 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystallizing hard-sphere glasses by doping with active particles
Ran
Ni
*ab,
Martien A.
Cohen Stuart
b,
Marjolein
Dijkstra
c and
Peter G.
Bolhuis
a
aVan ′t Hoff Institute for Molecular Sciences, Universiteit van Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
bLaboratory of Physical Chemistry and Colloid Science, Wageningen University, Dreijenplein 6, 6703 HB Wageningen, The Netherlands. E-mail: rannimail@gmail.com
cSoft Condensed Matter, Utrecht University, Princetonplein 5, 3584 CC Utrecht, The Netherlands
First published on 7th July 2014
Abstract
Crystallization and vitrification are two different routes to form a solid. Normally these two processes suppress each other, with the glass transition preventing crystallization at high density (or low temperature). This is even true for systems of colloidal hard spheres, which are commonly used as building blocks for novel functional materials with potential applications, e.g. photonic crystals. By performing Brownian dynamics simulations of glassy systems consisting of mixtures of active and passive hard spheres, we show that the crystallization of such hard-sphere glasses can be dramatically promoted by doping the system with small amounts of active particles. Surprisingly, even hard-sphere glasses of packing fraction up to ϕ = 0.635 crystallize, which is around 0.5% below the random close packing at ϕ ≃ 0.64. Our results suggest a novel way of fabricating crystalline materials from (colloidal) glasses. This is particularly important for materials that get easily kinetically trapped in glassy states, and the crystal nucleation hardly occurs.
A huge number of important applications associated with crystalline materials have made crystal fabrication a major research theme in the materials science community. The most common route toward the synthesis of a crystal is to supersaturate the corresponding fluid by increasing the density or lowering the temperature, after which the crystal may nucleate. With increasing supersaturation, the driving force for nucleation increases, which lowers the nucleation barrier.1 Simultaneously, however, the dynamics of the system also slows down, and at very high supersaturations the metastable fluid phase vitrifies into a glass before crystallization can occur.2 While there are some kinetically arrested glasses that can crystallize slowly via a sequence of stochastic micronucleation events,3 the glass transition generally remains a major obstacle for crystallization of highly supersaturated fluids.
We discuss here a way to circumvent the kinetic arrest, namely by using active matter. Active matter can be defined as a system of objects capable of continuously converting stored biological or chemical energy into motion. The interest in the dynamics of active matter stems from the wish to understand intriguing self-organization phenomena in nature as featured by bird flocks, bacterial colonies, tissue repair, and the cell cytoskeleton.4 The topic has also gained increasing research attention in chemistry: recent breakthroughs in particle synthesis have enabled the fabrication of artificial colloidal microswimmers that show high potential for applications in biosensing, drug delivery, etc.5 A number of different active colloidal systems have been realized in experiments, such as colloids with magnetic beads that act as artificial flagella,6 catalytic Janus particles,7–10 laser-heated metal-capped particles,11 light-activated catalytic colloidal surfers,12 and platinum-loaded stomatocytes.13 In contrast to passive colloidal particles that only undergo Brownian motion due to random thermal fluctuations of the solvent, active self-propelled colloids experience an additional force due to internal energy conversion. Recent theoretical14 and numerical15,16 work has shown that in systems of active hard spheres the glass transition is shifted to densities close to random close packing (RCP), which suggests that the role of activity is to de-vitrify glasses. In this work, we demonstrate that doping colloidal glasses with small amounts of active particles can significantly enhance the mobility of passive particles, and speed up the crystallization dynamics. Upon increasing the fraction of active particles, the crystallization pathway switches from spinodal decomposition to nucleation and growth, until too many active particles cause the system to adopt a non-equilibrium fluid state. Therefore, there is an optimal fraction of active particles for which the rate of crystallization is maximal.
