
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Are block copolymer worms more effective Pickering emulsifiers than block copolymer spheres?†
K. L.
Thompson
*,
C. J.
Mable
,
A.
Cockram
,
N. J.
Warren
,
V. J.
Cunningham
,
E. R.
Jones
,
R.
Verber
and
S. P.
Armes
*
Department of Chemistry, The University of Sheffield, Dainton Building, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@sheffield.ac.uk
First published on 12th September 2014
Abstract
RAFT-mediated polymerisation-induced self-assembly (PISA) is used to prepare six types of amphiphilic block copolymer nanoparticles which were subsequently evaluated as putative Pickering emulsifiers for the stabilisation of n-dodecane-in-water emulsions. It was found that linear poly(glycerol monomethacrylate)–poly(2-hydroxypropyl methacrylate) (PGMA–PHPMA) diblock copolymer spheres and worms do not survive the high shear homogenisation conditions used for emulsification. Stable emulsions are obtained, but the copolymer acts as a polymeric surfactant; individual chains rather than particles are adsorbed at the oil–water interface. Particle dissociation during emulsification is attributed to the weakly hydrophobic character of the PHPMA block. Covalent stabilisation of these copolymer spheres or worms can be readily achieved by addition of ethylene glycol dimethacrylate (EGDMA) during the PISA synthesis. TEM studies confirm that the resulting cross-linked spherical or worm-like nanoparticles survive emulsification and produce genuine Pickering emulsions. Alternatively, stabilisation can be achieved by either replacing or supplementing the PHPMA block with the more hydrophobic poly(benzyl methacrylate) (PBzMA). The resulting linear spheres or worms also survive emulsification and produce stable n-dodecane-in-water Pickering emulsions. The intrinsic advantages of anisotropic worms over isotropic spheres for the preparation of Pickering emulsions are highlighted. The former particles are more strongly adsorbed at similar efficiencies compared to spheres and also enable smaller oil droplets to be produced for a given copolymer concentration. The scalable nature of PISA formulations augurs well for potential applications of anisotropic block copolymer nanoparticles as Pickering emulsifiers.
Introduction
Pickering emulsions comprise oil or water droplets stabilised by solid particles and have been recognised for over a century.1,2 Many different types of solid particles have been utilised as Pickering emulsifiers, such as inorganic clays,3–6 silica7–11 and polymer latex particles.12 A layer of adsorbed particles surrounds each droplet to provide a strong steric barrier towards droplet coalescence, making Pickering emulsions far more stable than surfactant-stabilised emulsions. The energy of detachment for an individual particle adsorbed at the oil–water interface depends on both its three-phase contact angle and the particle radius, with significantly more energy being required to remove larger particles from the oil–water interface.13 In practice, this detachment energy is often so large (> 1000 kT) that in some cases certain types of nanoparticles can be considered to be irreversibly adsorbed.13,14Much of the growing literature on Pickering emulsions has focused on spherical particles. There are far fewer reports of the use of non-spherical Pickering emulsifiers based on rigid rods or flexible worms. Noble et al.15 prepared so-called ‘hairy’ colloidosomes using relatively large (10–70 μm) ‘microrods’ prepared from a photocurable epoxy resin as the droplet stabiliser. In this case the hot aqueous phase contained 1.5 wt% agarose that gelled upon cooling, trapping the ‘microrods’ at the interface. Vermant and co-workers16 used a multiple back-scattering technique to demonstrate that more stable Pickering emulsions are obtained when employing elongated polystyrene latexes with relatively high aspect ratios (with the original isotropic latex particles being used as a control). Similar results were also obtained with ellipsoidal hematite particles.16 Guevara et al.17 reported that 2D nano-sheets can produce highly stable foams. Cellulose fibres have also been used to stabilise both water-in-oil and oil-in-water emulsions.18–20 In this case the fibre aspect ratio strongly influenced the droplet surface coverage: short fibres led to densely-coated oil droplets (>80% coverage), whereas using longer fibres produced significantly lower surface coverages (∼40%).20
Recently, our group has reported the facile preparation of a range of diblock copolymer nanoparticles via polymerisation-induced self-assembly (PISA) using reversible addition–fragmentation chain transfer (RAFT) polymerisation in concentrated aqueous solution.21,22 In the prototypical formulation, a water-soluble poly(glycerol monomethacrylate) (PGMA) chain transfer agent is used to grow a water-insoluble poly(2-hydroxypropyl methacrylate) (PHPMA) block, leading to the in situ formation of PGMA–PHPMA diblock copolymer nanoparticles via RAFT aqueous dispersion polymerisation.23–25 In principle, the final particle morphology is simply dictated by the relative volume fractions of each block, although in practice the copolymer concentration can also play an important role.26 This versatile route to block copolymer nanoparticles enables the efficient synthesis of spheres, worms or vesicles directly in water at up to 25% solids. Moreover, the surface properties of these nanoparticles can be tuned by judicious selection of the stabiliser for a given core-forming block,27–31 which is likely to be of considerable interest when designing new bespoke Pickering emulsifiers.
Recently, we compared the Pickering emulsifier performance of linear and crosslinked PGMA–PHPMA block copolymer vesicles (also known as ‘polymersomes’) prepared using a PISA formulation.32 One important finding was that use of an ethylene glycol dimethacrylate (EGDMA) cross-linker was essential to preserve the relatively delicate vesicular morphology during the high-shear homogenisation required to generate the oil droplets. In the absence of any EGDMA, the linear vesicles simply dissociated during homogenisation and the resulting emulsion droplets became stabilised by individual copolymer chains, rather than vesicles. In contrast, EGDMA-crosslinked vesicles survived the emulsification conditions and were able to function as the desired ‘Pickering’ emulsifier. In the present work, we explore whether covalent cross-linking is also essential to prevent dissociation of PGMA-based block copolymer spheres and worms when employed as putative Pickering emulsifiers. In particular, we emphasise that the block copolymer worms described herein are much more readily accessible on a multi-gram scale than the various anisotropic nanoparticles that have been explored to date.15,16,18–20 Provided that conformal contact with the interface can be achieved, such worms are expected to be much more strongly adsorbed at the oil–water interface than the equivalent spherical nanoparticles. For example, if worm-like nanoparticles are sufficiently anisotropic (i.e. if their mean contour length L is much greater than the mean worm radius R), then their specific surface area, As, can be approximated by the equation: As ∼ 2/ρR, where ρ is the particle density and R is the mean worm radius (see ESI† for further details). In contrast, the spherical nanoparticles that undergo 1D fusion to form such worms during the PISA synthesis24,27 have a specific surface area given by As = 3/ρr, where r is the mean sphere radius and r ∼ R. Thus the reduction in specific surface area that occurs on fusing multiple spheres to form each individual worm is only around 33%. To a good first approximation, the energy of attachment of a worm of radius R comprising x fused spheres at the oil–water interface is expected to be x times higher than the energy of attachment of an individual spherical nanoparticle of radius r. Thus the worm morphology affords a specific surface area comparable to that of the original spheres, but the former particles are much more strongly adsorbed at the oil–water interface. Hence the present study is focused on addressing the following fundamental question: do worm-like nanoparticles offer significant advantages as Pickering emulsifiers over their spherical nanoparticle precursors?
For the sake of brevity, we introduce a shorthand notation to describe the various diblock and triblock copolymers synthesised in this study such that G, H, B and E stand for glycerol monomethacrylate, 2-hydroxypropyl methacrylate, benzyl methacrylate and ethylene glycol dimethacrylate, respectively. For example, Gx–Hy–Ez indicates a poly(glycerol monomethacrylate-block-2-hydroxypropyl methacrylate-block-ethylene glycol dimethacrylate) triblock copolymer, where x, y and z indicate the mean degrees of polymerisation (DP) of each block.
Experimental
Materials
Glycerol monomethacrylate was obtained from GEO Specialty Chemicals (Hythe, UK) and was used as received. 2-Hydroxypropyl methacrylate, ethylene glycol dimethacrylate, 4,4′-azobis(4-cyanopentanoic acid) (ACVA), n-dodecane, and tolylene 2,4-diisocyanate-terminated poly(propylene glycol) (PPG-TDI) were purchased from Aldrich (UK) and were used as received, unless otherwise stated. 4-Cyano-4-(2-phenyl-ethanesulfanylthiocarbonyl)sulfanylpentanoic acid (PETTC) was prepared in-house as reported previously and n-hexane was purchased from Fisher.31Synthesis of poly(glycerol monomethacrylate) macromolecular chain transfer agent (macro-CTA)
PGMA macro-CTAs with mean degrees of polymerisation of 37, 45, 51 and 100 (denoted G37, G45, G51 and G100, respectively) were synthesised via RAFT polymerisation of glycerol monomethacrylate in ethanol at 70 °C, as described previously.26Synthesis of linear G100–H200 nanoparticles via RAFT aqueous dispersion polymerisation
G100 macro-CTA (0.5 g, 0.031 mmol), HPMA monomer (0.89 g, 6.16 mmol) ACVA (2.9 mg, 0.010 mmol, CTA/ACVA molar ratio = 3.0) and previously degassed deionised water (13.59 g, 10% w/w), were weighed into a 50 mL round-bottomed flask. The reaction solution was purged with N2 for 30 min prior to immersion in an oil bath set at 70 °C. The reaction was stirred for 3 h at this temperature under a N2 atmosphere prior to being quenched by exposure to air. Linear G45–H100 diblock copolymer worms were prepared using the same protocol.Synthesis of crosslinked G100–H200–E20 nanoparticles via RAFT aqueous dispersion polymerisation
G100 macro-CTA (0.5 g, 0.031 mmol), HPMA monomer (0.89 g, 6.16 mmol) ACVA (2.9 mg, 0.010 mmol, CTA/ACVA molar ratio = 3.0) and previously degassed deionised water (13.59 g, 10% w/w), were weighed into a 50 mL round-bottomed flask. The reaction solution was purged with N2 for 30 min prior to immersion in an oil bath set at 70 °C. The reaction mixture was stirred for 3 h at this temperature under a N2 atmosphere prior to the addition of EGDMA cross-linker (0.122 g, 0.616 mmol). The colloidal dispersion was stirred overnight (16 h) to ensure complete EGDMA monomer conversion and the polymerisation was quenched by exposure to air. Crosslinked G45–H100–E10 worms were also prepared using the above protocol.Synthesis of linear G51–B250 nanoparticles via RAFT aqueous emulsion polymerisation
A typical protocol for the synthesis of PGMA51–PBzMA250 diblock copolymer was as follows: PGMA51 macro-CTA (0.1702 g), BzMA (0.8904 g, 5.05 mmol), ACVA (1.40 mg, 4.99 μmol; CTA/ACVA molar ratio = 4.0) and water (7.97 g, 10% w/w) were weighed into a 25 mL round-bottom flask and purged with nitrogen for 30 min, prior to immersion in an oil bath set at 70 °C for 20 h. The resulting copolymer was analysed by DMF GPC (Mn = 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 g mol−1, Mw/Mn = 1.25 vs. PMMA standards). 1H NMR spectroscopy analysis of the freeze-dried copolymer redissolved in d7-DMF indicated less than 1% residual BzMA monomer. DLS studies of a 0.20% w/w copolymer dispersion indicated an intensity-average particle diameter of 67 nm (DLS polydispersity = 0.08).
