts.
To summarize the experimental data: many different forms of Rh have been found or suggested for Rh in contact with ceria, with isolated Rh atoms at low coverages (below ∼0.3 ML), and agglomeration and eventually metallic Rh at higher coverages. In this paper we will present a systematic study to highlight the preferred state and behaviour of an isolated Rh atom on and in a CeO2(111) surface.
To our knowledge only three DFT studies have considered Rh doping of CeO2. The first15 did not contain any surfaces. The second16 described the effect of various transition metal dopants (including Rh) on dissociative methane adsorption and oxygen vacancy formation, using a p(2 × 2) supercell, hence 25% surface layer doping. However, it did not give details of the Rh–ceria system itself, except that the Rh is octahedrally coordinated with nearest neighbour distances of 2.16–2.49 Å. The third17 considered fifteen different transition metals doped into the second subsurface Ce layer in slab geometry models of the (111) and (110) surfaces. In comparison with experiment they showed that the oxygen vacancy formation energy gives a reasonable measure of the catalytic activity of doped ceria, and also that all fifteen of their dopants lowered the formation energy (increased the activity) relative to pure ceria – in the case of Rh by around 0.9 eV.
As far as computational Rh adatom–ceria (111) studies are concerned, a few18–24 have focused on chemical processes and/or Rh cluster formation but without presenting details for low coverage Rh/ceria structures, which are the focus of the present study (see also the recent review of defects on ceria25). Two have reported low coverage Rh adatom structures on ceria(111),21,26 showing that the most stable adatom site is in a three-fold hollow between three surface O ions. Although detailed electronic structure and analysis was not given, it was reported that adsorption involves charge transfer from the Rh to the surface, and that this transferred charge enters the Ce4f states of a single cerium ion, nominally changing it from Ce4+ to Ce3+. When similar calculations were instead performed in a larger p(3 × 3) surface supercell26 it was possible to show that these Ce3+ ions are located at the next nearest neighbour (NNN) location relative to the adsorbed Rh. However, we will show that several alternative Ce3+ locations exist, and these have not been reported or discussed earlier in the literature. This is an issue of considerable current interest, as it has recently been shown27–30 that Ce3+ ions formed together with oxygen vacancies exhibit a wide variety of competing low lying states, involving the nearest neighbour (NN), next nearest neighbour (NNN) and next next nearest neighbour (NNNN) and further shells of Ce ions around the vacancy, and that the resulting localization patterns are closely coupled to the structure and properties of the vacancies. In the present paper we will explore this issue in some detail, as well as the detailed electronic structure of the ground state.
In summary: some experimental studies suggest Rh doping, some suggest the presence of neutral Rh adatoms above Ce sites, and others suggest Rh adatoms above O, with charge transfer from Rh to ceria. Previous DFT results for Rh adatoms indicate an O-hollow site, with charge transfer, but other locations, such as surface doping and/or vacancy healing, have not been considered.
In our paper we provide answers to the following questions:
(i) If a Rh atom approaches an arbitrary ceria(111) surface containing both perfect regions and regions with O or Ce vacancies, where does it want to go and what does it want to do?
(ii) Which of the two – Rh adsorption or Rh doping – has the largest effect on the O vacancy formation energy (and thus also the oxygen storage capacity)?
(iii) If Rh adsorbs over surface O atoms, how much charge transfer is there from Rh and where do(es) the electron(s) go? Is there maybe a multitude of possible low energy Ce3+ localization sites, like that found for the electrons released via O vacancy formation?
(iv) And, on a more practical note: we use a p(3 × 3) cell in our calculations, but many calculations in the literature use (2 × 2) cells for metal/oxide interfaces. Are the consequences important?
2. Computational details
2.1 Calculation details and models used
We consider three modified CeO2(111) surface systems: (a) Rhx/CeO2 (Rh adatom), (b) Ce1−xRhxO2 with x = 0.111 in the surface layer (Rh doped onto the Ce sublattice), and (c) CeRhxO2–x with x = 0.111 in the surface layer (Rh doped onto the O sublattice), and will also report the formation energies of single surface oxygen vacancies in the presence of Rh.DFT calculations were carried out under periodic boundary conditions (PBCs), using the plane-wave based VASP code,31,32 with the PAW method.33 The Ce5s5p5d4f6s, O2s2p, and Rh4d5s electrons were treated as valence electrons and plane waves were included up to 30 Ry (408 eV). We treat the strong correlation effects amongst Ce4f electrons using the Dudarev form of the GGA+U functional34 together with the Perdew–Burke–Ernzerhof (PBE) form of GGA.35 We used U = 5 eV, based on the analysis made in ref. 4, 5 and 36–40.
CeO2 is a wide band gap semiconductor, with the cubic Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m structure of CaF2 and an experimental room temperature lattice parameter of a = 5.41 Å, or a0 = 5.39 Å when extrapolated to 0 K.40,41 Our PBE+U equilibrium lattice parameter is 5.48 Å.
m structure of CaF2 and an experimental room temperature lattice parameter of a = 5.41 Å, or a0 = 5.39 Å when extrapolated to 0 K.40,41 Our PBE+U equilibrium lattice parameter is 5.48 Å.
The (111) surface was modelled using a slab geometry comprising 4 (O–Ce–O) triple layers (altogether 108 atoms) and a 15 Å vacuum gap, see Fig. 1a. We will refer to the three atomic layers in the surface triple layer as T1a (O), T1b (Ce) and T1c (O), and those in the second triple layer as T2a (O), T2b (Ce) and T2c (O). The surface supercell chosen was a p(3 × 3) supercell, hence contained Ce36O72 for the clean slab. Some comparative results are given for p(2 × 2). The in-plane dimensions were fixed at the calculated equilibrium bulk lattice parameter (5.48 Å). In addition, the positions of ions in the bottom six atomic layers (dashed rectangular box in Fig. 1a) were fixed at their optimized bulk atomic positions to mimic the bulk. In all cases the remaining 6 atomic layers (and the Rh atom) were allowed to relax until the force on each atom was less than 0.02 eV Å−1. For Rh adsorption, one Rh atom was placed at different positions above one face of the slab while for Rh doping it was modelled by replacing either one oxygen (in layer T1a) or cerium (in layer T1b) atom by Rh, giving a doping concentration around 3% for the slab, or 11% for the surface triple layer.
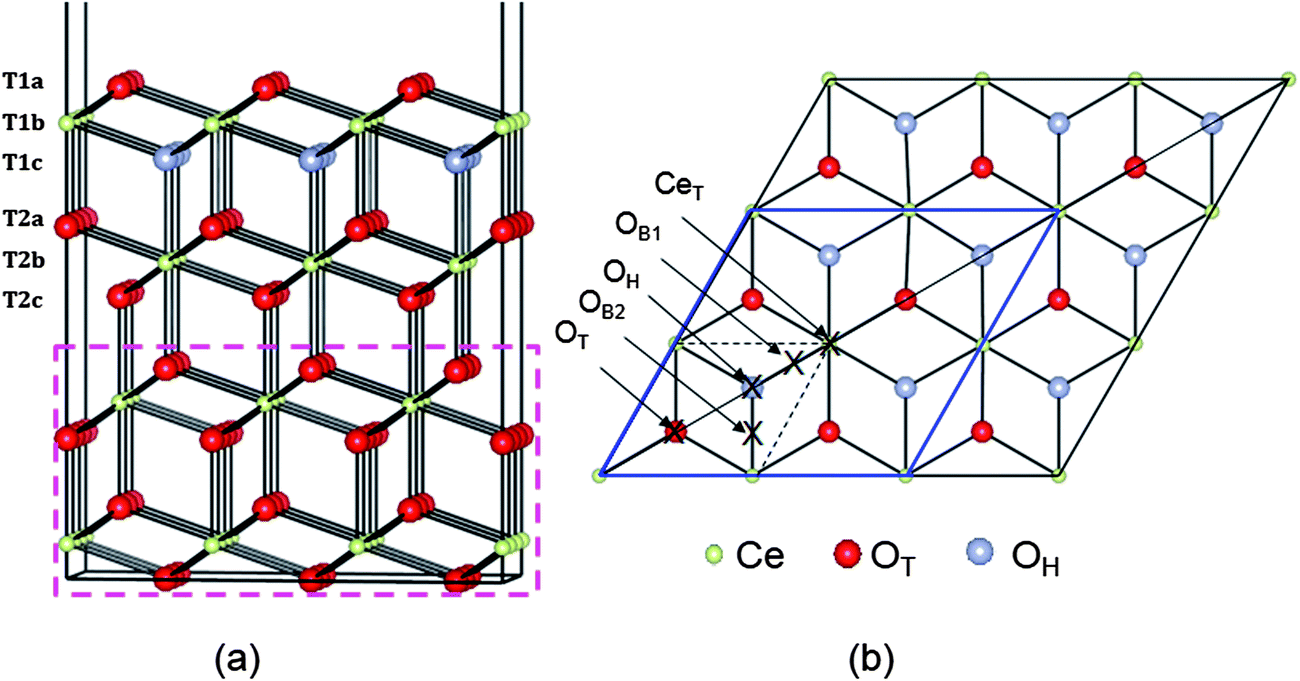 | ||
| Fig. 1 (a) Side view of the four O–Ce–O triple-layer p(3 × 3) slab model supercell used for the CeO2(111) surface. The bottom two triple layers (dashed rectangular box) are fixed at optimized bulk positions. (b) Top view of the supercell, with the labels for the five high symmetry adsorption sites marked to the left of the figure (B stands for bridge, H for hollow and T for top). | ||
Brillouin-zone integrations were performed using Monkhorst-Pack (MP) grids42 of (2 × 2 × 1) for the p(3 × 3) supercell and (4 × 4 × 1) for the p(2 × 2) supercell, together with a Gaussian smearing43 of 0.2 eV. Some checks on convergence will be presented in Section 2.3.