We performed event driven Brownian dynamics simulations of a colloidal glass system modeled by N monodisperse hard-sphere particles with diameter σ. The hard-sphere system is a simple model for colloidal systems forming glasses.17 Here, we focus on packing fractions above the glass transition, ϕg ≃ 0.58.2,17 To prepare the initial configuration, we use the Lubachevsky–Stillinger algorithm18 to grow the particles in a simulation box to the packing fraction of interest. The total number of particles in the system is fixed at N = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. We change the fraction of active particles α by randomly selecting Nα particles which are made active by applying a self-propelling force f on them in the simulations. Even though a number of particles in the system are driven and energy is continuously injected into the system, we assume the solvent to be at an equilibrium temperature T. The motion of particle i with position ri and orientation ûi can be described via the overdamped Langevin equation given by
000. We change the fraction of active particles α by randomly selecting Nα particles which are made active by applying a self-propelling force f on them in the simulations. Even though a number of particles in the system are driven and energy is continuously injected into the system, we assume the solvent to be at an equilibrium temperature T. The motion of particle i with position ri and orientation ûi can be described via the overdamped Langevin equation given by
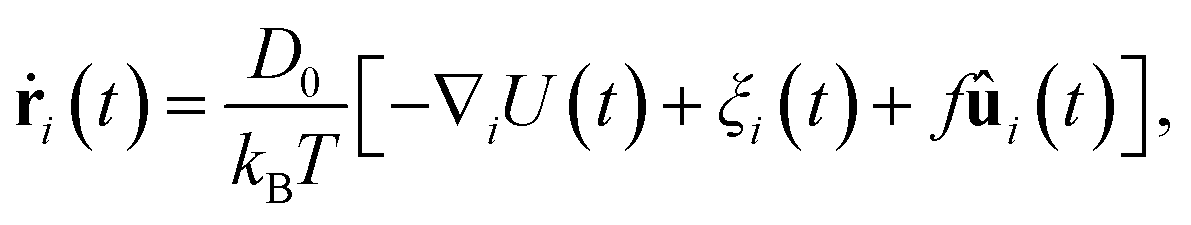 | (1) |
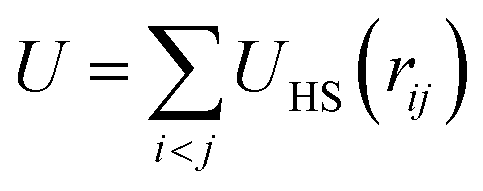 is the sum of excluded-volume interactions between all hard spheres with diameter σ, and D0 is the short-time self diffusion coefficient. A stochastic force with zero mean, ξi(t), describes the collisions with the solvent molecules, and satisfies 〈ξi(t)ξjT(t′)〉 = 2(kBT)21δijδ(t − t′)/D0 with 1 being the identity matrix. In addition, the self-propulsion of particle i is described by a constant force f in the direction ûi(t) at time t. Note that the free swimming speed of the self-propelled particles in dilute suspensions is given by fD0/kBT. In order to identify the crystalline clusters in the fluid phase, we employ the local bond-order parameter analysis.19–21
is the sum of excluded-volume interactions between all hard spheres with diameter σ, and D0 is the short-time self diffusion coefficient. A stochastic force with zero mean, ξi(t), describes the collisions with the solvent molecules, and satisfies 〈ξi(t)ξjT(t′)〉 = 2(kBT)21δijδ(t − t′)/D0 with 1 being the identity matrix. In addition, the self-propulsion of particle i is described by a constant force f in the direction ûi(t) at time t. Note that the free swimming speed of the self-propelled particles in dilute suspensions is given by fD0/kBT. In order to identify the crystalline clusters in the fluid phase, we employ the local bond-order parameter analysis.19–21