700 g mol−1, Mw/Mn = 1.25 vs. PMMA standards). 1H NMR spectroscopy analysis of the freeze-dried copolymer redissolved in d7-DMF indicated less than 1% residual BzMA monomer. DLS studies of a 0.20% w/w copolymer dispersion indicated an intensity-average particle diameter of 67 nm (DLS polydispersity = 0.08).
RAFT seeded emulsion polymerisation of linear G37–H60–B30 worms
G37 macro-CTA (1.00 g, 0.16 mmol), HPMA monomer (1.41 g, 9.76 mmol), ACVA (9.11 mg, 0.033 mmol, CTA/ACVA molar ratio = 5.0) and previously degassed deionised water (21.75 g, 10% w/w), were weighed into a 50 mL round-bottomed flask. The reaction solution was purged with N2 for 30 min prior to immersion in an oil bath set at 70 °C. The reaction solution was stirred for 2 h at this temperature under a N2 atmosphere prior to the addition of BzMA (0.86 g, 0.616 mmol). The colloidal dispersion was stirred for a further 4 h to ensure complete BzMA monomer conversion and the polymerisation was quenched by exposure to air.Preparation of Pickering emulsions and colloidosomes
Either n-dodecane or n-hexane (2.0 mL) was homogenised with 2.0 mL of a 0.05–2.00% w/w aqueous copolymer dispersion for 2 min at 20 °C using a IKA Ultra-Turrax T-18 homogeniser with a 10 mm dispersing tool operating at 12000 rpm. All emulsions proved to be stable with respect to droplet coalescence for months on standing at 20 °C. To prepare the colloidosomes, PPG-TDI (8.0 mg) crosslinker was weighed into a sample vial and then dissolved in n-hexane (2.0 mL) prior to homogenisation with the aqueous copolymer dispersion. The resulting stable milky-white emulsion was allowed to stand at 20 °C for 1 h to allow urethane bond formation between the terminal isocyanate groups on the cross-linker and the hydroxyl groups on the PGMA (and/or PHPMA) chains.
DMF GPC
Molecular weights and polydispersities were assessed using a gel permeation chromatography (GPC) instrument equipped with a Varian 290-LC pump injection module, a Varian 390-LC refractive index detector, two Polymer Laboratories PL gel 5 μm Mixed-C columns with a DMF mobile phase containing 0.01 M LiBr operating at 60 °C with a constant flow rate of 1.0 mL min−1. DMSO was used as a flow rate marker and calibration was achieved using a series of near-monodisperse poly(methyl methacrylate) standards.1H NMR spectroscopy
1H NMR spectra were recorded in either D2O or CD3OD using a Bruker Avance 400 spectrometer operating at 400 MHz.Dynamic light scattering (DLS)
Intensity-average hydrodynamic diameters were obtained by DLS using a Malvern Zetasizer NanoZS instrument at a fixed scattering angle of 173°. Aqueous dispersions of 0.01% w/w particles were analysed using disposable cuvettes and the results were averaged over three consecutive runs. The deionised water used to dilute each sample was ultra-filtered through a 0.20 μm membrane in order to remove extraneous dust.Transmission electron microscopy
Aqueous particle dispersions were diluted fifty-fold at 20 °C to generate 0.20% w/w dispersions for transmission electron microscopy (TEM) studies. Copper/palladium TEM grids (Agar Scientific) were surface-coated in-house to produce a thin film of amorphous carbon. The grids were then plasma glow-discharged for 30 seconds to create a hydrophilic surface. Individual samples (0.20% w/w, 12 μL) were adsorbed onto the freshly glow-discharged grids for one minute and then blotted with filter paper to remove excess solution. To stain the copolymer aggregates, uranyl formate solution (0.75% w/w; 9 μL) was soaked on the sample-loaded grid for 20 seconds and then carefully blotted to remove excess stain. The grids were then dried using a vacuum hose. Imaging was performed at 100 kV using a Phillips CM100 instrument equipped with a Gatan 1 k CCD camera.Optical microscopy
Optical microscopy images of Pickering emulsion droplets were recorded using a Motic DMBA300 digital biological microscope equipped with a built-in camera and analysed using Motic Images Plus 2.0 ML software.Laser diffraction
Each emulsion was sized using a Malvern Mastersizer 2000 instrument equipped with a small volume Hydro 2000SM sample dispersion unit (ca. 50 mL), a HeNe laser operating at 633 nm and a solid-state blue laser operating at 466 nm. The stirring rate was adjusted to 1000 rpm in order to avoid creaming of the emulsion during analysis. After each measurement, the cell was rinsed once with ethanol, followed by three rinses with doubly-distilled water; the glass walls of the cell were carefully wiped with lens cleaning tissue to avoid cross-contamination and the laser was aligned centrally to the detector prior to data acquisition.Determination of Pickering emulsifier efficiency via either turbidimetry or UV spectroscopy
The nanoparticle adsorption efficiency at the oil–water interface was determined by analysis of the lower aqueous phase after creaming of the oil droplets had occurred on standing. The remaining non-adsorbed nanoparticles were detected and thus the fraction of adsorbed nanoparticles was calculated indirectly by difference. Turbidimetry was used for the crosslinked Gx–Hy–Ez nanoparticles, with scattering curves being determined for known concentrations of aqueous copolymer dispersions in order to produce a linear calibration plot of absorbance versus concentration at an arbitrary wavelength of 750 nm (see ESI, Fig. S3†). For the linear PBzMA-based nanoparticles (G51–B250 and G37–H60–B30), calibration plots were constructed in DMF, which leads to molecular dissolution of the copolymer chains. The UV absorbance at 269 nm assigned to the aromatic benzyl groups on the PBzMA blocks was determined as a function of copolymer concentration to obtain a linear calibration plot (see ESI, Fig. S3†). The lower aqueous phases of these creamed Pickering emulsions were then dried at 50 °C for 120 h prior to molecular dissolution of the copolymer in DMF. [This protocol was essential to avoid UV scattering problems, but it could not be utilised for the crosslinked worms described above since these nanoparticles cannot be molecularly dissolved.] The fraction of nanoparticles adsorbed onto the oil droplets was then calculated by difference.Results and discussion
The synthetic route used to prepare Pickering emulsions and colloidosomes using either spherical or worm-like PGMA–PHPMA nanoparticles is shown in Fig. 1. We have previously reported that the analogous block copolymer vesicles can stabilise n-dodecane emulsions.32 However, it was found that an EGDMA cross-linker was essential to retain the vesicular morphology during high shear homogenisation in order to obtain genuine Pickering emulsions. In contrast, linear vesicles prepared in the absence of any EGDMA did not survive the high-shear emulsification conditions. Instead, vesicular dissociation occurred to produce individual copolymer chains which subsequently acted as macromolecular surfactants for the emulsion droplets.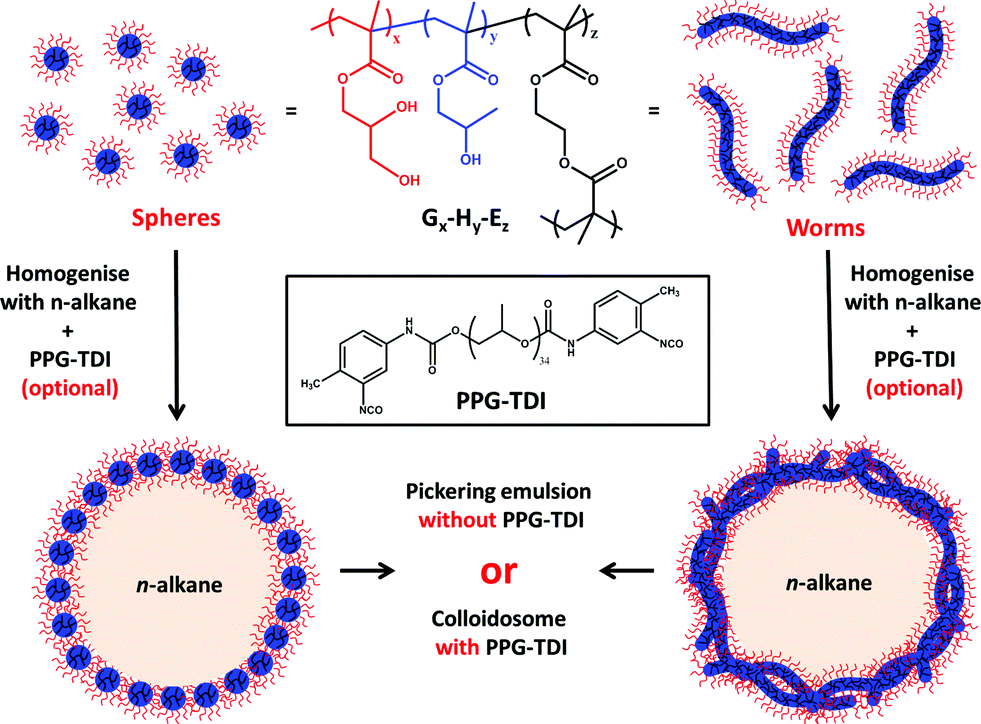 | ||
| Fig. 1 Synthetic route to Pickering emulsions using (left) spherical or (right) worm-like triblock copolymer nanoparticles prepared by RAFT aqueous dispersion polymerisation of HPMA at 70 °C using a PGMA macro-CTA stabiliser and an EGDMA cross-linker. The relative mean degrees of polymerisation of the PGMA and PHPMA blocks (x or y, respectively) dictate their relative volume fractions and hence the final nanoparticle morphology. Colloidosomes are obtained simply by addition of PPG-TDI cross-linker to the oil phase prior to homogenisation, which was conducted at 12000 rpm for 2 min at 20 °C. | ||
In the present work, we wished to investigate whether the problem of shear-induced particle disintegration was restricted to vesicles or also applied to spherical nanoparticles and worm-like micelles. It has been previously shown that linear PGMA–PHPMA vesicles are relatively delicate: they undergo partial collapse under ultrahigh vacuum conditions27 and do not tolerate the addition of ionic surfactants.33 Thus it was conceivable that the vesicular copolymer morphology alone was the primary reason for the disintegration observed during high-shear emulsification. On the other hand, it is also known that the PHPMA block is only weakly hydrophobic. For example, variable temperature 1H NMR studies indicate that significant plasticisation of the core-forming PHPMA block occurs on cooling an aqueous dispersion of PGMA–PHPMA worms from 25 °C to 5 °C, which is sufficient to induce a worm-to-sphere order–order transition.34 Thus the specific block composition (rather than the vesicular morphology per se) could be the main reason for the vesicle dissociation observed in the absence of chemical cross-linker. If this were the case, then linear PGMA–PHPMA spheres and worms might also be expected to break up when subjected to the same high shear conditions.