2.2 Energies calculated
For the adatom system, we evaluate the adsorption energy:| Eads = E(Rh/CeO2) − [E(CeO2) + Eref(Rh)] | (1) |
For Rh incorporated into the Ce sublattice, we calculate the substitution energy as
| Edoping at Ce site = [E(RhxCe1−xO2) + Eref(Ce)] − [E(CeO2) + Eref(Rh)] | (2a) |
For Rh incorporated into the O sublattice, we calculate the substitution energy as
| Edoping at O site = [E(RhxCeO2−x) + Eref(O)] – [E(CeO2) + Eref(Rh)] | (2b) |
In many cases, substitutional doping can formally be seen as two distinct processes: forming a vacancy and healing it using Rh. The first is connected with the vacancy formation energies
| Evac at the Ce site = [E(Ce1−xO2) + Eref(Ce)] − [E(CeO2)] | (3a) |
and
| Evac at the O site = [E(CeO2−x) + Eref(O)] − [E(CeO2)] | (3b) |
The second stage, healing the vacancy, is interesting in its own right, since surfaces sometimes contain a significant concentration of vacancies. The healing energy can be defined as
| Ehealing at the Ce vacancy site = [E(RhxCe1−xO2)] − [E(Ce1−xO2) + Eref(Rh)] | (4a) |
and
| Ehealing at the O vacancy site = [E(RhxCeO2−x)] − [E(CeO2−x) + Eref(Rh)] | (4b) |
2.3 Convergence checks
In order to assess the accuracy of our results, we have performed some additional calculations at the relaxed structure of our most stable Rh adatom configuration (see Section 3.2 for the structure), but with increased basis set, k-point integration, etc. (additional relaxation was not performed). We find that the adsorption energy Eads changes by only 0.003 eV if we increase the k-point integration grid to 4 × 4 × 1. If we increase the plane wave cutoff to 700 eV the adsorption energy increases by 0.02 eV, but if we add an additional 5th triple layer on the bottom of the slab (i.e. 15 atomic layers) then it decreases by 0.02 eV (note that Krcha et al.16 found a need for 5 or more triple layers for some adatoms, e.g., Ag, V, W, but not for Rh). Finally, adding dipole corrections perpendicular to the surface alters the energies by 0.001 eV. Overall, our main results are therefore expected to be accurate to around 0.01 eV, apart from errors arising from the choice of DFT functional itself.3. Results
3.1 The fate of a Rh atom arriving at the ceria(111) surface
If a Rh atom approaches an arbitrary ceria(111) surface, one of five things might be expected:1. It can adhere as an adatom (and then possibly diffuse into the material as an interstitial).
2. It can “heal” a cerium vacancy.
3. It can “heal” an oxygen vacancy.
4. It can kick out and replace an existing cerium ion.
5. It can kick out and replace an existing oxygen ion.
Options 2 and 4, and options 3 and 5 lead to the same end-points, namely Rh doped substitutionally into the cerium or oxygen sublattices, respectively (assuming subsequent desorption where applicable). Table 1 shows the limiting energies for each process: any value in between is possible, depending on the experimental conditions (the partial pressures of oxygen, Rh and Ce, the physical state of reactants etc.). Clearly, any cerium vacancies present will be filled first (line 2 in Table 1), leading to Rh doping of the Ce sublattice; details in Section 3.3 below. After that, Rh is most likely to attach as an adatom (line 1 in Table 1), as discussed in Section 3.2. Replacement of a cerium ion (line 4) is energetically disfavoured. Replacement of an oxygen ion (line 5) or filling an oxygen vacancy (line 3) turns out to be unstable. In all attempts to examine this structure the Rh instead relaxes back out of the oxygen site, to leave a Rh adatom next to an oxygen vacancy. This resulting structure will be discussed in Section 3.4, together with vacancy formation next to Rh adatoms and Rh dopants in the Ce sublattice.
| E ads, Ehealing or Edoping (eV) w.r.t.a | ||||
|---|---|---|---|---|
| Rh gas | Rh solid | |||
| Ce solid | Ce gas | Ce solid | Ce gas | |
| a Energies given with respect to (w.r.t.) all four extreme combinations of Rh and Ce reference energies. b With NNN localization of Ce4f electrons, since this is the most favourable. c Equivalent to Ce vacancy formation + Rh healing. d Equivalent to O vacancy formation + Rh healing. e See text. | ||||
| Rh adatom at OH on CeO2(111)b | −3.37 | −3.37 | +2.53 | +2.53 |
| Rh-healing a Ce vacancy in layer T1b | −10.22 | −10.22 | −4.32 | −4.32 |
| Rh-healing an O vacancy in layer T1a | Unstablee | Unstablee | Unstablee | Unstablee |
| Rh-doping@Ce sitec | +5.44 | +8.45 | +11.34 | +14.35 |
| Rh-doping@O sited | Unstablee | Unstablee | Unstablee | Unstablee |
3.2 Rh adatoms on ceria(111)
Several possible adsorption sites for the Rh adatom were investigated, including sites above layer 1a anions and layer 1b cations, all hollow sites (i.e. sites above layer 1c anions) and bridge sites between any two of them; see Fig. 1b. Two stable structures were found. In the most stable structure, denoted OH, the Rh is in a three-fold hollow between layer 1a anions and above a layer 1c anion. At the less stable “bridge” site, denoted OB, the Rh coordinates to two layer 1a anions. In the p(3 × 3) supercell, there are two inequivalent nearest-neighbour O–O bridge sites, denoted OB1, and OB2. This supercell contains a mirror plane along its long axis (and perpendicular to the plane of the surface itself), as shown in Fig. 1b. Adsorption at OB1 maintains this mirror plane, while adsorption at OB2 breaks it, so we find slightly different calculated energies for the two sites, although in the large supercell limit they should be equivalent.In keeping with previous studies, we always find electron transfer from Rh to the surface, with the excess charge localized on a single Ce ion, which becomes nominally Ce3+, and gains a spin of 1 μB. This 4f electron can localize on a number of different Ce ions, with the adsorption site (OH or OB) interrelated with the localization site. We compare the possibilities in Section 3.2.1 below, before describing the geometric structure and DOS of the most stable one in more detail in 3.2.2. It has been shown elsewhere that there is a hopping barrier for movement of this Ce4f electron through the lattice.44–46 Hence, in semiconductor terms, Rh adatoms behave as single donors, in the sense of having a single electron that can conduct an electric current in response to an applied electric field.
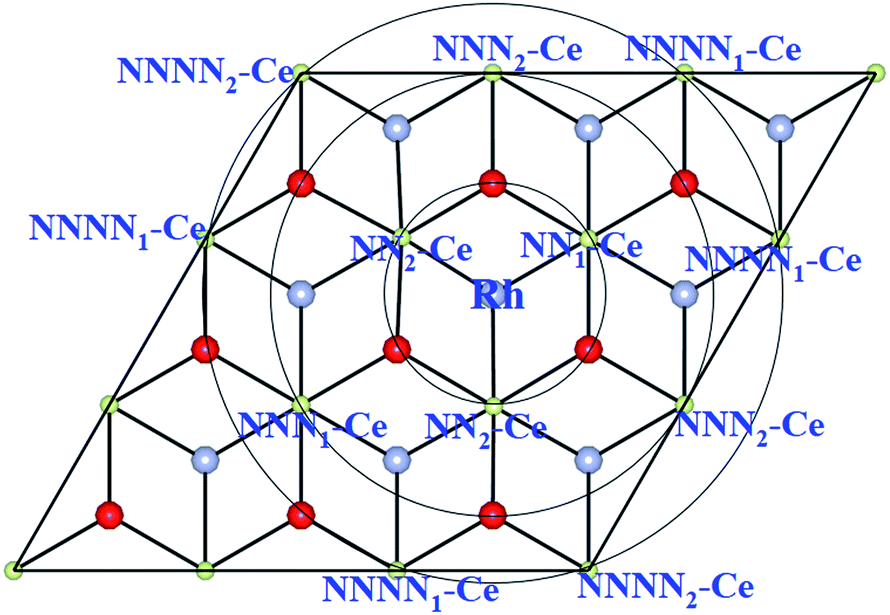 | ||
| Fig. 2 Schematic top view showing Ce ions within NN, NNN and NNNN shells around Rh adsorbed at the OH site on the ceria (111) surface. | ||
We find all six charge localization positions to be stable. We also find that these particular localization patterns are intimately linked to the adsorption structure. For Rh at the OH site, we find only localization on NNN sites or further way (attempts to produce, say, OH-NN2 relax back to OB1-NN2). For Rh at the OB1 site, we find only NN2 localization, while Rh adsorbed at the OB2 site can occur with either NN1 or NN2 localization (again, attempts to produce OB1-NNN2, for example, always relax back to OH-NNN2). This appears to be a steric effect due, at least in part, to the larger ionic radius of Ce3+ as compared to Ce4+. Altogether this gives seven different stable combinations of the Rh position and the Ce3+ localization pattern. The relaxed structures and spin densities (ρspin up–ρspin down) are plotted in Fig. 3a–g, and some energy and structural information is given in Table 2. The most stable has Egasads = −3.37 eV, with the 4f charge localized at a NNN2 site, see Fig. 3d. This is in agreement with the results obtained by Song et al.26, and is very similar to the results found recently for Ce3+ localization associated with gold adatoms on a ceria(111) surface,47 and around oxygen vacancies.27–29 In the latter case, the preference seems to be for one NN Ce3+ and one NNN Ce3+. However, what we show here is that the previously reported26 structure (Rh at the OH site on ceria (111) with NNN2 localization) is only one possibility out of several, with all the energies lying within 0.4 eV of one another (see Table 2). What is more, the energies of the two NNNN localizations (which are almost degenerate) lie between NNN1 and NNN2, and much closer to the lower energy NNN2. This means that, as NNN1 and NNN2 approach each other in the large supercell limit, the NNNN localization may in fact turn out to be the most stable.