Fig. 1a shows the time evolution of the crystalline fraction in hard-sphere glasses for 0.58 ≤ ϕ ≤ 0.635, initiated in the disordered state. For packing fractions just above the hard-sphere glass transition ϕ ≃ 0.58, crystallization to a face-centered-cubic (fcc) structure occurs almost immediately via spinodal-decomposition, which is indicated by the immediate increase in crystalline particles without any waiting time. With increasing packing fraction, the crystallization of the hard-sphere glass slows down dramatically due to the emerging glassy dynamics. When ϕ ≥ 0.61, the crystallization is severely suppressed by the slow dynamics. Typical snapshots in Fig. 1b exemplify the slow growth of crystalline clusters in a dense hard-sphere glass at ϕ = 0.61, in agreement with previous simulation3 and experimental results.22
 | ||
| Fig. 1 (a) The fraction of crystalline particles Xf as a function of time tD0/σ2 in systems of passive Brownian hard-sphere glasses with various packing fractions 0.58 ≤ ϕ ≤ 0.635. D0 is the short-time translational self diffusion coefficient. (b) Three snapshots from a typical crystallization trajectory of a passive Brownian hard-sphere glass with ϕ = 0.61 at tD0/σ2 = 2, 20 and 40 (from left to right), respectively, where only the particles in crystalline clusters are shown and different colors denote different crystalline clusters. The fluid-like particles are shown as small spheres. | ||
Next, we activate a randomly selected small amount of particles in the hard-sphere glass by equipping them with a self-propelling force f. This force is applied on the center-of-mass of the particle in the direction û. The magnitude of the self-propelling force f is constant, while the orientation û undergoes a free Brownian rotation with a rotational diffusion coefficient Dr = 3D0/σ2 according to the Stokes–Einstein relationship. Fig. 2a shows the time evolution of the crystalline fraction in a hard-sphere glass of packing fraction ϕ = 0.61 containing various number fractions of active hard spheres, i.e. 0 ≤ α ≤ 0.15, with fσ/kBT = 80. Clearly, by doping the glass with only 1% of active hard spheres, the crystallization dynamics speeds up significantly. Similar to the passive hard-sphere glass, crystallization of the hard-sphere glass with α = 0.01 occurs immediately without any waiting time via spinodal decomposition as shown in Fig. 2a, which can be seen from the coupling between the increase of the largest cluster nmax and the decrease of the number of clusters Nclust. In addition, as shown in Fig. 2b, the mean square displacement of passive hard spheres increases faster in systems with more active particles, which is in agreement with ref. 16. This suggests that the existence of active particles in the glass enhances the mobility of the passive particles, which assists the coalescence of the clusters and speeds up the crystallization dynamics. We define the “crystallization time” τX as the time to reach a fraction of crystalline particles Xf = 0.2 as in ref. 23. Fig. 2e shows that with increasing fraction of active particles α the crystallization time τX decreases to a minimum at α = 0.02, after which τX increases again with α. This increase is due to the fact that the presence of active particles lowers the stability of the resulting crystal phase, thus decreasing the driving force of the phase transition.24 Therefore, the nucleation rate, i.e. the number of critical nuclei per unit volume and time, decreases. Indeed, at α = 0.1, as shown in Fig. 2a, the hard-sphere glass first melts into a non-equilibrium fluid phase, which is indicated by the large diffusivity of the particles without