In order to further investigate the possible loss of copolymer morphology during high shear emulsification, six examples of linear and crosslinked block copolymer worms and spheres were prepared using RAFT-mediated PISA formulations. Table 1 summarises the copolymer morphologies, molecular weight data and DLS diameters obtained for the various G–H, G–H–E, G–Bz and G–H–B nanoparticles used in this investigation. It is emphasised that DLS reports a spherical-average hydrodynamic diameter based on the Stokes–Einstein equation; thus this parameter should be treated with some caution when considering highly anisotropic worm-like particles.
| Target block copolymer composition | Copolymer morphology | Linear or crosslinked? | M n b/g mol−1 | M w/Mn | DLS diameter (PDI)/nm | Average worm contour length (nm) | Droplet diameter at 0.06% w/w (μm) | Pickering adsorption efficiencya (%) | Droplet surface coverage at maximum efficiencya |
|---|---|---|---|---|---|---|---|---|---|
| a Calculated at 0.06% w/w copolymer concentration. | |||||||||
| G100–H200 | Spheres | Linear | 82800 |
1.20 | 50 (0.14) | — | 48 ± 14 | — | — |
| G100–H200–E20 | Spheres | Crosslinked | — | — | 47 (0.13) | — | 237 ± 129 | 90 | 0.93 |
| G45–H140 | Worms | Linear | 39300 |
1.08 | 315 (0.34) | 176 ± 115 | 46 ± 14 | — | — |
| G45–H100–E10 | Worms | Crosslinked | — | — | 122 (0.28) | 172 ± 117 | 179 ± 50 | 90 | 0.47 |
| G51–B250 | Spheres | Linear | 51100 |
1.19 | 81 (0.10) | — | 418 ± 117 | 80 | 0.91 |
| G37–H60–B30 | Worms | Linear | 21000 | 1.16 | 72 (0.21) | 120 ± 90 | 219 ± 73 | 100 | 0.71 |
Representative TEM images obtained for the six types of block copolymer nanoparticles summarised in Table 1 are shown in Fig. 2. The synthesis of crosslinked spherical nanoparticles (G100–H200–E20) was relatively straightforward, with 10 mol% EGDMA (based on HPMA monomer) simply being added at the end of the HPMA polymerisation.23 In this case, the cross-linker has minimal effect on both the particle size and morphology, with uniform spheres of around 50 nm diameter being produced both in the presence and absence of EGDMA. In contrast, the preparation of crosslinked worms was rather more problematic. This is presumably because the pure worm phase occupies a relatively narrow region of the phase diagram.27 Highly anisotropic flexible worms were obtained when targeting a G45–H140 diblock (see Fig. 2C). However, replacing 10 units of HPMA with the same number of EGDMA units (i.e. targeting G45–H130–E10) only resulted in a mixed vesicle/worm phase (see Fig. S1 in the ESI†). It seems that the addition of cross-linker shifts the vesicle/worm phase boundary, at least for this particular formulation. In view of this problem, a shorter PHPMA block was targeted to afford an overall triblock composition of G45–H100–E10. Fortunately, this formulation yielded a pure phase comprising crosslinked worms (Fig. 2D). Recently, we have found that RAFT aqueous emulsion polymerisation of BzMA using a PGMA macro-CTA invariably gives a purely spherical morphology, regardless of the target block DPs or copolymer concentration.35 This also proved to be the case in the present work, with G51–B250 forming well-defined, near-monodisperse spherical nanoparticles with a DLS diameter of 67 nm. In striking contrast, growing a relatively short PHPMA block from the PGMA macro-CTA prior to BzMA polymerisation leads to well-defined worms when targeting a G37–H60–B30 composition. This particular triblock copolymer formulation is perhaps best considered as an example of a RAFT aqueous emulsion polymerisation of BzMA using a G37–H60 diblock copolymer precursor. DLS analysis of this precursor indicates the presence of ill-defined, weakly scattering nano-objects with a sphere-equivalent diameter of 141 nm. This suggests that this diblock precursor has not actually undergone micellar nucleation prior to BzMA addition and is therefore likely to comprise weakly interacting molecularly-dissolved copolymer chains. This unusual RAFT PISA formulation clearly warrants further work, but in the present study it is simply exploited as a convenient route to linear G37–H60–B30 worms that comprise a strongly hydrophobic PBzMA block in addition to the weakly hydrophobic PHPMA block.
 | ||
| Fig. 2 Representative transmission electron microscopy images obtained for: (A) linear G100–H200 spherical nanoparticles, (B) crosslinked G100–H200–E20 spherical nanoparticles, (C) linear G45–H140 worms, (D) crosslinked G45–H100–E10 worms, (E) linear G51–B250 spheres and (F) linear G37–H60–B30 worms. | ||
Previously, Blanazs et al. reported that G54–H140 linear worms exhibit thermo-responsive behaviour in aqueous solution at 10% w/w solids, undergoing a reversible morphological transition to form spheres on cooling from 25 °C to 5 °C as judged by TEM, SAXS and rheology studies.34 Variable temperature DLS studies confirmed that the G45–H140 linear worms prepared in this work undergo a similar thermal transition (see Fig. 3A). Thus the apparent spherical-average hydrodynamic diameter decreases from 315 nm at 25 °C to just 25 nm at 4 °C, with a concomitant reduction in scattered light from approximately 3 × 105 to 6 × 103 kilocounts per second (kcps). These observations are fully consistent with the worm-to-sphere thermal transition reported previously.34 Interestingly, further cooling from 4 to 2 °C led to a further reduction in size from 25 nm to 12 nm and a further drop in count rate to 1 × 103 kcps, which suggests further disintegration of the spherical nanoparticles to give either molecularly dissolved or very weakly interacting copolymer chains. A similar second transition has been inferred for a G49–H130 diblock copolymer by Kocik et al. on the basis of small-angle X-ray scattering studies.36 Moreover, it is worth emphasising that the corresponding sphere-to-worm transition does not occur on returning to 25 °C for this 0.25% w/w aqueous copolymer solution. Such irreversible behaviour is presumably observed because the highly cooperative 1D fusion of multiple spheres to form worms becomes infinitely slow at such high dilution. In contrast, the dissociation of worms to give spheres is not a cooperative process, so it is relatively unaffected at such low copolymer concentrations. Fig. S2† indicates that copolymer concentrations of at least 1.0% w/w are required for the linear G45–H145 worms to regain their original sphere-equivalent diameter after a cooling/warming cycle.