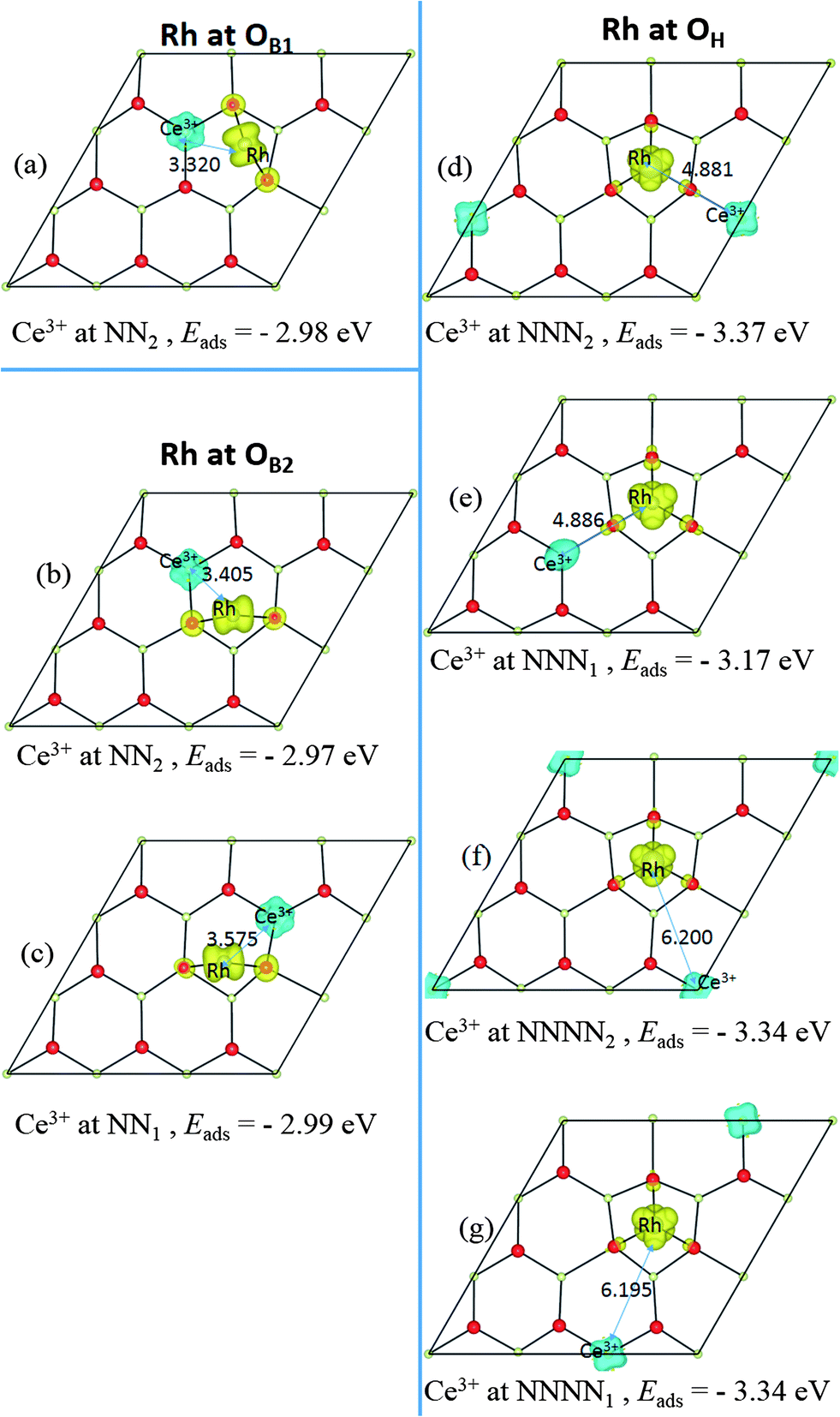 | ||
| Fig. 3 Top views of the relaxed geometric structures and the spin density (ρspin up–ρspin down) for a Rh adatom at the OB1 site in (a), OB2 site in (b) and (c), OH site in (d)–(g). In each image, the location of the nominal Ce3+ ion from the Ce4f charge localization (at NN1, NN2, NNN2, NNNN1 or NNNN2) is indicated below the image, as is the adsorption energy. | ||
| Site and structurea | Egasadsb (eV) | E mag (eV) | d(Rh–Ce3+)d (Å) | q(Rh)e (e) | σ Rh (μB) | Δq(Ce3+)g (e) | σ Ce (μB) |
|---|---|---|---|---|---|---|---|
| a Adsorption sites OB1, OB2 and OH are defined in Fig. 1. The alternative locations of the (nominal) Ce3+ ion with respect to the Rh atom are labelled NN1, NN2, NNN1 and so on, as defined in Fig. 2. b Egasads is the adsorption energy with respect to a gas phase Rh source (negative value means stabilizing). c E mag is the energy difference between FM and AFM couplings of the Rh and Ce3+ spins. Note that since the structures in the two are almost identical, the errors due to plane-wave cut-off, k-point integration, supercell approximation etc. are almost identical. Hence cancellation of errors allows the difference in energy to be much better converged than Egasads itself. d d(Rh–Ce3+) is the distance between the Rh atom and the (nominal) Ce3+ ion. e q(Rh) is the net charge of the Rh adatom from Bader electron density analysis. f σ(Rh) is the net spin magnetic moment of the Rh adatom from Bader electron density analysis. g Δq(Ce3+) is the change in the charge of the reduced Ce ion compared to its charge before the Rh adsorption (+2.38 e) from Bader electron density analysis. h σ(Ce) is the net spin magnetic moment of the Ce ion from Bader electron density analysis. i Note: FM ground state not AFM. | |||||||
| p(3 × 3) supercell | |||||||
| OB1-NN2 | −2.98 | 0.003 | 3.32 | +0.61 | 1.45 | −0.29 | −0.97 |
| OB2-NN1 | −2.99 | 0.003 | 3.58 | +0.50 | 1.45 | −0.27 | −0.97 |
| OB2-NN2 | −2.97 | 0.005 | 3.41 | +0.50 | 1.46 | −0.28 | −0.95 |
| OH-NNN1 | −3.17 | 0.002 | 4.87 | +0.62 | 1.64 | −0.27 | −0.97 |
| OH-NNN2 | −3.37 | 0.004 | 4.88 | +0.62 | 1.64 | −0.28 | −0.98 |
| OH-NNNN1 | −3.34 | 0.0001 | 6.20 | +0.64 | 1.64 | −0.26 | −0.98 |
| OH-NNNN2 | −3.34 | 0.0004 | 6.20 | +0.62 | 1.60 | −0.28 | −1.00 |
| p(2 × 2) supercell | |||||||
| OH-NNN1 | −3.62 | −0.002i | 4.83 | +0.62 | 1.67 | −0.29 | −0.99 |
In all of the states shown in Fig. 3 for p(3 × 3), the spin on the Rh and the spin on the Ce are antiferromagnetically (AFM) coupled. However, we have also found ferromagnetically (FM) coupled versions of each one. Apart from the signs, the spin densities of these FM versions (not shown) are almost indistinguishable from the AFM versions, and the energy difference between the FM and AFM versions is very small (0.0001–0.005 eV, see Table 2) and generally decreases with increasing Rh–Ce separation (see footnote c of Table 2 for comments on convergence).
In the smaller p(2 × 2) cell there are only three possible localizations: NNN1 (= NNN2via the PBCs for this supercell), NN1 and NN2. The NNNN site is indistinguishable from the NN2 site via the PBCs, and neither NN1 nor NN2 localization appears to be stable. NNN is stable, with an Egasads value of −3.62 eV, i.e. it is energetically over-stabilized relative to p(3 × 3) by being NNN in three different directions at once (due to the PBCs). We also find that in p(2 × 2) the Rh and Ce3+ are very weakly FM coupled rather than AFM. The alternative localizations are missing, since p(2 × 2) is too small to hold them. Moreover, we find four stable adsorption sites (OH, OB, OT and CeT) in p(2 × 2) rather than just two (OH, OB) in p(3 × 3). As a result, issues of magnetic coupling, optimal localization, and available adsorption sites for low densities of Rh adatoms cannot be addressed properly in p(2 × 2). This would affect the predicted thermodynamics, amongst other things.
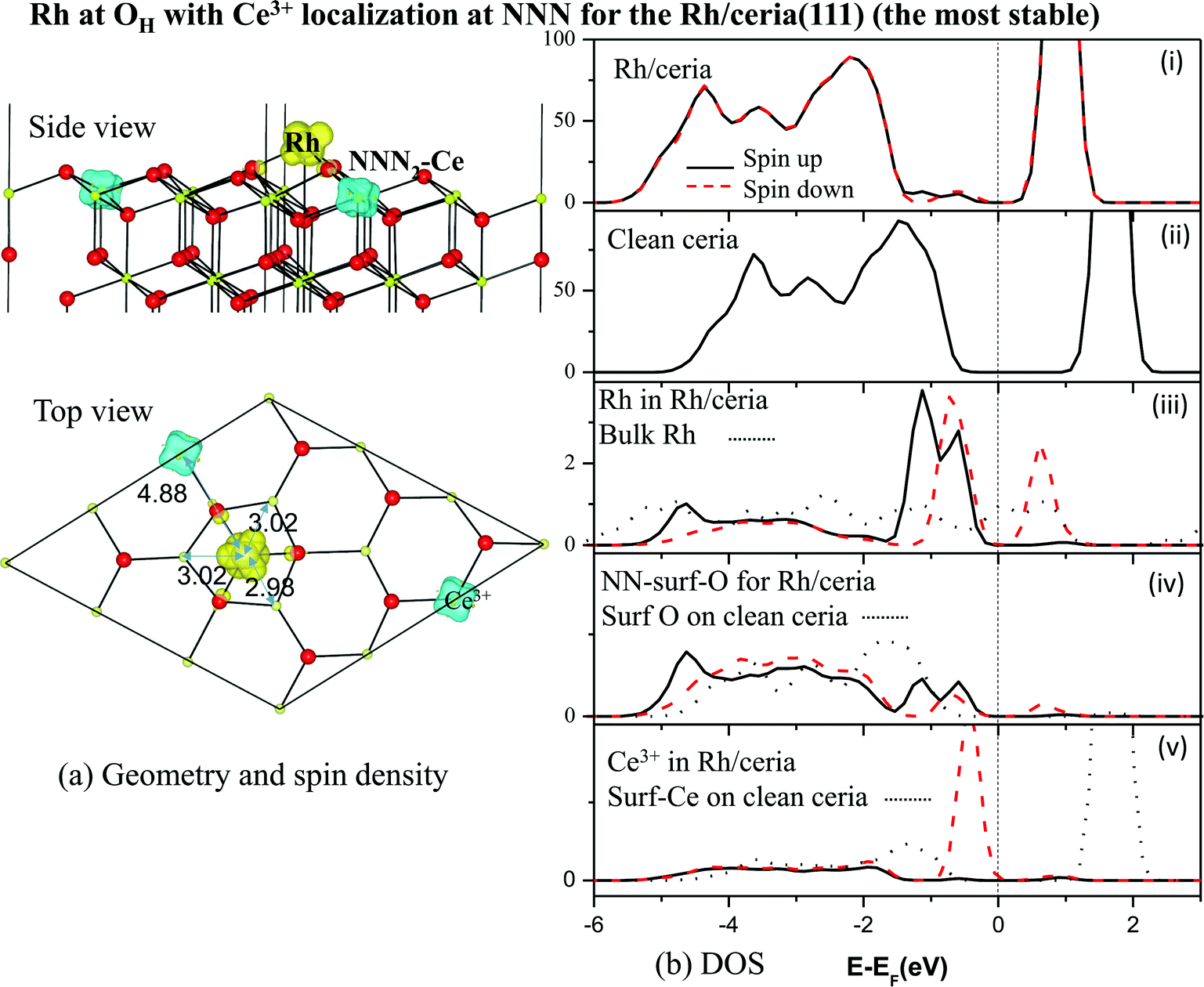 | ||
| Fig. 4 Rh adatom adsorbed at the OH site with Ce3+ at NNN2: (a) geometric structures and spin density (ρspin up–ρspin down), and (b) density of states (DOS) in panels (i) and (ii), and Partial DOS (PDOS) in panels (iii)–(v). The vertical dashed line represents the Fermi energy. Thicker black solid and dashed red curves are for spin up and spin down, respectively. The DOS of all other OH cases are very similar regardless of Ce3+ location. | ||
A Bader charge analysis48 of the total charge density, given in Table 3, confirms that the Rh adatom has been partially oxidized, losing about 0.6 electrons to the substrate (as also found in the p(2 × 2) cell21). By far the most significant change in the substrate occurs for the single NNN Ce ion, which receives about 0.3 electrons. This value for a nominal Ce4+ → Ce3+ reduction may seem small, but it is very close to that seen in similar calculations for Ce4+ → Ce3+ reductions associated with vacancy creation.5,36–40 In those studies the change in Ce4f charge (as opposed to the total charge) is around 0.9 electrons,40 but is partially compensated by an outward redistribution of non-Ce4f charge. In the current case, the degree of transfer assessed by magnetisation is really very complete, with a spin of 0.98 μB appearing on the Ce3+ ion (Table 3).
| Atomic layersa | Groups of atoms in each layerb | Rh adatom at the OH site (with one NNN Ce3+), cf.Fig. 4 | Rh substitution on a Ce site in layer T1b, (with no Ce3+), cf.Fig. 5 | ||||
|---|---|---|---|---|---|---|---|
| # | Δqd (e) | Spine (μB) | # | Δqd (e) | Spine (μB) | ||
| a Data given for the uppermost four atomic layers of ceria and for Rh (as an adatom or as a dopant) in the slab (as shown in Fig. 1), plus the sum over these four ceria layers and the Rh, the sum of the remaining 8 layers. b The different categories of near neighbours (NNs) refer to the closest O and Ce shells surrounding the Rh, and are listed separately for each layer. c Columns labelled “#” give the total number of atoms in each group. d The net charge redistribution (per atom of the type in question) induced by the Rh adsorption, compared to the atomic charges in the stoichiometric ceria(111) slab and a lone Rh atom, obtained from Bader analysis. Hence reference values used: Rh(0.00 e), O(−1.19 e) for layer T1a, and Ce(+2.38 e) for layer T1b, O(−1.16 e) for layer T1c and O(−1.20 e) for layer T2a. e Atomic spins, obtained from the Bader analysis. Note: a free Rh(g) atom has a spin of 2.18 μB. | |||||||
| Rh adatom | Rh adatom | 1 | +0.62 | +1.64 | |||
| O in T1a | NNs | 3 | −0.00 ⇒ +0.07 | −0.00 ⇒ +0.01 | 3 | +0.16 ⇒ +0.17 | +0.04 ⇒ +0.06 |
| Others | 6 | −0.01 ⇒ +0.00 | +0.00 ⇒ +0.12 | 6 | −0.01 ⇒ +0.01 | −0.00 ⇒ +0.00 | |
| Rh dopant in T1b | Rh dopant | 1 | +1.54 | +0.72 | |||
| Ce in T1b | Ce III | 1 | −0.28 | −0.98 | 0 | N/A | N/A |
| Ce IV NNs | 3 | −0.09 ⇒ −0.01 | −0.02 ⇒−0.00 | 6 | −0.01 ⇒ +0.00 | +0.00 ⇒ +0.00 | |
| Other Ce | 5 | −0.03 ⇒ −0.00 | +0.00 ⇒ +0.02 | 2 | −0.05, +0.01 | −0.00, +0.00 | |
| O in T1c | NNs | 0 | N/A | N/A | 3 | +0.08 ⇒ +0.10 | −0.01 ⇒ −0.00 |
| Others | 9 | −0.07 ⇒ +0.02 | −0.00 ⇒ +0.00 | 6 | −0.02 ⇒ −0.01 | +0.00 ⇒ +0.00 | |
| O in T2a | NN | 0 | N/A | N/A | 1 | +0.10 | +0.13 |
| Others | 9 | −0.02 ⇒ +0.02 | −0.00 ⇒ +0.00 | 8 | −0.01 ⇒ +0.03 | −0.00 ⇒ +0.00 | |
| Σ Over all the above | 37 | −0.15 | +1.01 | 36 | +2.25 | +0.98 | |
| Σ All other layers | 72 | +0.15 | −0.01 | 72 | +0.14 | +0.02 | |
| The removed Ce | −2.38 (Ce+2.38 ⇒ Ce0) | ||||||
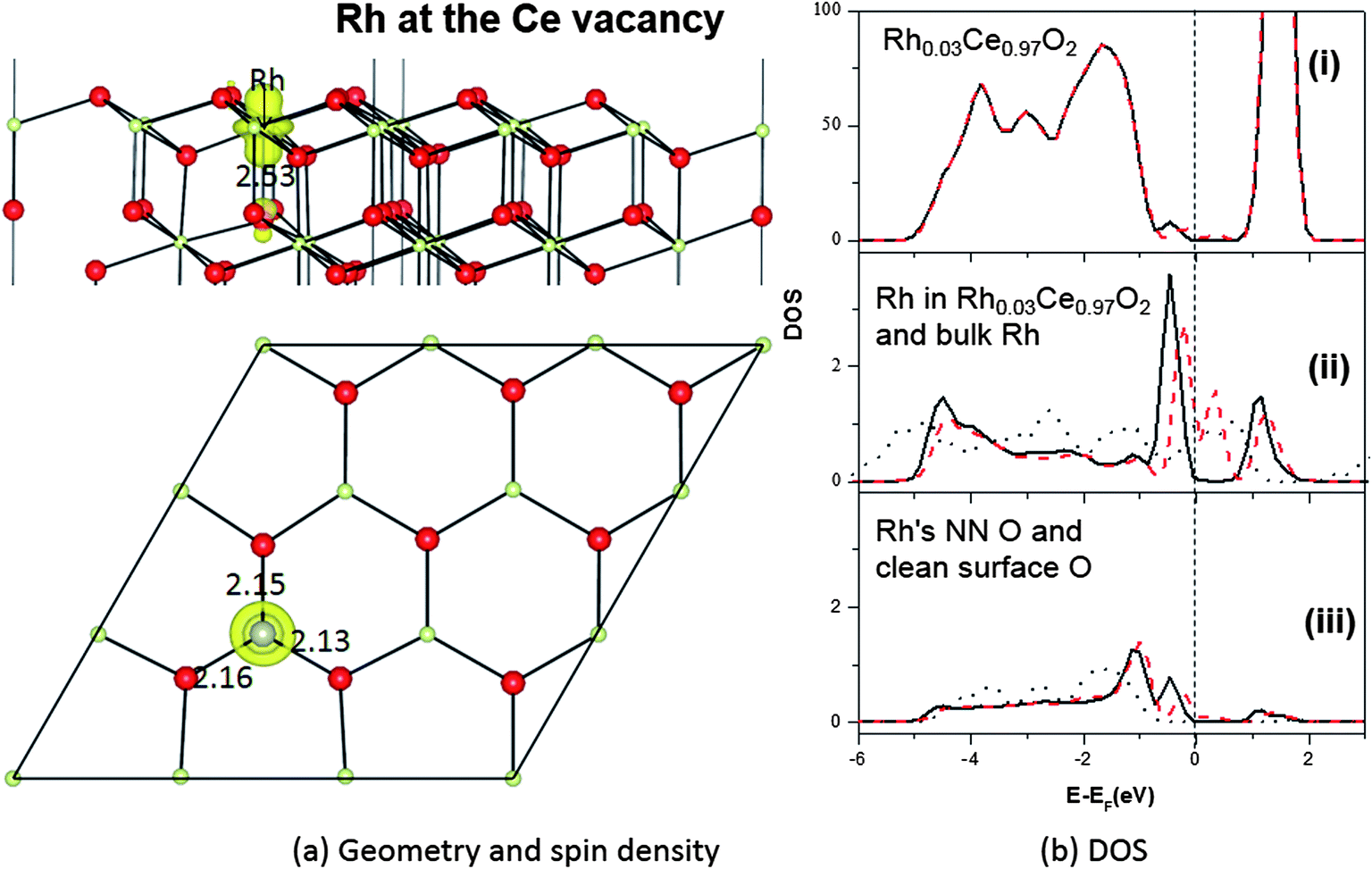 | ||
| Fig. 5 Rh-doped CeO2(111) system with Rh on a Ce site in the T1b layer: (a) top and side views of the geometric structures and spin density (ρspin up–ρspin down), and (b) DOS curves for (i) total DOS, (ii) bulk Rh (the dotted thin curve) and PDOS for the Rh dopant (the thicker black solid and dashed red curves for spin up and spin down, respectively, here and in other panels), (iii) PDOS of the Rh's NN O and for the corresponding O in the clean ceria(111) surface (the dashed thin curve). | ||
Evidently, the Rh adatom is mainly oxidized through charge transfer to the NNN2 Ce ion, as previously reported,21,26 but with significant contributions from other nearby ions. Hence at low coverage, we agree with the experimental picture of Ce3+ creation via charge transfer11–13 from Rh to Ce, rather than the picture of a neutral adsorbed Rh only indirectly causing the presence of Ce3+via induced oxygen vacancy formation.10 We note that the latter picture was based upon the values of the bulk work function of Rh, which will likely be very different from that for mono- or nano-dispersed Rh. The situation can be expected to change when the coverage is sufficient to allow, say, metallic Rh islands or nanoclusters to form.
To summarize: the Rh adatom prefers to adsorb above a subsurface O (OH site), pulling its three surface layer O neighbours outwards, and transferring charge primarily to a NNN Ce ion on the surface. There are many competing locations for this Ce3+ ion, all very close in energy.
3.3 Rh doped ceria surface: Ce site substitution
The geometric structure of a Rh dopant on the Ce sublattice is shown in Fig. 5a. When a nominal Ce4+ ion in the T1b layer is replaced by the smaller Rh, the nearest O ions move towards the Rh, with Rh–O bonds of ∼2.13 Å. This is smaller than the Ce–O bonds they replace (2.37 Å), but larger than the Rh–O bonds in RhO249,50 and Rh2O351,52 (∼1.97 Å and ∼1.80 Å). The strain in the resulting Rh–O and neighbouring O–Ce bonds thus destabilizes the local surface structure, potentially aiding catalytic activity. Similar results have been seen for Au doped ceria.14In its two common bulk oxides, Rh2O3 and RhO2, Rh has (nominal) oxidation states of +III and +IV, respectively, so, from a semiconductor doping point of view, one would therefore naïvely expect Rh when replacing the nominally +IV Ce ion to behave as either (i) an isoelectronic substitution, possibly introducing local distortions to band edges but not to acceptor or donor levels, or (ii) a single acceptor, perhaps able to act as a p-type dopant.
The results of a Bader analysis48 of our calculated total charge distribution are listed in Table 3, and show no significant extra Ce4f charge or spin density on any Ce ion. The Rh dopant has lost 1.5 electrons and is therefore in a higher oxidation state now as a dopant than when it was as an adatom, but in a lower oxidation state than the Ce it replaced, which had a charge of +2.4 e on the clean CeO2(111) surface. Since the difference in charge here is 0.9 e, the oxidation state of Rh may be considered to be +III. As a result, the Bader charge of the three NN oxygen ions is only about −1.0 e for each (Table 3), rather than the −1.2 e on the clean surface. These charge transfers and oxidation assignments are also reflected in the total spin of the doped system, which is about 1.0 μB, mainly localized on the 4d states of the Rh dopant (0.7 μB) and its neighbouring oxygen ions (Table 3). Hence Rh is behaving roughly as a single acceptor.
The acceptor state itself can be seen in the DOS (Fig. 5b), where a single empty spin down state lies above the Fermi level. However, rather than lying just above the valence band edge it is rather high in the O2p–Ce4f band gap, with filled states in the lower half of the gap. Indeed, there are now DOS features covering almost the entire O2p–Ce4f gap. A Bader analysis (see Table 4) of the partial charge for the filled states (energy window −0.7 eV to 0.0 eV) shows that they are mostly shared between the Rh dopant (2.47 electrons) and its three NN surface O ions (1.71 electrons together), with very little elsewhere (this is similar to what was found in the bulk,15 except that in the bulk the gap states covered a narrower energy window). Bader analysis of the empty gap states above the Fermi level (energy window 0.0 to 0.7 eV) shows space for exactly one electron, projected mostly on the Rh itself (0.62 electrons) and the four nearest neighbour O ions (0.27 electrons together) (see Table 4). The O and Rh PDOS for the gap states contain similar features, indicating Rh–O bond reorganization, and indeed the gap states resemble filled bonding and partially filled antibonding levels, with strong interactions between Rh4d and O2p.
| Atomic layersa | Groups of atoms in each layera | Number of the ions in the groupa | n electron in filled gap statesb | n electron in empty gap statesb |
|---|---|---|---|---|
| a Atomic layers and groups of neighbours are the same as those used in Table 3. b Charges obtained from Bader analyses of the partial charge densities. The arrow indicates a range. | ||||
| Rh | Rh | 1 | 2.47 | 0.62 |
| O in T1a | NNs | 3 | 0.55 ⇒ 0.60 | 0.04 ⇒ 0.06 |
| Others | 6 | 0.01 ⇒ 0.02 | 0.00 ⇒ 0.00 | |
| Ce in T1b | Ce III | 0 | N/A | N/A |
| Ce IV NNs | 6 | 0.04 ⇒ 0.05 | 0.01 ⇒ 0.01 | |
| Others | 2 | 0.00, 0.01 | 0.00, 0.00 | |
| O in T1c | NNs | 3 | 0.04 ⇒ 0.04 | 0.01 ⇒ 0.01 |
| Others | 6 | 0.01 ⇒ 0.02 | 0.00 ⇒ 0.00 | |
| NNs | 1 | 0.15 | 0.11 | |
| O in T2a | Others | 8 | 0.00 ⇒ 0.00 | 0.00 ⇒ 0.00 |
| Σ Over all the above | 37 | 4.92 | 0.95 | |
| Σ Rest of slab | 72 | 0.08 | 0.05 | |
Putting the Bader and DOS analyses together, it can be inferred that the filled states in the lower part of the band gap would allow Rh on the CeO2(111) surface to act as a deep multiple donor, while the empty state in the mid-gap would allow it to behave as a single deep acceptor. Though useless as a semiconductor dopant, the presence of this empty DOS state lowers the formation energy for oxygen vacancies, by accepting an extra electron at an energy level below that of the Ce4f state. We will show in Section 3.3 that this does indeed lower the formation energy of vacancies neighbouring Rh dopants in the Ce sublattice.
In the case of Rh doping on the Ce site, there is very little difference at all between the results in p(2 × 2) and p(3 × 3), with very similar geometric and electronic structures. The doping energies are also similar, both being very large in comparison with, say, thermal energies under either experimental or growth conditions: Edoping = +5.79 eV in p(2 × 2) and +5.44 eV in p(3 × 3), using gas reference energy for Rh and solid for Ce (the lowest energy combination).
3.4 Oxygen vacancy formation in the vicinity of Rh
Rh substitution of a surface O ion, (possibilities 3 and 5 in the list at the start of Section 3.1), may seem like an esoteric construction, but can be viewed as one possible result of Rh adsorption on a partially reduced ceria surface. However, attempts to place a Rh atom on the O sublattice do not succeed for CeO2(111). Instead, the Rh leaves the O site, and moves to the nearest neighbouring OB adatom site, distorted by the presence of the oxygen vacancy left behind. The structure and spin density are shown in Fig. 6. We here find three nominal Ce3+ ions, rather than one. This five-component complex (one vacancy, one adatom and three Ce3+ ions) barely fits inside this p(3 × 3) supercell, so that defect–image interactions etc. lead to a rather untidy looking structure. This also leaves little scope for a realistic comparison of different potential configurations of the Rh and Ce3+ ions like that reported in Section 3.2.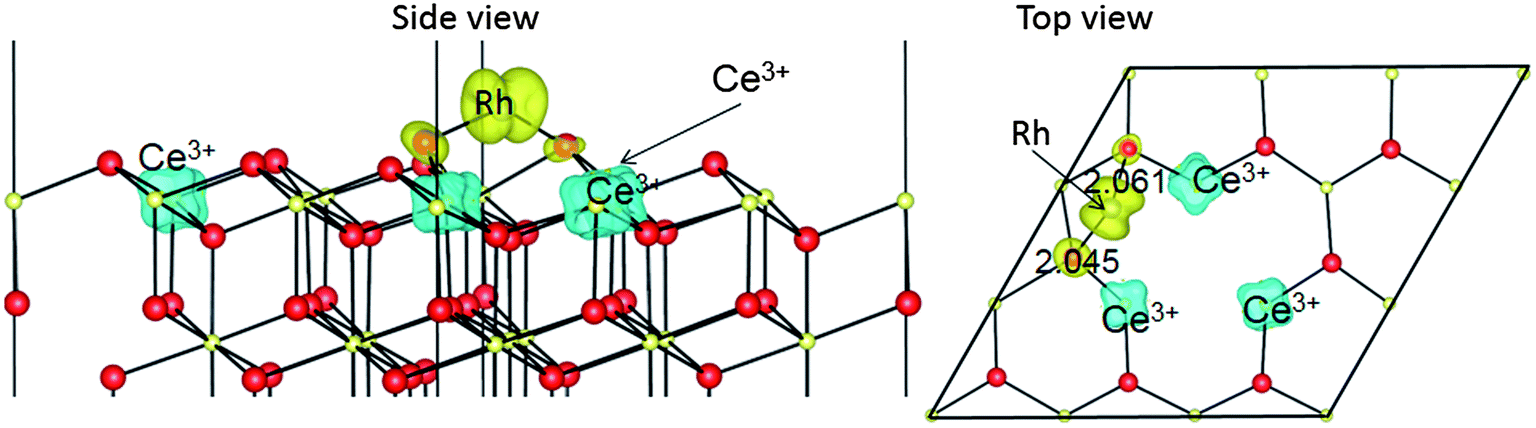 | ||
| Fig. 6 The geometric structures and spin density (ρspin up–ρspin down) for a Rh adatom neighbouring an oxygen vacancy site on CeO2(111), the result of attempting to dope the Rh onto a surface O site on CeO2(111). | ||
The Edoping value we obtain (−0.54 eV with respect to a gas phase Rh atom source) is meaningless, since the doped structure relaxes back into a vacancy and an adatom (see Fig. 6). However we can extract some useful information from our calculation in two different ways:
We can evaluate the adsorption energy for Rh next to an oxygen vacancy in a (3 × 3) cell:
| Eads next to VO = [E(Rh/CeO2−x)] − [E(CeO2−x) + Eref(Rh)] | (5) |
We can also evaluate the formation energy for an oxygen vacancy next to an adsorbed Rh.
| Evac next to Rh-adatom = [E(Rh/CeO2−x) + Eref(Rh)] − [E(Rh/CeO2)] | (6) |
This turns out to be +2.83 eV. If we calculate the energy for vacancy creation on the clean surface in the same p(3 × 3) supercell (calculated with the same plane-wave cut-off, k-point grid etc.), then we obtain a value of +2.65 eV, which is thus 0.17 eV less than in the vicinity of adsorbed Rh. This corresponds to a slight suppression of VO formation next to the adsorbed Rh, as calculated in this p(3 × 3) supercell. In the p(2 × 2) cell, we find slightly less suppression. This may perhaps imply that in a larger supercell than p(3 × 3) we would find even more suppression. However, in p(2 × 2) every single surface ion is part of the vacancy–Rh adatom complex, or is a nearest neighbour to it, with a very distorted position. In other words, there is no undisturbed CeO2(111) surface left within the supercell, so the p(2 × 2) cell is really too small for these calculations, hence the extrapolation may be unreliable.
We can also consider oxygen vacancy formation next to a Rh ion doped onto the Ce sublattice, as shown in Fig. 7. Vacancy creation in ceria normally involves the transfer of two electrons to localized Ce4f states, leading to two (nominally) Ce3+ ions. In this case, we find only one. The second electron has localized on the Rh dopant ion. In other words, it has entered the empty state in the upper part of the O2p ⇒ Ce4f gap which we previously found in the PDOS of the Rh dopant, as shown in Fig. 5b.
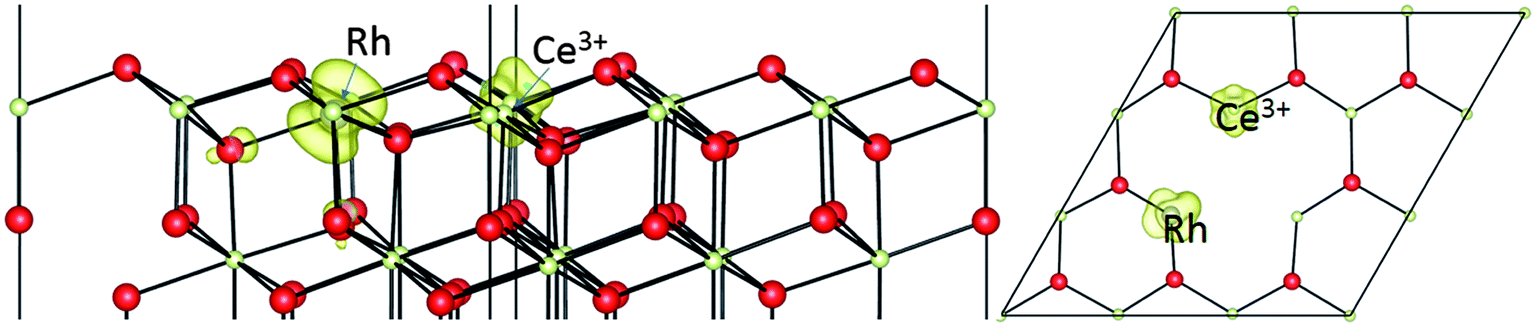 | ||
| Fig. 7 The geometric structures and spin density (ρspin up–ρspin down) for an oxygen vacancy neighbouring a Rh ion doped onto a surface Ce site on CeO2(111). | ||
Since this gap state lies below the Ce4f states, it provides an additional stabilization to the vacancy, so we find that the formation energy for a vacancy nearest neighbouring a Rh doped onto the Ce sublattice is +1.63 eV. This is 1.2 eV lower (less expensive) than in the absence of the Rh. Our 1.63 eV is in reasonable agreement with the value of 1.52 eV reported by Krcha et al.16 using a mirrored slab and a p(2 × 2) supercell. The reduction in formation energy relative to pure ceria (111) is also similar to the 0.9 eV reduction oberved by Aryanpour et al.17 It is interesting to note, however, that we find here a larger increase in activity when the Rh is doped into the surface Ce layer (T1b) rather than the subsurface Ce layer (T2b) as in the calculations of Aryanpour et al.
Hence we find a mild suppression of oxygen vacancy creation in the presence of low coverage, atomically dispersed Rh adatoms on the ceria (111) surface, which would adversely affect the oxygen storage capacity. However, this is countered by a significant enhancement of oxygen vacancy creation (and hence storage capacity) in the presence of single Rh dopants in the surface layer. On the other hand, as discussed in Section 3.3 above, in the absence of cerium vacancies, the formation of Rh dopants on the Ce sublattice is disfavoured at equilibrium, and Rh adatoms should be expected, unless non-equilibrium processes dominate. As a result, the effect of low coverage atomically dispersed Rh on the oxygen storage capacity of ceria (111) is likely to vary, being a complex interplay of the sample history and environment. It should, however, be emphasised that other effects will come into play at larger Rh coverages, especially if the formation of metallic islands and/or nanoclusters is considered.
4. Discussion
4.1 Comparison between the Rh doped and Rh adatom systems
We have found two stable locations for atomically dispersed Rh on the ceria(111) surface: doped Ce1−xRhxO2(111), and Rh adatom/CeO2(111) (the latter with numerous low energy configuration variations). The key differences between the two may be understood as follows: in the Rh adatom–ceria(111) system, the Rh at the centre of the three O anions is mainly oxidized by a Ce4+ → Ce3+ reduction process on a NNN Ce ion. Meanwhile in the doped Ce1−xRhxO2 system, the Rh atom is oxidized by its NN O ions, and to a higher oxidation state than in the adatom case, although to a lower oxidation state than that of the Ce ion it replaces.Comparing the two structures, in both cases (adatom or doped), Rh has only O neighbours, in agreement with Hosokawa et al.,11 rather than the Ce-top site suggested by Pfau et al.10 (note also that Rh adatoms lead to the presence of Ce3+ ions, even without the vacancy creation assumed by Pfau et al.). When comparing our calculated distances with those coming from EXAFS (Table 5),13 we must bear in mind that the DFT functional we are using, PBE+U, always under-binds, producing overly long bond lengths, and the under-binding worsens with increasing U > 0 eV.3 We obtained a 1.7% overestimation of the bulk lattice parameter, so our calculated Rh–O and Rh–Ce distances must be rescaled for comparison with the experiment. We thus compare Rh–O distances of 2.13 and 2.11 Å for the adatom and doped systems, respectively, with the EXAFS value of 2.05 Å. Neither of the theoretical structures match exactly, but neither is inconsistent. However, the experimental coordination number of 2.2 ± 0.6 is more in keeping with the adatom (3 closest neighbours) than the doped system (3 + 3). The Rh–Ce distances differ more. Taking the under-binding into account, we predict 2.94 Å for the adatom and 3.76 Å for the doped Rh, compared to 3.16 Å from the experiment (the coordination numbers in this case are harder to compare, as the models used to fit the experimental data mixed Ce and Rh). Neither of the comparisons are convincing, so overall, we cannot say whether the EXAFS better supports the adatom model or the Ce site doping model, or perhaps a mixture.
| Calculations (this work).a All Rh–X distances < 4 Å are included | Experimental EXAFSd (13) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Rh adatom at the OH site | Rh doped onto the Ce site | Assuming 1% Rh | Assuming 2% Rh | ||||||||
| Raw dist.b | Scaled dist.c | Rh coord. | Raw dist.b | Scaled dist.c | Rh coord. | Distance | Rh Coord. | Distance | Rh Coord. | ||
| a Calculated values are from the p(3 × 3) supercell. Distances, given in Ångströms, grouped by atomic layers, with labels T1a, T1b etc. following Fig. 1. All distances below 4 Å are included. b “Raw distances” are those obtained directly in the PBE+U calculations. c “Scaled distances” have been reduced to correct for PBE+U's 1.7% overestimation of the lattice parameter. This allows easier comparison with experiments. d Two sets of EXAFS distances were presented in ref. 13, both from the same set of experimental data, one through fitting under the assumption of 1% Rh doping of the cerium sublattice, the other under the assumption of 2% Rh doping. The EXAFS results find the first shell of O ions around the Rh, followed by a cation shell which contains neighbouring Rh substitutions as well as Ce. | |||||||||||
| Rh–O | T1a | 2.166 | 2.130 | 3 | 2.149 | 2.113 | 3 | 2.045 ± 0.003 | 2.2 ± 0.6 | 2.060 ± 0.005 | 2.3 ± 0.4 |
| T1c | 2.883 | 2.835 | 1 | 2.210 | 2.173 | 3 | |||||
| T2a | N/A | N/A | N/A | 2.529 | 2.487 | 1 | |||||
| Rh–Ce | T1b | 2.985 | 2.935 | 3 | 3.822 | 3.758 | 6 | 3.163 ± 0.004 | 5.4 ± 0.7 | 3.157 ± 0.006 | 5.1 ± 0.5 |
| T2b | N/A | N/A | N/A | 3.820 | 3.756 | 3 | |||||
| Rh–Rh | N/A | N/A | 2.722 ± 0.003 | 5.7 ± 0.3 | 2.733 ± 0.003 | 5.4 ± 0.5 | |||||
Regarding the energetics, the relative stability of the two sites, and the various possible interactions of lone Rh atoms approaching the ceria(111) surface, we find a wide variation in the environment, conditions and sample history. We find that the Rh will never heal an existing oxygen vacancy, or replace a surface oxygen ion, but instead moves to an adatom position, most likely at more than the nearest neighbour distance from the vacancy. For the lone adatom in a vacancy-free region of the surface, the most stable configuration has Eads = −3.37 eV relative to Rh vapour. However, if the opposite environmental extreme is considered (hence adsorption relative to Rh metal) Eads = +2.53 eV, so that Rh adatoms are then not stable.
For the doped systems, the energy gained from Rh healing of cerium vacancies is always significant (−10.22 eV up to −4.32 eV), but replacement of an existing surface Ce ion always costs energy, with Edoping ranging from +5.44 eV up to +14.35 eV. However, these calculated energies each assume some form of equilibrium, so the fact that Rh doping is believed to have been seen experimentally,13 may be due to non-equilibrium formation processes (such as Ce deficient growth), and kinetic limitations. Indeed, Gayen et al.13 saw indications of three separate Rh sites in their samples, which were prepared by non-equilibrium methods. So, in summary, we may assume that both Rh adatoms and Rh surface doping do occur, depending upon the conditions and sample history.
4.2 Comparison between the p(2 × 2) and p(3 × 3) supercells
In most cases we find little difference in electronic or geometric structures between the p(2 × 2) and p(3 × 3) supercells. However, some of the structures we have found simply cannot exist in the smaller supercell, including the second most stable structure overall, namely a Rh adatom with Ce4f charge localization at the NNNN distance from the Rh. This inability to tell different Ce4f charge localization patterns apart is obviously important in many situations. Similarly, we find weak AFM coupling to the spin on the Ce3+ ion in the p(3 × 3) cell, but weak FM coupling in p(2 × 2). Obviously, if these structures or properties are under examination, the larger p(3 × 3) cell is needed.Regarding the energetics, we found significant differences between the two cells in some cases, but small changes in others. The problem is that there is currently no known a priori way to tell when the difference will be small, or when the larger cell is needed in order to study isolated defects, apart from simply trying it. Indeed, even p(3 × 3) is a little small: the energies of the NNN1 and NNN2 configurations should be equivalent in a large enough supercell, but differ here by ∼0.21 eV, with the two NNNN configurations in between. Indeed, we see indications that in a large enough supercell, NNNN might be more stable.
5. Conclusions
Using plane wave DFT with the PBE+U functional, we have studied the geometric and electronic structures and energetics of lone Rh atoms interacting with stoichiometric and non-stoichiometric ceria(111) surfaces. We find that Rh will readily heal any cerium vacancies present, leading to Rh doping of the Ce sublattice, but it is much less likely to eject and replace an existing Ce ion. Rh healing of oxygen vacancies, or Rh doping into the oxygen sublattice, is never stable. Rh adatoms form readily under most, though not all conditions. In keeping with previous studies,21,26 we find that the most stable Rh adatom configuration has the Rh in a three-fold hollow site with only oxygen nearest neighbours (three in the T1a surface layer and one in the T1c subsurface layer), together with one Ce3+ ion located at the next nearest neighbour (NNN) distance. However, we have shown here that this is only one of a large number of possible configurations, all very close in energy (within 0.4 eV), suggesting that at experimentally relevant temperatures the adatom structure may appear much more dynamic and variable.We have found some evidence suggesting that, at low coverage, atomically dispersed Rh adatoms would mildly suppress the oxygen storage capacity, while atomically dispersed Rh dopants in the surface layer, which should be much less common than Rh adatoms, would strongly enhance it. Hence the oxygen storage capacity in low coverage Rh–ceria(111) systems should be strongly dependent on sample preparation, history and conditions. At higher (at least local) coverages, other processes may be expected, such as metallic Rh island growth, or Rh nano-cluster growth.
For the Rh-doped ceria(111) system, it is found that the Rh dopant is oxidized mainly by its O neighbours, while at the adatom site it is oxidized mainly by a NNN Ce ion, which converts Ce4+ to Ce3+, as also observed experimentally. Hence charge transfer from Rh to CeO2 occurs in both cases, but in one the transfer is to O2p states and in the other it is to Ce4f.
We have also compared results from the p(2 × 2) and p(3 × 3) supercells, and found that in most cases the results are quite similar, although some structures are too large to fit properly into the p(2 × 2) supercell, and some, but not all, calculated energies can differ significantly between the two supercells. In these cases larger supercells are essential for the study of lone defects.
Returning to our original set of questions:
(i) First it heals Ce vacancies, and then it forms adatoms. It can not easily eject and replace an existing Ce ion, and will never replace an oxygen ion, or heal an oxygen vacancy.
(ii) Rh adatoms have a mild suppressing effect, but Rh dopants strongly enhance it.
(iii) About 0.5–0.7 e, and yes, there is a multitude!
(iv) The results are generally similar, but only when the structure will fit into p(3 × 3).
Acknowledgements
Support for this work from the National Natural Scence Foundation of China (Grant no. 11147006, 11174070 and U1304518), China Postdoctoral Science Foundation funded project (Grant no. 2012M521399) and Postdoctoral Research sponsorship in Henan Province (Grant no. 2011038), and the Foundation for the Key Young Teachers of Henan Normal University and Start-up Foundation for Doctors of Henan Normal University is gratefully acknowledged. On the Swedish side, we are thankful for support from the Swedish Research Council (VR), the Swedish National Strategic e-Science program eSSENCE, the Swedish Foundation for International Cooperation in Research and Higher Education (STINT) and the COST Action CM1104 (Reducible oxide chemistry, structure and functions). Part of the calculations were performed with resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC and UPPMAX.References
- A. Trovarelli, Catalysis by ceria and related materials, Imperial College Press, 2002 Search PubMed.
- H. C. Yao and Y. F. Yu Yao, J. Catal., 1984, 86, 254–265 CrossRef CAS.
- Z. Yang, T. K. Woo, M. Baudin and K. Hermansson, J. Chem. Phys., 2004, 120, 7741 CrossRef CAS PubMed.
- M. Nolan, S. Grigoleit, D. C. Sayle, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 576, 217–229 CrossRef CAS PubMed.
- M. Nolan, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 595, 223–232 CrossRef CAS PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- S. Bernal, J. J. Calvino, M. A. Cauqui, J. M. Gatica, C. Larese, J. A. Perez Omil and J. M. Pintado, Catal. Today, 1999, 50, 175–206 CrossRef CAS.
- I. K. Naik and T. Y. Tien, J. Phys. Chem. Solids, 1978, 39, 311–315 CrossRef CAS.
- A. Pfau and K. D. Schierbaum, Surf. Sci., 1994, 321, 71–80 CrossRef CAS.
- A. Pfau, K. D. Schierbaum and W. Göel, Surf. Sci., 1995, 331–333, 1479–1485 CrossRef CAS.
- S. Hosokawa, M. Taniguchi, K. Utani, H. Kanai and S. Imamura, Appl. Catal., A, 2005, 289, 115–120 CrossRef CAS PubMed.
- R. Wang, H. Xu, X. Liu, Q. Ge and W. Li, Appl. Catal., A, 2006, 305, 204–210 CrossRef CAS PubMed.
- A. Gayen, K. R. Priolkar, P. R. Sarode, V. Jayaram, M. S. Hegde, G. N. Subbanna and S. Emura, Chem. Mater., 2004, 16, 2317–2328 CrossRef CAS.
- V. Shapovalov and H. Metiu, J. Catal., 2007, 245, 205–214 CrossRef CAS PubMed.
- Z. Yang, G. Luo, Z. Lu, T. K. Woo and K. Hermansson, J. Phys.: Condens. Matter, 2008, 20, 035210 CrossRef PubMed.
- M. D. Krcha, A. D. Mayernick and M. J. Janik, J. Catal., 2012, 293, 103–115 CrossRef CAS PubMed.
- M. Aryanpour, A. Khetan and H. Pitsch, ACS Catal., 2013, 3, 1253–1262 CrossRef CAS.
- H. L. Chen, S. H. Liu and J. J. Ho, J. Phys. Chem. B, 2006, 110, 14816–14823 CrossRef CAS PubMed.
- H.-L. Chen, W.-T. Peng, J.-J. Ho and H.-M. Hsieh, Chem. Phys., 2008, 348, 161–168 CrossRef CAS PubMed.
- S. Nokbin, K. Hermansson and J. Limtrakul, Abstr. Papers Am. Chem. Soc., 2006, 231 Search PubMed , INOR 247.
- Z. Lu and Z. Yang, J. Phys.: Condens. Matter, 2010, 22, 475003 CrossRef PubMed.
- B. Li, O. K. Ezekoye, Q. Zhang, L. Chen, P. Cui, G. Graham and X. Pan, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125422 CrossRef.
- W. Song, A. P. J. Jansen and E. J. M. Hensen, Faraday Discuss., 2013, 162, 281–292 RSC.
- D. Gerçeker and I. Önal, Appl. Surf. Sci., 2013, 285P, 927–936 CrossRef PubMed , in press.
- J. Paier, C. Penschke and J. Sauer, Chem. Rev., 2013, 113, 3949–3985 CrossRef CAS PubMed.
- W. Song, C. Popa, A. P. J. Jansen and E. J. M. Hensen, J. Phys. Chem. C, 2012, 116, 22904–22915 CAS.
- M. V. Ganduglia-Pirovano, J. L. F. Da Silva and J. Sauer, Phys. Rev. Lett., 2009, 102, 026101 CrossRef.
- J. Kullgren, K. Hermansson and C. Castleton, J. Chem. Phys., 2012, 137, 044705 CrossRef PubMed.
- H.-Y. Li, H.-F. Wang, X.-Q. Gong, Y.-L. Guo, Y. Guo, G. Lu and P. Hu, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 193401 CrossRef.
- G. E. Murgida and M. V. Ganduglia-Pirovano, Phys. Rev. Lett., 2013, 110, 246101 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS.
- J. L. F. Da Silva, M. V. Ganduglia-Pirovano, J. Sauer, V. Bayer and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 045121 CrossRef.
- D. A. Andersson, S. I. Simak, B. Johansson, I. A. Abrikosov and N. V. Skorodumova, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035109 CrossRef.
- L. Christoph, C. Javier, M. N. Konstantin and I. Francesc, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035115 CrossRef.
- S. Fabris, S. de Gironcoli, S. Baroni, G. Vicario and G. Balducci, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71 CrossRef CAS , 041102(R).
- C. W. M. Castleton, J. Kullgren and K. Hermansson, J. Chem. Phys., 2007, 127, 244704 CrossRef CAS PubMed.
- S. Rossignol, F. Gerard, D. Mesnard, C. Kappenstein and D. Duprez, J. Mater. Chem., 2003, 13, 3017–3020 RSC.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS.
- H. L. Tuller and A. S. Nowick, J. Phys. Chem. Solids, 1977, 38, 859–867 CrossRef CAS.
- C. Castleton, A. Green, J. Kullgren and K. Hermansson, 2014, in preparation.
- T. Zacherle, A. Schriever, R. A. De Souza and M. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 134104 CrossRef.
- N. C. Hernandez, R. Grau-Crespo, N. H. de Leeuw and J. F. Sanz, Phys. Chem. Chem. Phys., 2009, 11, 5246–5252 RSC.
- G. Henkelman, A. Arnaldsson and H. Jonsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef PubMed.
- M. E. Grillo, Comput. Mater. Sci., 2005, 33, 83–91 CrossRef CAS PubMed.
- G. Demazeau, A. Baranov, R. Pottgen, L. Kienle, M. H. Moller, R.-D. Hoffmann and M. Valldor, Z. Naturforsch., A: Phys. Sci., 2006, 61B, 1500 Search PubMed.
- A. Wold, R. J. Arnott and W. J. Croft, Inorg. Chem., 1963, 2, 972–974 CrossRef CAS.
- Y. D. Scherson, S. J. Aboud, J. Wilcox and B. J. Cantwell, J. Phys. Chem. C, 2011, 115, 11036–11044 CAS.
| This journal is © The Royal Society of Chemistry 2014 |