any crystalline clusters (Fig. 2a and b). Then the system stays in the non-equilibrium fluid state for a time t*D0/σ2 ≃ 20, followed by nucleation and growth of a single critical cluster (see a typical sequence of snapshots in Fig. 2d). It is surprising that the nucleation and growth of a single nucleus can occur in such a dense hard-sphere glass. This finding allows us to further study the role of active particles in the crystal nucleation. In Fig. 2c, we map the configurations onto the nmax–xα plane, in which nmax and xα are the size of the largest cluster and the fraction of active particles in this cluster, respectively. For small clusters, the fraction of active particles fluctuates strongly between 0 and 0.4, but when it increases to around the critical size, i.e. 100 < n*max < 200, the fraction of active particles in the nuclei remains below α, suggesting that the critical nucleus comprises mainly passive hard spheres. Therefore, the active particles in the system mainly act as “stirrers” or “mixers”, speeding up the mobility of the passive particles without actually initiating the crystal nucleation.
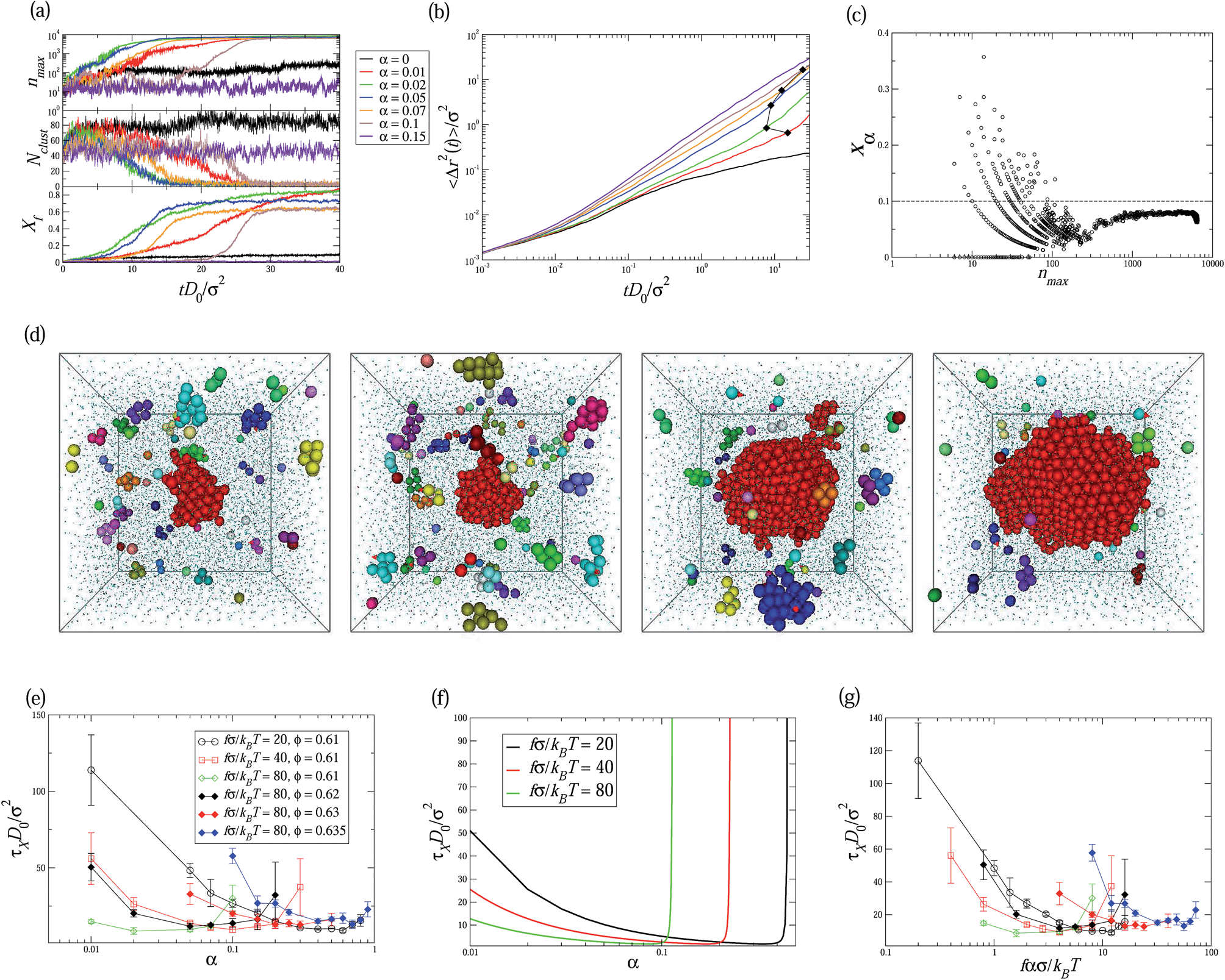 | ||
| Fig. 2 (a) The largest crystalline cluster size nmax, the number of crystalline clusters Nclust, and the fraction of crystalline particles Xf as a function of time tD0/σ2 in systems of hard-sphere glasses at packing fraction ϕ = 0.61 doped by various number fractions of active hard spheres α with self-propulsion fσ/kBT = 80. (b) The mean square displacement 〈Δr2(t)〉/σ2 for the passive particles in the system. The filled diamonds denote the 〈Δr2(t)〉/σ2 at t = τX. (c) Projection of configurations on the nmax and xα plane for systems of hard-sphere glasses at packing fraction ϕ = 0.61 doped by α = 0.1 active hard spheres with self-propulsion fσ/kBT = 80. The horizontal dashed line denotes the composition of active particles α = 0.1 in the whole system. (d) Snapshots from a typical trajectory of the nucleation of a single cluster at ϕ = 0.61 with α = 0.1 and fσ/kBT = 80. From left to right, the time and the corresponding largest cluster size in the system are (tD0/σ2, nmax) = (17.4, 83), (20, 168), (22.8, 1008) and (24.2, 1844), respectively. The red arrows on the spheres denote the direction of the self-propelling force on the active particles. (e) Crystallization time τXD0/σ2 as a function of the composition of active particles α in hard-sphere glasses of various packing fractions. (f) Theoretical prediction of the crystallization time τXD0/σ2 as a function of α for different self-propulsions f at ϕ = 0.61. (g) Crystallization time τXD0/σ2 as a function of fασ/kBT, where the legends of the symbols are the same as in (e). | ||
After further increasing the active particle fraction to α > 0.1 the system stays in the non-equilibrium fluid phase, and nucleation remains a rare event. In fact, it may even be hampered completely, since the large amount of active particles can also melt the crystal into a fluid phase.24 Hence, the enhanced crystallization of hard-sphere glasses by doping with active particles is the result of a competition between mobilizing (stirring) the glass and destabilizing the resulting crystals, yielding a maximal crystallization speed at around α* ≃ 0.02 with fσ/kBT = 80 at ϕ = 0.61.