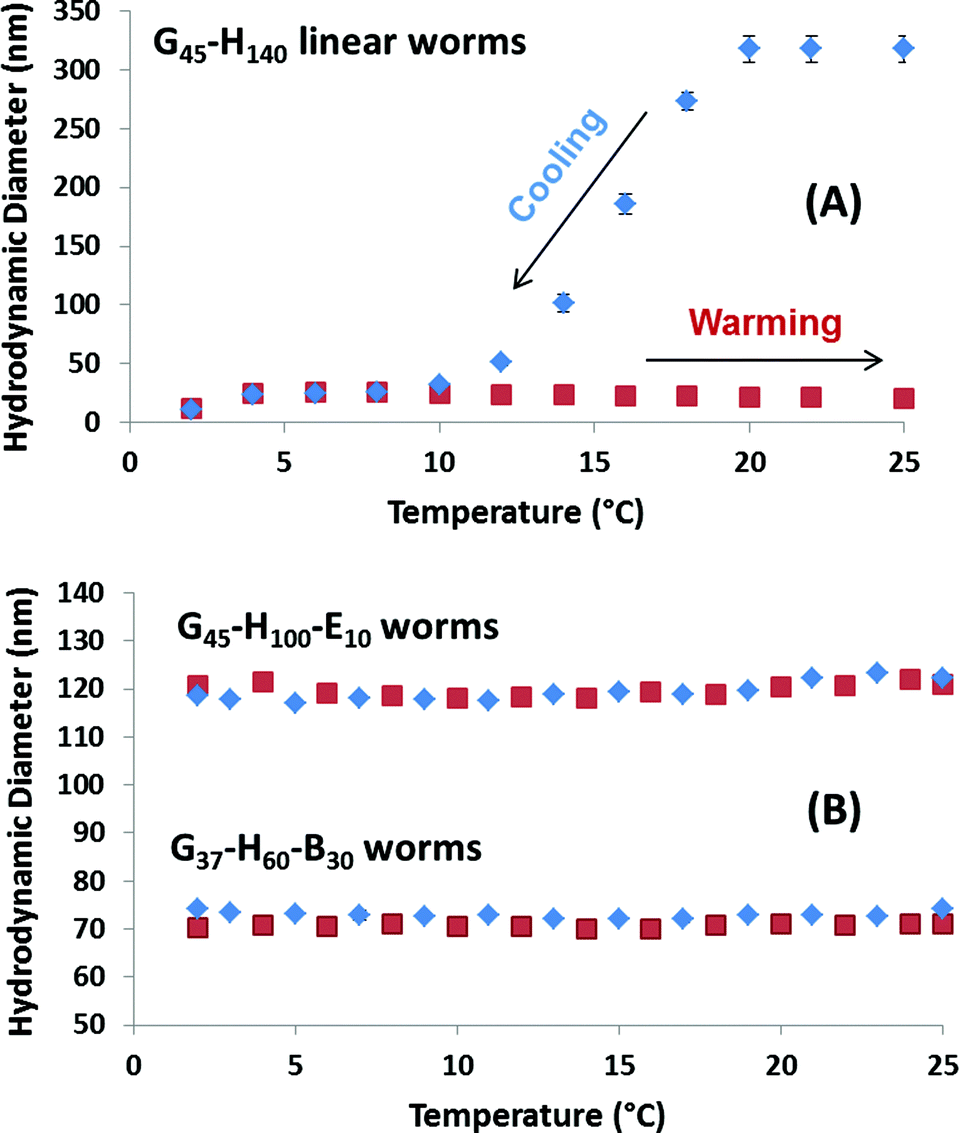 | ||
| Fig. 3 Variable temperature DLS studies obtained for 0.25% w/w aqueous dispersions of linear (A) G45–H140 worms, and (B) crosslinked G45–H100–E10 worms and linear G37–H60–B30 worms (see Table 1). The blue and red data points indicate the change in sphere-equivalent diameter on cooling and heating, respectively. Note the irreversible thermo-responsive behaviour observed for the linear G45–H140 worms at this relatively low copolymer concentration, and also the lack of any thermo-responsive behaviour found for the other two types of worms. Error bars represent the standard deviation determined for three separate measurements. | ||
In contrast, the crosslinked G45–H100–E10 worms exhibit qualitatively different behaviour on cooling a 0.25% w/w aqueous dispersion from 25 °C to 5 °C, see Fig. 3B. There is essentially no change in the apparent sphere-equivalent diameter on either cooling or heating, indicating that no worm-to-sphere transition occurs in this case. These negative observations are of course fully consistent with the additional covalent stabilisation conferred by the EGDMA cross-linker. Similarly, there is no discernible change in particle size during the same thermal cycle for the G37–H60–B30 worms. Thus it appears that the PBzMA block is sufficiently hydrophobic to suppress the thermo-responsive nature of the PHPMA block, with the former conferring additional physical stabilisation via enhanced hydrophobic interactions between the core-forming blocks. In principle, the greater stability indicated by the lack of thermal response observed for the chemically or physically crosslinked worms is likely to enhance the ability of such nanoparticles to survive high-shear emulsification (see later).
Initially, the linear G100–H200 and crosslinked G100–H200–E20 spherical nanoparticles were compared as putative Pickering emulsifiers.
Fig. 4 shows how the mean droplet diameter varies with copolymer concentration for these two dispersions. For the chemically crosslinked G100–H200–E20 nanoparticles, smaller oil droplets are obtained at higher copolymer concentrations. This is because there are more nanoparticles available to coat (and hence) stabilise the oil droplet surface. A minimum mean droplet diameter of approximately 50 μm is obtained at between 0.5 and 1.0% w/w copolymer concentration, which corresponds to the optimum conditions required for emulsification (i.e. minimum droplet diameter and maximum adsorption efficiency). This behaviour is typical of a Pickering emulsifier and has been reported for both PGMA-stabilised polystyrene latexes prepared by conventional aqueous emulsion polymerisation37 and also for the crosslinked G53–H350–E20 vesicles discussed earlier.32 The maximum adsorption efficiency for these G100–H200–E20 spheres was calculated to be 90% based on turbidimetry studies of the lower aqueous phase of the emulsion after creaming of the oil droplets. This makes them rather more efficient than the previously reported crosslinked G53–H350–E20 vesicles,32 but less efficient than the (larger) PGMA50–PS latex particles.37
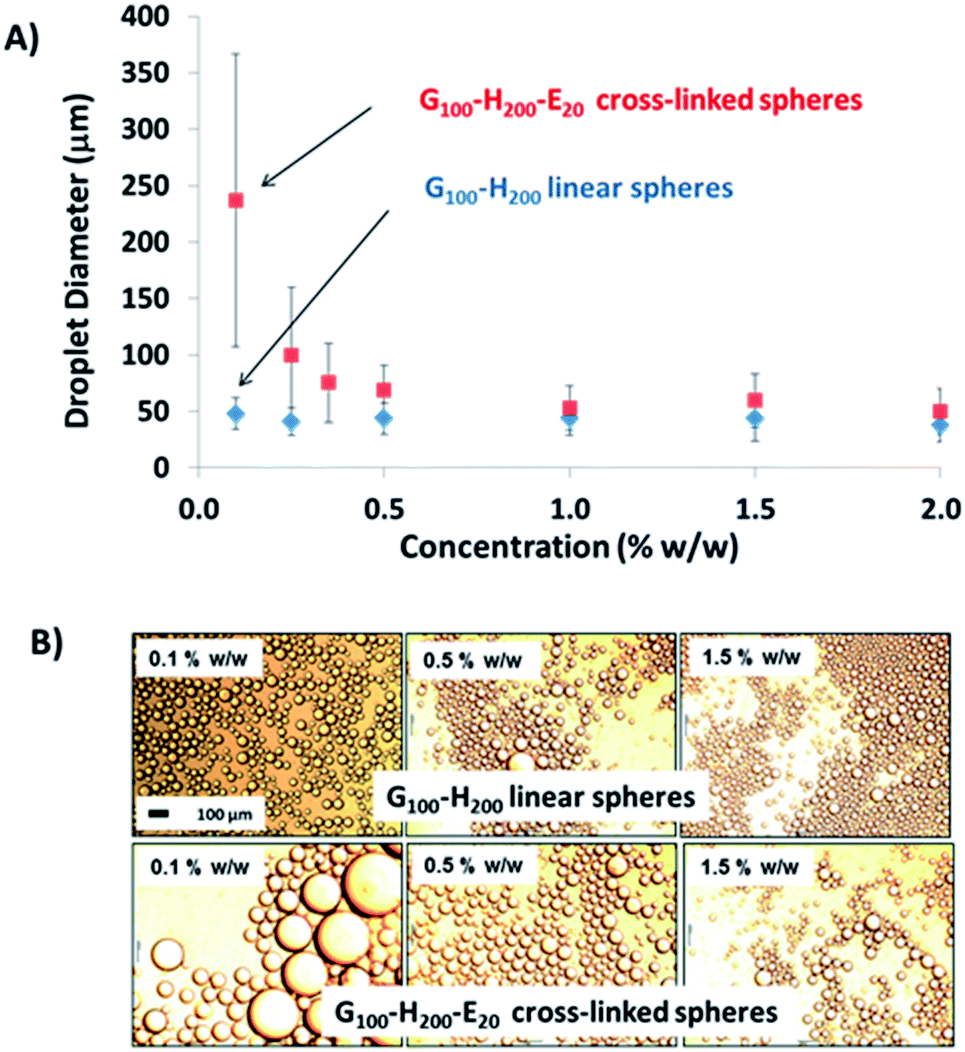 | ||
| Fig. 4 (A) Mean laser diffraction droplet diameter versus copolymer concentration for both linear G100–H200 spheres and crosslinked G100–H200–E20 spheres. The error bars represent the standard deviation of each mean volume-average droplet diameter, rather than the experimental error. (B) Optical microscopy images recorded for selected emulsions corresponding to the data points shown in (A). The 100 μm scale bar in the first image applies to all images. | ||
In contrast, no significant change in the mean oil droplet diameter occurs when varying the concentration of the linear G100–H200 spherical nanoparticles. Taken at face value, these data suggest that the linear spheres can stabilise smaller oil droplets than the crosslinked spheres at a given copolymer concentration (<1.0% w/w). Given that these nanoparticles are essentially the same size, this should not be the case if they adsorb with similar packing densities at the surface of the oil droplets. In principle, the surface coverage (Cs) for these spherical nanoparticles packed around large spherical oil droplets can be calculated using eqn (1) below, as reported previously.11
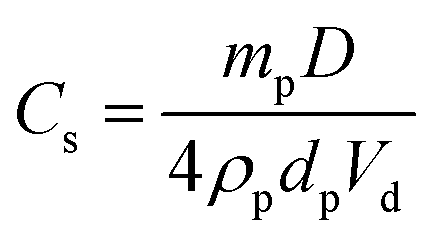 | (1) |
This equation is simply the surface area of the adsorbed particles divided by the surface area of the droplets, where Cs is the surface coverage of the droplets by the spherical nanoparticles, D is the mean droplet diameter (as determined by laser diffraction), mp is the mean nanoparticle mass, ρp is the nanoparticle density, dp is the mean nanoparticle radius (as determined by DLS) and Vd is the total volume of the oil droplet phase. In the case of the crosslinked G100–H200–E20 nanoparticles of 47 nm diameter, oil droplets with a mean diameter of 237 μm are produced at a copolymer concentration of 0.10% w/w (assuming an adsorption efficiency of 90%, see Table 1). This suggests a Cs value of approximately 0.93, indicating that the entire oil droplet surface is uniformly coated with close-packed nanoparticles. This is certainly consistent with a stable emulsion, since such a dense nanoparticle layer should prevent droplet coalescence. Indeed, TEM provides good evidence for a close-packed monolayer of spherical nanoparticles for this particular system (see later). In principle, P should not exceed 0.84 for small monodisperse spheres packed as a monolayer on larger spheres.38 In practice, this modest discrepancy simply reflects the various approximations that are inherent in such calculations.
In the case of the linear G100–H200 spherical nanoparticles, the same 0.10% w/w copolymer concentration affords stable oil droplets of approximately 48 μm diameter. Using eqn (1), we calculate Cs = 0.19 under these conditions, which suggests that only 19% of the droplet surface is coated by the nanoparticles. Given an oil volume fraction of 0.50, this Cs value seems to be rather low to account for the excellent long-term droplet stability that is observed experimentally, particularly given that the almost identical crosslinked G100–H200–E20 nanoparticles exhibit such a comparatively high surface coverage. Instead, a more likely scenario is that these oil droplets are actually stabilised by individual G100–H200 copolymer chains, which are generated via dissociation of the linear G100–H200 spherical nanoparticles during high shear homogenisation. Thus, as found for the crosslinked G53–H350–E20 vesicles discussed earlier,32 the EGDMA comonomer is essential to preserve the original copolymer morphology generated during the PISA synthesis and hence ensure that a true Pickering emulsion is obtained (as opposed to emulsion droplets stabilised by a molecularly-dissolved G100–H200 diblock copolymer surfactant).
The same experiments were performed on the linear G45–H140 and crosslinked G45–H100–E10 worms, see Fig. 5. An aqueous dispersion of the linear worms was diluted immediately prior to emulsion preparation at 20 °C in order to avoid inadvertently triggering the worm-to-sphere transition. Thus such nanoparticles should be present in their original worm morphology (and this was confirmed by TEM studies). As a comparison, emulsions were also prepared by conducting high-shear homogenisation at 0 °C with the aid of an ice bath. The crosslinked worms displayed similar behaviour to the crosslinked spherical nanoparticles discussed above, and also the crosslinked vesicles reported previously.32 The mean size of the oil droplets is gradually reduced at higher copolymer concentrations until a limiting minimum diameter of around 49 μm is attained. The maximum adsorption efficiency is again approximately 90%, making the affinity of these crosslinked worms for the n-dodecane–water surface comparable to that of the crosslinked spheres. This is perfectly reasonable given that they are both Gx–Hy–Ez copolymers, which should hence exhibit similar particle wettabilities at the oil–water interface. However, it is worth emphasising that, although the same Pickering adsorption efficiency (90%) is observed in both cases, the worms produce significantly smaller oil droplets at approximately half the surface coverage (0.47 vs. 0.93; see Table 1). For example, when using an initial copolymer concentration of 0.10% w/w (for which the final supernatant concentration of non-adsorbed copolymer is ∼0.01% w/w in both cases), the mean droplet diameter is 237 μm for the crosslinked spheres, but only 131 μm for the crosslinked worms. Hence the total surface area of the latter oil droplets is significantly higher. This is because the high aspect ratio of the worms allows the droplet surface to become sufficiently coated to prevent coalescence at a somewhat lower packing fraction than that of the spherical particles, particularly when working at such low copolymer concentrations. It is hypothesised that the nanoparticle packing fraction on the oil droplet surface gradually increases when using higher concentrations of the crosslinked worms, instead of remaining essentially constant as found for the spherical nanoparticles.
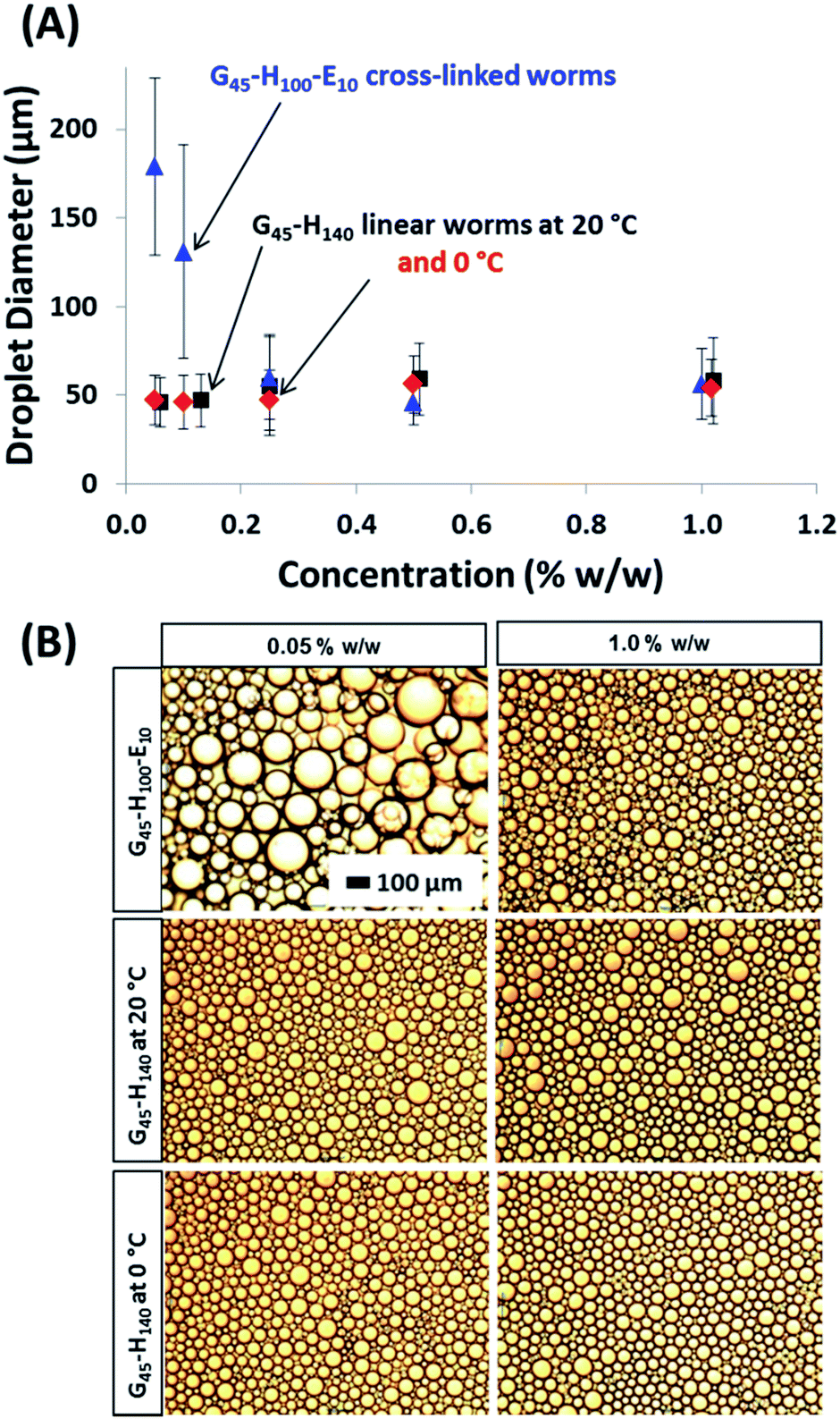 | ||
| Fig. 5 (A) Mean laser diffraction droplet diameter versus copolymer concentration for both linear G45–H140 and crosslinked G45–H100–E10 worms. The corresponding data obtained for the linear G45–H140 worms utilised for high-shear homogenisation conducted at 0 °C is also shown. The error bars represent the standard deviation of each mean volume-average droplet diameter, rather than the experimental error. (B) Optical microscopy images for selected emulsions corresponding to data points shown in (A). The 100 μm scale bar shown in the top left image applies to all six images. | ||
Using the linear worms produced oil droplet diameters of approximately 50 μm, regardless of the copolymer concentration. This indicates that the linear worm morphology is not sufficiently robust to survive the high-shear homogenisation conditions, thus the oil droplets formed in this case are actually stabilised by individual copolymer chains. This interpretation is supported by the data obtained using the linear worms for emulsifications conducted at around 0 °C. Under these conditions, DLS studies suggest that linear worms dissociate to produce essentially molecularly-dissolved copolymer chains, see Fig. 3. Hence this is consistent with the almost identical droplet diameter vs. copolymer concentration data sets obtained when using the linear worms at 25 °C and 0 °C, see Fig. 5A.