The influence of self-propulsion on the crystallization time is summarized in Fig. 2e. Increasing the fraction of active particles in hard-sphere glasses from α = 0 always decreases the crystallization time τX, reaching a minimum at some optimal doping fraction, after which τX increases again with α. The open symbols in Fig. 2e show that the optimal composition of active particles decreases with increasing magnitude f of the self-propulsion force of active particles, i.e. induction of coalescence of the crystalline clusters in the glass requires a decreasing amount of stronger active particles. Similarly, as shown by the solid symbols in Fig. 2e, for fσ/kBT = 80 the optimal composition of active particles increases with packing fraction because strong stirring by active particles is required to crystallize denser glasses. Strikingly, by doping with only 10% active particles with fσ/kBT = 80, we have succeeded in crystallizing a hard-sphere glass at ϕ = 0.635, a packing fraction for which crystallization has never been observed before.2,3 The measured crystallization time is also shown in Fig. 2e.
We can rationalize the above behaviour by adapting classical nucleation theory (CNT) to our system. While CNT is clearly derived for quasi-equilibrium systems, we assume that nucleation in a non-equilibrium system is still governed by the same fundamental physics. The two main factors in CNT, the nucleation barrier and the kinetic prefactor, will be influenced by the presence of active particles. The kinetic prefactor will increase, because of the active particles, and it is likely that the nucleation barrier will increase, as the driving force for crystallization will be lower due to the active particles. A more quantitative analysis can be put forward as follows. According to CNT, the crystallization rate as a function of α and f has the form
| k(α, f) = D(α, f)e−ΔG(α, f)/kBT ≃ 1/τX, | (2) |
 | (3) |
In conclusion, by performing event-driven Brownian dynamics simulations, we systematically studied the crystallization of hard-sphere glasses consisting of a mixture of passive and active hard spheres. Our results can be summarized as follows: (1) doping hard-sphere glasses with a small amount of active particles enhances the mobility of the passive particles, which assists the coalescence of the crystalline clusters and speeds up the crystallization dynamics via spinodal decomposition; (2) upon increasing the fraction of active particles further, the crystallization speed reaches a maximum, beyond which the glass first melts into a non-equilibrium fluid, and the crystal may form via nucleation and growth afterwards. These results can be reasonably well explained by a modified CNT. By doping with active particles, we are able to crystallize hard-sphere glasses up to ϕ = 0.635, around 0.5% below the RCP limit ∼0.64, for which no crystallization has ever been observed before. When the density of a colloidal glass approaches the RCP, the pressure of the system diverges, and the driving force of the crystallization increases to infinity. Since we find that the fraction of active particles in the critical nuclei is lower than that in the bulk phase, doping with a tiny amount of strong active particles enhances the mobility of passive particles without changing significantly the stability of the crystal phase, which enables the crystallization of glasses at densities close to the RCP. Hence, we expect that this crystallization method can be employed for most colloidal glasses as long as the thermodynamically stable phase is a crystal. For instance, in systems of binary hard spheres, the crystallization has become highly challenging, and we hope our method can be further employed to help the crystallization of binary hard-sphere crystals. Moreover, we wish to note that if the active particles are much smaller than the passive particles, the situation can be much more complicated, since it has been found that the small active particles can produce giant long range effect interactions between the large passive particles in the system.28 Although most experimental studies correspond to low activities fσ/kBT < 10,7,9,11,13,29 light-activated colloids,12 catalytic Janus particles,10 and particles with artificial magnetic flagella6 are capable of producing self-propulsions as high as fσ/kBT ≃ 20, 50, 80, respectively, making our findings highly relevant for these systems. Moreover, one can also use optical tweezers to actively move small amounts of colloidal particles in the glass with large forces,30 which should have a profound effect on the crystallization dynamics of colloidal glasses. In this work, we neglected the effect of hydrodynamics, which could be an important direction for future investigation. However, it has been found that Brownian dynamics simulations without explicit hydrodynamics can reproduce most of the structures and patterns observed in experiments,12,24 which suggests that the method used in this work can capture the essential physics in the dynamic assembly of active colloidal swimmers.
Our results suggest a new way of fabricating crystalline materials from glasses, and it may be particularly important for the crystallization of photonic crystals.31,32
Acknowledgements
R.N. and M.A.C.S acknowledge financial support from the European Research Council through Advanced Grant 267254 (BioMate). This work is part of the research programme VICI 700.58.442, which is financed by the Netherlands Organization for Scientific Research (NWO).References
- S. Auer and D. Frenkel, Nature, 2001, 409, 1020 CrossRef CAS PubMed.