PGMA-stabilised polystyrene latex particles have been previously employed by Thompson et al. for the preparation of covalently crosslinked colloidosome microcapsules.37 The hydroxyl groups on the PGMA steric stabiliser chains are readily amenable to crosslinking using an oil-soluble polymeric diisocyanate (PPG-TDI), which leads to the formation of colloidosomes. Importantly, these covalently-stabilised latex superstructures can survive an ethanol challenge intact, even after complete removal of the oil droplet phase. In the present work, we used the same approach to cross-link the various linear and crosslinked PGMA–PHPMA nanoparticles (or linear copolymer chains) at the oil–water interface in order to study the copolymer morphology via TEM. However, ethanol is a good solvent for the core-forming PHPMA block and is thus likely to swell or even dissolve the copolymer nanoparticles. Therefore an alternative approach was required: n-hexane was employed instead of n-dodecane to allow convenient removal of the oil phase via evaporation, rather than via an ethanol challenge. The PPG-TDI crosslinker was dissolved in n-hexane prior to homogenisation and the resulting emulsion was allowed to stand at 20 °C for 1 h to enable cross-linking to occur. The n-hexane was then evaporated by magnetically stirring the diluted aqueous colloidosome suspension exposed to the atmosphere at 20 °C (at the back of a fume hood). Intact microcapsules were observed by optical microscopy for both worms and spheres prepared either with or without the EGDMA crosslinker. These microcapsules were imaged by TEM to assess the copolymer morphology. Fig. 6 shows the microcapsules prepared using the linear G100–H200 spheres and the linear G45–H140 worms. The microcapsule surface is completely smooth and featureless in each case, with no evidence for any adsorbed copolymer particles. Thus the microcapsule shell appears to comprise a molecular film of PPG-TDI crosslinked PGMA–PHPMA copolymer chains. These observations are fully consistent with the concentration-independent droplet diameters observed when employing such linear nanoparticles as putative Pickering emulsifiers. Moreover, similar TEM observations were reported for linear PGMAx–PHPMAy vesicles in our earlier study.32 Thus all the experimental evidence suggests that linear PGMA–PHPMA nanoparticles are not sufficiently robust to survive high shear homogenisation.
 | ||
| Fig. 6 TEM images of the edge and surface of various microcapsules prepared at 20 °C using: (A) linear G100–H200 spheres, (B) linear G45–H145 worms, (C) crosslinked G100–H200–E20 spheres, (D) crosslinked G45–H100–E10 worms, (E) linear G51–B250 spheres and (F) linear G37–H60–B30 worms. Note the smooth, featureless surface with no indication of any adsorbed nanoparticles in both (A) and (B). This suggests that these linear nanoparticles have each dissociated during high shear homogenisation and are actually present as adsorbed copolymer chains, rather than as nanoparticles. In contrast, the morphology of the original nanoparticles is clearly visible in images (C) to (F). [N.B. images (A) to (E) were obtained for PPG-TDI crosslinked colloidosomes, whereas image (F) was obtained for a dried Pickering emulsion droplet.] | ||
In contrast, Fig. 6C and D shows TEM images obtained for colloidosomes prepared using crosslinked G100–H200–E20 spherical nanoparticles and crosslinked G45–H100–E10 worms, respectively. Clearly, the former emulsifier leads to the formation of colloidosomes that comprise densely-packed spherical nanoparticles. This image is consistent with the high packing efficiency calculated above, but is in striking contrast to the smooth, featureless microcapsule surface obtained when using the corresponding linear spherical nanoparticles. Moreover, it is perhaps worth noting that the inter-particle separation distance within the colloidosome wall is significantly smaller than in our previous work,37 which in principle could lead to enhanced retention of macromolecules or nanoparticles encapsulated within such colloidosomes.39
A densely-packed layer of nanoparticles is also observed on the surface of colloidosomes prepared using the crosslinked G45–H100–E10 worms (see Fig. 6D). In the literature, it is generally assumed that highly anisotropic rod-like particles such as ellipsoidal polystyrene particles lie predominately flat at both the oil–water and air–water interfaces.16,40,41 Similar behaviour is also observed for cellulose nanofibers.18,19 In the present study, this also seems to be the case for the majority of worms, although some worms do appear to protrude out from the edge of the colloidosome surface. However, it is not clear whether this is actually the case in the ‘wet’ emulsion, or if this is simply a drying artefact that arises during TEM grid preparation. In principle, it may be feasible for some fraction of a relatively long worm to become adsorbed within the plane of the droplet surface, with the remainder of the worm extending out into the aqueous phase. Given the relatively flexible nature of these worms, this hypothesis is not unreasonable (particularly in the limit of high worm coverage).
Overall, it is clear that linear PGMA–PHPMA diblock copolymers in the form of either spherical nanoparticles or worms dissociate during homogenisation (just like the analogous vesicles reported earlier32) to produce individual copolymer chains. Under such high-shear emulsification conditions, the EGDMA cross-linker is essential to preserve the original copolymer morphology and hence ensure genuine Pickering stabilisation of the oil droplets, as opposed to emulsion stabilisation via molecularly-dissolved block copolymer surfactant. However, PHPMA is known to be only weakly hydrophobic,42 so we decided to examine whether a more hydrophobic core-forming block could enhance the stability of such nano-objects when evaluated as putative Pickering emulsifiers.
To address this important question, linear G51–B200 spherical nanoparticles of approximately 67 nm diameter were prepared via RAFT aqueous emulsion polymerisation for assessment as a Pickering emulsifier. Accordingly, these nanoparticles were homogenised with n-dodecane at various copolymer concentrations and the resulting emulsions were characterised using optical microscopy and laser diffraction (see Fig. 7).43 Using the G51–B200 spheres leads to a strongly concentration-dependent droplet diameter, unlike the earlier data set obtained for the linear G100–H200 spherical nanoparticles (which is included in Fig. 7 to aid direct comparison). The increase in mean droplet diameter observed on lowering the concentration of the linear G51–B250 spheres is fully consistent with the behaviour of the EGDMA crosslinked nanoparticles discussed above, as well as the PGMA–PS latexes reported earlier.37 TEM imaging of the colloidosome surface prepared with these G51–B250 spheres also shows fully intact spheres adsorbed at the droplet interface. Thus it appears that chemical cross-linking is not a pre-requisite to preserve the original copolymer morphology during homogenisation provided that the core-forming block is sufficiently hydrophobic to stabilise the nanoparticles with respect to their shear-induced dissociation.
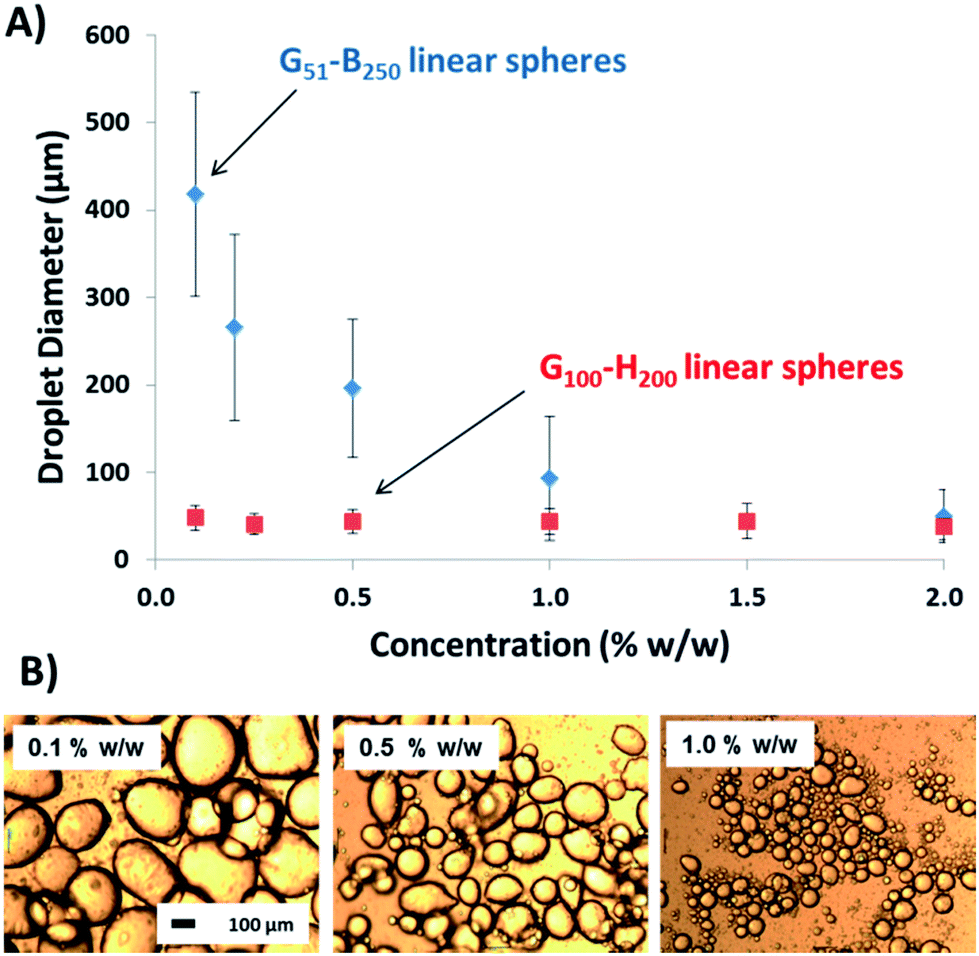 | ||
| Fig. 7 (A) Laser diffraction volume-average droplet diameter against copolymer concentration for G51–B250 spheres and G100–H200 linear spheres for comparison. The error bars represent the standard deviation of the mean droplet diameter, rather than the experimental error. (B) Optical microscopy images obtained for selected emulsions stabilised using the G51–B250 spheres. The 100 μm scale bar shown in the left-hand image applies to all three images. | ||
Finally, high shear emulsification of n-dodecane was conducted in the presence of the linear G37–H60–B30 worms. The important question here is: does a relatively short PBzMA block confer sufficient stability to the worms so as to enable the formation of a genuine Pickering emulsion? Fig. 8 confirms that larger droplets are formed as the worm concentration is gradually reduced. In view of the data sets discussed above, this strongly suggests that these anisotropic nanoparticles adsorb intact at the oil–water interface, in contrast to the molecularly-dissolved copolymer chains obtained when examining the linear G45–H140 worms. As indicated by the absence of a worm-to-sphere transition for G37–H60–B30 (see Fig. 3), even a relatively short PBzMA block is sufficient to stabilise these worms during high-shear emulsification; in this case nanoparticle dissociation is prevented as a result of the significantly stronger hydrophobic interactions between the core-forming blocks. This is further confirmed by TEM analysis of the colloidosome surface in Fig. 6F, which clearly shows that the original worm-like morphology is preserved at the droplet surface. It is also emphasised that these G37–H60–B30 worms proved to be extremely efficient Pickering emulsifiers: no worms could be detected in the supernatant phase after droplet creaming, suggesting essentially 100% adsorption at the oil–water interface. We speculate that this enhanced efficiency compared to the EGDMA crosslinked worms may be a result of the more hydrophobic PBzMA block increasing the particle contact angle at the oil–water interface, hence leading to stronger adsorption.
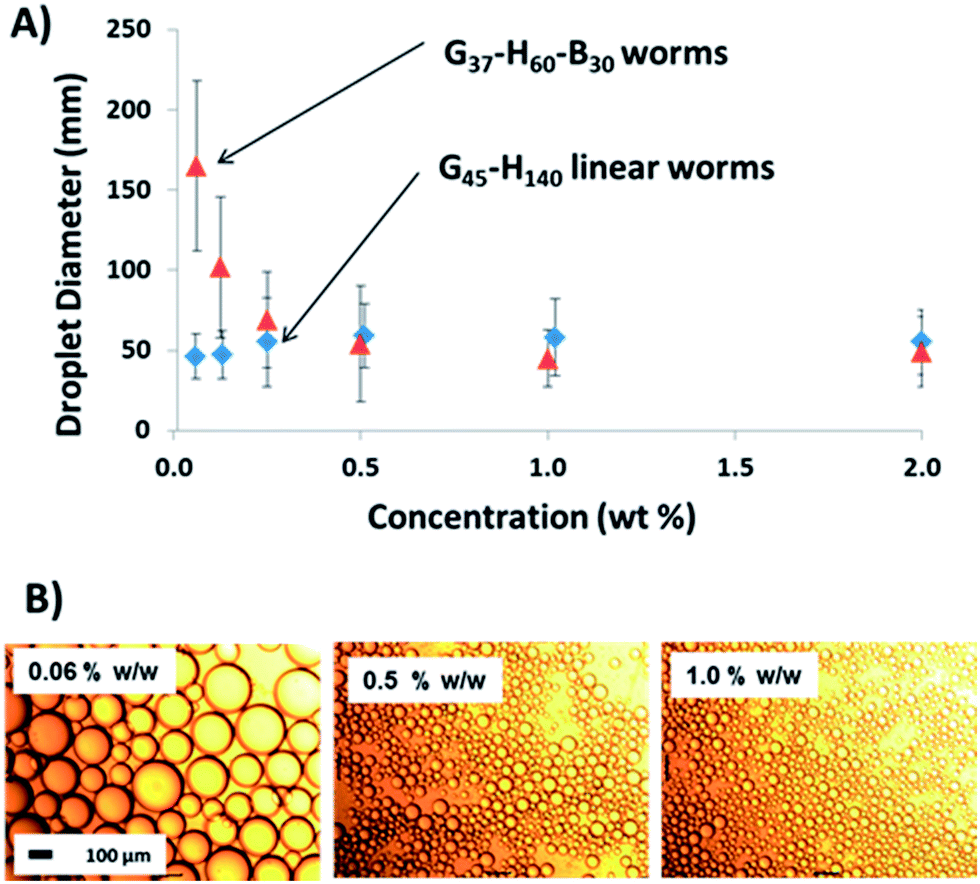 | ||
| Fig. 8 (A) Laser diffraction volume-average droplet diameter against copolymer concentration for two sets of linear worms (G37–H60–B30 and G45–H140). Only the former worms act as a genuine Pickering emulsifier; the latter worms undergo molecular dissolution during high shear emulsification and instead act as a block copolymer surfactant. The error bars represent the standard deviation of the mean droplet diameter, rather than the experimental error. (B) Optical microscopy images obtained for selected emulsions stabilised using the G37–H60–B30 linear worms. The 100 μm scale bar applies to all images. | ||
It is worth emphasising that, in general, the worms can be considered to be more effective Pickering emulsifiers than the equivalent spherical nanoparticles. More specifically, they are at least as efficiently (and almost certainly rather more strongly) adsorbed at the droplet interface. Moreover, smaller oil droplets are consistently produced when worms are used as the Pickering emulsifier when compared to an equal mass of spherical nanoparticles. The surface coverage (Cw) of the worms adsorbed on the surface of the oil droplets can be estimated using a modified version of eqn (1), which was derived for spherical nanoparticles.
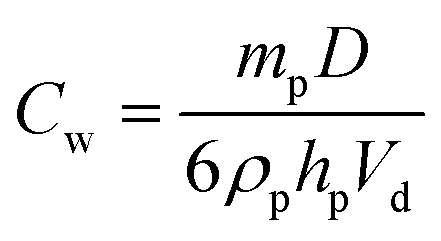 | (2) |
The main difference between eqn (1) and (2) is that hp now represents the mean worm thickness (estimated from TEM) rather than the sphere diameter. The surface coverages calculated for the lower worm concentrations are shown in Table 1. Worm surface coverages, Cw, of 0.47 and 0.63, are calculated for the EGDMA and BzMA stabilised worms respectively. These values are significantly lower than those obtained for oil droplets stabilised using spherical nanoparticles. The reciprocal of the mean oil droplet diameter, D, is plotted against the mass of adsorbed worms, mp, in Fig. 9a. According to eqn (2), the gradient should be inversely proportional to the worm surface coverage (Cw).19,20
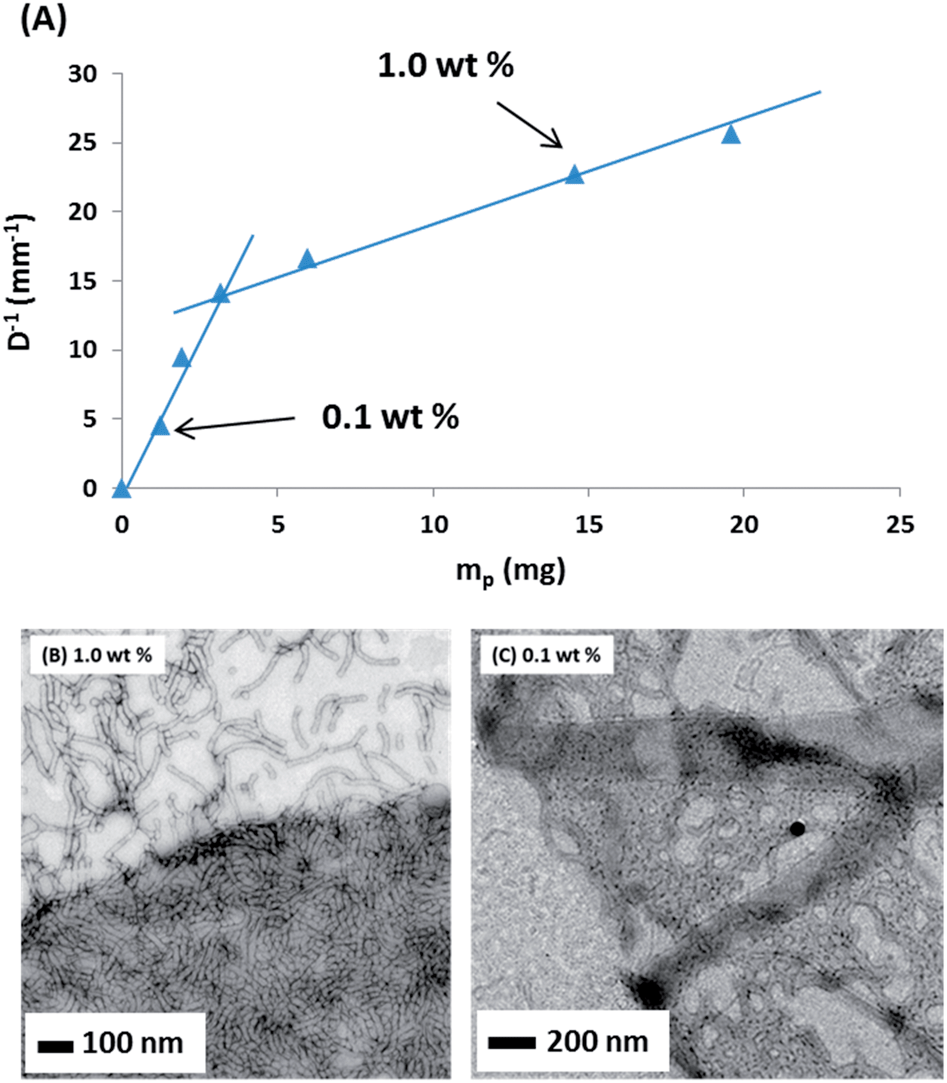 | ||
| Fig. 9 (A) Variation of the reciprocal of the mean oil droplet diameter (D−1) with nanoparticle mass (mp) for linear G37–H60–B30 worms (top). The gradient is inversely proportional to the surface coverage, hence the change in slope indicates two different modes of coverage. Representative TEM images obtained for n-hexane droplets stabilised using G37–H60–B30 worms prepared at copolymer concentrations of (B) 1.0% w/w and (C) 0.1% w/w. A significantly higher worm surface coverage is observed when using the higher copolymer concentration (see image (B)). | ||
Thus two distinct surface coverage regimes can be obtained, depending on the copolymer concentration used to prepare the Pickering emulsions. Very similar behaviour has been recently reported by Kalashnikova et al.19 for anisotropic Pickering emulsifiers based on bacterial cellulose nanofibres. At a relatively high copolymer concentration (e.g. 1.0% w/w) a high surface coverage is obtained, while at a relatively low copolymer concentration (e.g. 0.10% w/w) a significantly lower surface coverage is observed. [In contrast, a linear plot – indicating a single ‘high surface coverage’ regime - is obtained for the crosslinked spheres, see Fig. S4 in the ESI.†] This was confirmed by TEM analysis of dried Pickering emulsion droplets (prepared using n-hexane as the oil phase and in the absence of any PPG-TDI crosslinker) for both copolymer concentrations (i.e. 0.10 and 1.0% w/w) of the G37–H60–B30 triblock copolymer worms (see representative images shown in Fig. 9b and c, respectively). Clearly, the Pickering emulsion droplet surface is much more densely packed with worms when using the higher copolymer concentration. At the lower copolymer concentration, the worms appear to adjust their surface packing density by forming ‘islands’, rather than individual worms becoming more evenly spaced. This cooperative behaviour allows them to stabilise somewhat smaller oil droplets than the equivalent spherical nanoparticles, while at the same time being much more strongly adsorbed at the oil–water interface. It is worth noting that these triblock copolymer linear worms are much more flexible than the bacterial cellulose nanofibres reported by Kalashnikova et al.19 Thus it is the particle anisotropy, rather than the particle stiffness, that appears to be responsible for their strikingly similar Pickering emulsifier behaviour. In summary, the highly anisotropic character of these copolymer nanoparticles directly enhances their ability to act as an efficient Pickering emulsifier.
Conclusions
PISA offers a versatile and highly convenient route for the synthesis of a range of sterically-stabilised block copolymer nanoparticles with either spherical or worm-like morphologies in concentrated aqueous solution. In principle, such nanoparticles can act as novel Pickering emulsifiers for the stabilisation of oil-in-water emulsions. However, in practice the core-forming PHPMA block is only weakly hydrophobic, hence linear PGMA–PHPMA diblock copolymer nano-objects do not survive the high shear conditions required for homogenisation of the oil phase. This problem is observed for both spherical and worm-like copolymer morphologies. Fortunately, covalent stabilisation of both types of nanoparticles is readily achieved via chemical crosslinking with EGDMA, which enables their survival during emulsification. Whether such nanoparticles remain intact or undergo dissociation during homogenisation is readily assessed by examining the mean oil droplet diameter as a function of copolymer concentration. For soluble block copolymer chains, the mean droplet diameter exhibits essentially no concentration dependence, whereas the crosslinked nanoparticles exhibit strong concentration dependence (as expected for genuine Pickering emulsifiers). These findings are corroborated by TEM studies of colloidosome microcapsules prepared by introducing an oil-soluble polymeric crosslinker into the oil phase, since only those spherical or worm-like nanoparticles that were crosslinked prior to homogenisation are observed intact at the colloidosome surface. However, if the weakly hydrophobic PHPMA is either replaced or supplemented by a relatively hydrophobic core-forming block (e.g. PBzMA), the resulting linear spherical PGMA–PBzMA or worm-like PGMA–PHPMA–PBzMA nanoparticles are sufficiently stable to be utilised as Pickering emulsifiers. Hence chemical cross-linking is not mandatory for Pickering emulsification: sufficient stabilisation can be conferred simply by introducing stronger hydrophobic interactions between the core-forming blocks. Worm-like nanoparticles are shown to be much more effective Pickering emulsifiers compared to their spherical counterparts, since they are: (i) at least as efficiently adsorbed; (ii) almost certainly much more strongly adsorbed; and (iii) also enable smaller droplets to be produced for a given nanoparticle concentration. It appears that an anisotropic morphology also enables stable Pickering emulsions to be produced with relatively low surface coverage of the oil droplets when homogenisation is conducted at relatively low copolymer concentrations. Given the potentially scalable synthesis of these copolymer worms via RAFT-mediated PISA formulations, these findings suggest new research avenues for the rapidly-growing field of bespoke polymer-based Pickering emulsifiers.Acknowledgements
EPSRC and The University of Sheffield are thanked for funding a Post-doctoral Prize Fellowship for KLT. SPA and KLT also acknowledge EPSRC for a Platform grant (EP/J007846/1) and SPA is a recipient of a five-year ERC Advanced Investigator grant (PISA 320372). SPA thanks Ashland (USA) for CASE support of an EPSRC PhD studentship for VJC.Notes and references
- S. U. Pickering, J. Chem. Soc., 1907, 91, 2001–2021 RSC.
- W. Ramsden, Proc. R. Soc. London, 1903, 72, 156–164 CrossRef CAS.
- Y. Cui, M. Threlfall and J. S. van Duijneveldt, J. Colloid Interface Sci., 2011, 356, 665–671 CrossRef CAS PubMed.
- S. A. F. Bon and P. J. Colver, Langmuir, 2007, 23, 8316–8322 CrossRef CAS PubMed.
- S. Cauvin, P. J. Colver and S. A. F. Bon, Macromolecules, 2005, 38, 7887–7889 CrossRef CAS.
- Y. Cui and J. S. van Duijneveldt, Langmuir, 2012, 28, 1753–1757 CrossRef CAS PubMed.
- S. Levine, B. D. Bowen and S. J. Partridge, Colloids Surf., 1989, 38, 325–343 CrossRef CAS.
- B. P. Binks and S. O. Lumsdon, Phys. Chem. Chem. Phys., 1999, 1, 3007–3016 RSC.
- B. P. Binks and S. O. Lumsdon, Phys. Chem. Chem. Phys., 2000, 2, 2959–2967 RSC.
- B. P. Binks and C. P. Whitby, Langmuir, 2004, 20, 1130–1137 CrossRef CAS.
- F. Gautier, M. Destribats, R. Perrier-Cornet, J.-F. Dechezelles, J. Giermanska, V. Heroguez, S. Ravaine, F. Leal-Calderon and V. Schmitt, Phys. Chem. Chem. Phys., 2007, 9, 6455–6462 RSC.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2001, 17, 4540–4547 CrossRef CAS.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2000, 16, 8622–8631 CrossRef CAS.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21–41 CrossRef CAS.
- P. F. Noble, O. J. Cayre, R. G. Alargova, O. D. Velev and V. N. Paunov, J. Am. Chem. Soc., 2004, 126, 8092–8093 CrossRef CAS PubMed.
- B. Madivala, S. Vandebril, J. Fransaer and J. Vermant, Soft Matter, 2009, 5, 1717–1727 RSC.
- J. S. Guevara, A. F. Mejia, M. Shuai, Y.-W. Chang, M. S. Mannan and Z. Cheng, Soft Matter, 2013, 9, 1327–1336 RSC.
- I. Kalashnikova, H. Bizot, B. Cathala and I. Capron, Biomacromolecules, 2012, 13, 267–275 CrossRef CAS PubMed.
- I. Kalashnikova, H. Bizot, B. Cathala and I. Capron, Langmuir, 2011, 27, 7471–7479 CrossRef CAS PubMed.
- I. Kalashnikova, H. Bizot, P. Bertoncini, B. Cathala and I. Capron, Soft Matter, 2013, 9, 952–959 RSC.
- X. Zhang, S. Boisse, W. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS.
- S. Boisse, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M.-H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC.
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581 CrossRef CAS PubMed.
- L. P. D. Ratcliffe, A. J. Ryan and S. P. Armes, Macromolecules, 2013, 46, 769–777 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- C. Grazon, J. Rieger, N. Sanson and B. Charleux, Soft Matter, 2011, 7, 3482–3490 RSC.
- Polymer Handbook, 2nd edn., ed. J. Brandrup and E. H. Immergut, Wiley Interscience, New York, 1974 Search PubMed.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2011, 28, 914–922 CrossRef PubMed.
- K. L. Thompson, P. Chambon, R. Verber and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 12450–12453 CrossRef CAS PubMed.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Langmuir, 2012, 28, 1196–1205 CrossRef CAS PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett and S. P. Armes, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- M. K. Kocik, O. O. Mykhaylyk and S. P. Armes, Soft Matter, 2014, 10, 3984–3992 RSC.
- K. L. Thompson, S. P. Armes, J. R. Howse, S. Ebbens, I. Ahmad, J. H. Zaidi, D. W. York and J. A. Burdis, Macromolecules, 2010, 43, 10466–10474 CrossRef CAS.
- J. A. Balmer, S. P. Armes, P. W. Fowler, T. Tarnai, Z. Gaspar, K. A. Murray and N. S. J. Williams, Langmuir, 2009, 25, 5339–5347 CrossRef CAS PubMed.
- O. J. Cayre, J. Hitchcock, M. S. Manga, S. Fincham, A. Simoes, R. A. Williams and S. Biggs, Soft Matter, 2012, 8, 4717–4724 RSC.
- M. G. Basavaraj, G. G. Fuller, J. Fransaer and J. Vermant, Langmuir, 2006, 22, 6605–6612 CrossRef CAS PubMed.
- B. Madivala, J. Fransaer and J. Vermant, Langmuir, 2009, 25, 2718–2728 CrossRef CAS PubMed.
- J. Madsen, S. P. Armes and A. L. Lewis, Macromolecules, 2006, 39, 7455–7457 CrossRef CAS.
- In this particular case, relatively non-spherical n-dodecane droplets were produced, whereas spherical droplets were obtained for each of the other five Pickering emulsions. The former observation is reproducible but not currently understood. However, we note that spherical droplets can be achieved by using alternative oils (e.g. sunflower oil) – see ref. 35.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sm01724b |
| This journal is © The Royal Society of Chemistry 2014 |