- E. Zaccarelli, C. Valeriani, E. Sanz, W. C. K. Poon, M. E. Cates and P. N. Pusey, Phys. Rev. Lett., 2009, 103, 135704 CrossRef CAS.
- E. Sanz, C. Valeriani, E. Zaccarelli, W. C. K. Poon, P. N. Pusey and M. E. Cates, Phys. Rev. Lett., 2011, 106, 215701 CrossRef.
- D. Gonzalez-Rodriguez, K. Guevorkian, S. Douezan and F. Brochard-Wyart, Science, 2012, 338, 910 CrossRef CAS PubMed.
- S. Ebbens and J. Howse, Soft Matter, 2010, 6, 726 RSC.
- R. Dreyfus, J. Baudry, M. Roper, M. Fermigier, H. Stone and J. Bibette, Nature, 2005, 437, 862 CrossRef CAS PubMed.
- J. R. Howse, R. A. L. Jones, A. J. Ryan, T. Gough, R. Vafabakhsh and R. Golestanian, Phys. Rev. Lett., 2007, 99, 048102 CrossRef.
- A. Erbe, M. Zientara, L. Baraban, C. Kreidler and P. Leiderer, J. Phys.: Condens. Matter, 2008, 20, 404215 CrossRef.
- J. Palacci, C. Cottin-Bizonne, C. Ybert and L. Bocquet, Phys. Rev. Lett., 2010, 105, 088304 CrossRef.
- L. Baraban, M. Tasinkevych, M. Popescu, S. Sanchez, S. Dietrich and O. Schmidt, Soft Matter, 2012, 8, 48 RSC.
- G. Volpe, I. Buttinoni, D. Vogt, H. Kümmerer and C. Bechinger, Soft Matter, 2011, 7, 8810 RSC.
- J. Palacci, S. Sacanna, A. P. Steinberg, D. J. Pine and P. M. Chaikin, Science, 2013, 339, 936 CrossRef CAS PubMed.
- D. A. Wilson, R. J. M. Nolte and J. C. M. van Hest, Nat. Chem., 2012, 4, 268 CrossRef CAS PubMed.
- L. Berthier and J. Kurchan, Nat. Phys., 2013, 9, 310 CrossRef CAS.
- L. Berthier, 2013, arXiv:1307.0704.
- R. Ni, M. Cohen-Stuart and M. Dijkstra, Nat. Commun., 2013, 4, 2704 Search PubMed.
- G. Parisi and F. Zamponi, Rev. Mod. Phys., 2010, 82, 789 CrossRef.
- B. D. Lubachevsky and F. H. Stillinger, J. Stat. Phys., 1990, 60, 561 CrossRef.
- P. J. Steinhardt, D. R. Nelson and M. Ronchetti, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 784 CrossRef CAS.
- R. Ni and M. Dijkstra, J. Chem. Phys., 2011, 134, 034501 CrossRef PubMed.
- L. Filion, M. Hermes, R. Ni and M. Dijkstra, J. Chem. Phys., 2010, 133, 244115 CrossRef CAS PubMed.
- W. van Megen and S. M. Underwood, Nature, 1993, 362, 616 CrossRef CAS.
- C. Valeriani, E. Sanz, E. Zaccarelli, W. Poon, M. Cates and P. Pusey, J. Phys.: Condens. Matter, 2011, 23, 194117 CrossRef CAS PubMed.
- J. Bialké, T. Speck and H. Löwen, Phys. Rev. Lett., 2012, 108, 168301 CrossRef.
- R. Ni, Ph.D. thesis, Utrecht University, 2012.
- A. Jepson, V. A. Martinez, J. Schwarz-Linek, A. Morozov and W. C. K. Poon, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2013, 88, 041002 CrossRef.
- K. Hall, J. Chem. Phys., 1972, 57, 2252 CrossRef CAS PubMed.
- R. Ni, M. Cohen-Stuart and P. Bolhuis, arXiv:1403.1533, submitted.
- J. Schwarz-Linek, C. Valeriani, A. Cacciuto, M. E. Cates, D. Marenduzzo, A. N. Morozov and W. C. K. Poon, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4052 CrossRef CAS PubMed.
- B. van der Meer, R. Fokkink, J. van der Gucht and J. Sprakel, 2013, arXiv:1309.5837.
- A.-P. Hynninen, J. H. J. Thijssen, E. C. M. Vermolen, M. Dijkstra and A. Van Blaaderen, Nat. Mater., 2007, 6, 202 CrossRef CAS PubMed.
- M. Eldridge, P. Madden and D. Frenkel, Nature, 1993, 365, 35 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |