 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Binding of amphiphilic and triphilic block copolymers to lipid model membranes: the role of perfluorinated moieties
Christian
Schwieger
*a,
Anja
Achilles
b,
Sven
Scholz
a,
Jan
Rüger
a,
Kirsten
Bacia
c,
Kay
Saalwaechter
b,
Jörg
Kressler
a and
Alfred
Blume
a
aInstitute of Chemistry, Martin Luther University Halle-Wittenberg, 06120 Halle (Saale), Germany. E-mail: christian.schwieger@chemie.uni-halle.de
bInstitute of Physics, Martin Luther University Halle-Wittenberg, 06120 Halle (Saale), Germany
cZIK HALOmem, Martin Luther University Halle-Wittenberg, 06120 Halle (Saale), Germany
First published on 4th June 2014
Abstract
A novel class of symmetric amphi- and triphilic (hydrophilic, lipophilic, fluorophilic) block copolymers has been investigated with respect to their interactions with lipid membranes. The amphiphilic triblock copolymer has the structure PGMA20-PPO34-PGMA20 (GP) and it becomes triphilic after attaching perfluoroalkyl moieties (F9) to either end which leads to F9-PGMA20-PPO34-PGMA20-F9 (F-GP). The hydrophobic poly(propylene oxide) (PPO) block is sufficiently long to span a lipid bilayer. The poly(glycerol monomethacrylate) (PGMA) blocks have a high propensity for hydrogen bonding. The hydrophobic and lipophobic perfluoroalkyl moieties have the tendency to phase segregate in aqueous as well as in hydrocarbon environments. We performed differential scanning calorimetry (DSC) measurements on polymer bound lipid vesicles under systematic variation of the bilayer thickness, the nature of the lipid headgroup, and the polymer concentration. The vesicles were composed of phosphatidylcholines (DMPC, DPPC, DAPC, DSPC) or phosphatidylethanolamines (DMPE, DPPE, POPE). We showed that GP as well as F-GP binding have membrane stabilizing and destabilizing components. PPO and F9 blocks insert into the hydrophobic part of the membrane concomitantly with PGMA block adsorption to the lipid headgroup layer. The F9 chains act as additional membrane anchors. The insertion of the PPO blocks of both GP and F-GP could be proven by 2D-NOESY NMR spectroscopy. By fluorescence microscopy we show that F-GP binding increases the porosity of POPC giant unilamellar vesicles (GUVs), allowing the influx of water soluble dyes as well as the translocation of the complete triphilic polymer and its accumulation at the GUV surface. These results open a new route for the rational design of membrane systems with specific properties.
Introduction
Amphiphilic block copolymers have been shown in numerous studies to influence the properties and integrity of lipid membranes. Research in this field has been stimulated by their potential use in pharmaceutical and medical applications. Poloxamers (also known under the trade names Pluronics® or Synperonics®) are in this respect the most extensively studied block copolymers. They have a symmetric ABA architecture and consist of a hydrophobic poly(propylene oxide) (PPO) middle block, flanked by a hydrophilic poly(ethylene oxide) (PEO) block on either side. Depending on the length of the PPO and PEO blocks they show very different behaviour. On the one hand they have been shown to disturb the membrane integrity leading to an increased permeability and facilitated translocation of dyes1 or drugs2 across the membrane; on the other hand they can also act as membrane sealants, which reduce membrane leakage or even help to restore the permeation barrier of damaged membranes.3 Both effects are of potential pharmaceutical interest, e.g. as chemo-sensitizer for drug uptake or as healing agent for injured cell membranes.4,5 Key parameters that regulate the type of interaction are the hydrophilic/hydrophobic balance (HLB), i.e. the ratio of PPO and PEO block lengths6 on the one hand and the absolute length of the PPO block on the other hand.7 In general, a higher hydrophobicity leads to enhanced insertion of the copolymers into the membrane. The length of the hydrophobic block defines the pathway of hydrophobic insertion.7,8 Copolymers with longer PPO chains can span the membrane, with their hydrophilic blocks being directed to either side of the bilayer. Copolymers with shorter PPO block insert only partially from one side.8,9A sufficient hydrophilicity is necessary to render the polymers water soluble and to prevent a strong self-aggregation, i.e. micellization of the polymers in the aqueous phase. In case of poloxamers this hydrophilicity is imposed by the PEO blocks. However, PEO interacts only marginally with the lipid bilayer2 and counteracts the propensity of insertion. To overcome this drawback, it was proposed to substitute the PEO blocks by poly(glycerol monomethacrylate) (PGMA) blocks.10,11 Due to the high propensity of PGMA to act as hydrogen bond donor and acceptor, the interaction with the lipid headgroups should be markedly increased. Moreover, the bulkiness of the PGMA side chains might increase the membrane disturbing effects, as it was reported for copolymers containing hyperbranched polyglycerols.12 The self-aggregation behaviour of such amphiphilic PGMA-PPO-PGMA triblock copolymers in aqueous solution11 as well as their behaviour at the air/water interface13 and their interactions with lipid monolayers14 have been studied before. It was shown that PGMA-PPO-PGMA inserts into lipid monolayers above the monolayer-bilayer equivalence pressure and affects the organization of the lipid molecules. We now present the first study on the interaction of these novel triblock copolymers with lipid bilayers, servings as model for biological membranes. The PPO block of the investigated copolymers has a degree of polymerization (DP) of 34 which is sufficient to potentially span a lipid bilayer. The PGMA blocks have each a DP of 20, which makes the polymers water soluble.
This triblock copolymer was investigated along with one of its derivatives, which was synthesized by terminally attaching perfluoroalkyl chains to both PGMA blocks.15 The resulting polymer has the structure F9-PGMA20-PPO34-PGMA20-F9, where F9 stands for a perfluorononyl moiety. The chemical structures of both polymers under investigation along with their approximate contour block length are given in Fig. 1. Throughout this report the non-fluorinated triblock copolymer will be abbreviated with GP and the fluorinated derivative with F-GP. The fluorinated chains are hydrophobic, but not lipophilic,16i.e. they are fluorophilic and not miscible with n-alkanes (C6 and above) and separate into an own phase. Therefore, the fluorinated copolymers are called triphilic or polyphilic.17 This triphilicity leads to interesting self-aggregation in aqueous environment into multi-compartment micelles.18,19 It might also lead to interesting new structures of the inserted triphilic polymer within the lipid bilayers. It is one aim of this work to investigate the influence of the fluorinated chains on the interaction of the block copolymers with lipid bilayers.
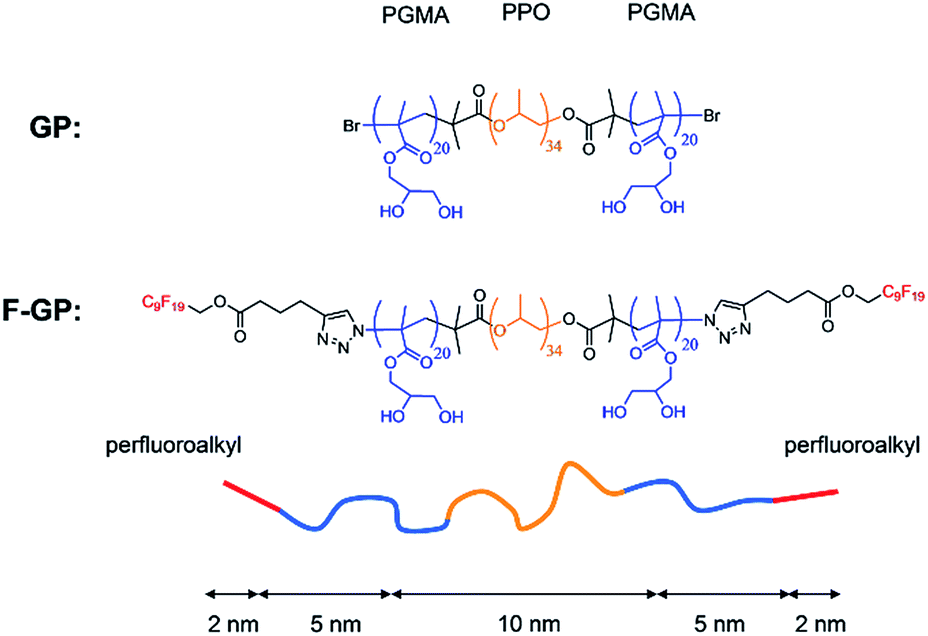 | ||
| Fig. 1 Structures of the studied block copolymers GP and F-GP and schematic representation of a polymer chain with its hydrophobic (yellow), hydrophilic (blue) and fluorophilic (red) building blocks and their approximate contour lengths in fully stretched all-trans conformation. | ||
The influence of GP and F-GP adsorption on the properties of lipid bilayers was investigated by differential scanning calorimetry (DSC) experiments. DSC monitors the gel to liquid phase transition temperature (Tm) of the membranes. Tm is a thermodynamic property that is influenced by any changes in the free energy of the gel or liquid crystalline phase of the bilayer. Therefore, it is sensitive to polymer adsorption and/or insertion which lead to changes in enthalpy and/or entropy of the system. However, systematic experiments and thorough analyses are necessary to conclude on a binding situation. Some DSC studies have been performed on the interaction of poloxamers with lipid membranes with different, partly conflicting results.7,9,20–22 This might be due to the variety of studied poloxamers but also to the difficulty to disentangle the possible effects on Tm. In this study systematic variation of bilayer thickness, nature of the lipid headgroup and polymer concentration allowed us to unravel different modes of interaction between the polymers and the lipid membrane and their different effects on Tm. In addition, we used 2D-NOESY NMR spectroscopy to investigate the insertion of the block copolymers into the bilayer by monitoring the spatial proximity of lipid acyl chains and PPO units. On a larger length scale we used fluorescence microscopy to show the effect of GP and F-GP binding on the membrane integrity. We examined the capacity of both polymers to facilitate dye influx into giant unilamellar vesicles (GUVs) as well as the ability of the complete copolymers to translocate across the bilayer.
The combination of these methods together with the systematic variation of the experimental parameters give a comprehensive view of the way the PGMA containing copolymers bind to lipid membranes and on the influence of the addition of fluorinated moieties to the chain ends.
Materials and methods
Materials
Methods
For the CLSM experiments giant unilamellar vesicles (GUVs) were prepared by a modified electroformation method. POPC was dissolved in chloroform at a total concentration of 10 mg mL−1. For the polymer co-localization experiments the membrane label BODIPY (0.5 mol%) was added. After manual spreading of this solution onto two indium-tin-oxide (ITO) coated glass slides (GeSiM, Großerkmannsdorf, Germany) the lipid film was dried at 60 °C. Both slides separated by a 3 mm thick silicon spacer and held together with office clips were used to form a chamber that was filled with sucrose solution (530 mOsmol kg−1). Via copper conductive tape (3M, SPI supplies) the slides were connected to a pulse generator (Conrad, Germany) and a 3 V, 10 Hz sinusoidal voltage was applied for 5 h. The formed GUVs were collected overnight by sedimentation in an iso-osmolar glucose solution.
For the dye influx experiments 20 μL of the copolymer solution (0.8 mmol L−1 GP; 0.9 mmol L−1 F-GP) and 4 μL of an aqueous dye solution (Alexa Fluor® 488 hydrazide, 500 nmol L−1) were pre-mixed in a 96 well plate. 1 μL of the respective GUV suspension was then carefully injected from above and allowed to settle to the well's ground. To prevent osmotic pressure differences, all solutions contained iso-osmolar amounts of glucose. After addition of the GUV suspension the well plate was immediately mounted on the microscope. After focusing on selected GUVs, images were recorded in a time series with a camera exposure time of 300 ms. Due to this procedure there is a lag time between mixing of the GUVs with the polymer solution and the beginning of the time series of maximal 10 s. The recorded images were processed with the ZEN microscope software (Carl Zeiss Microimaging). To evaluate the amount of dye influx, integral fluorescence intensities of selected regions of interest were determined and normalized to the average intensity of the respective image.
Colocalization experiments of membrane stained GUVs and rhodamine-labelled polymer were performed in an analogous manner.
In the resulting two-dimensional spectrum S(ω1,ω2), ω1 characterizes the resonance frequency of a given nucleus before, and ω2 the frequency after the mixing time tm, i.e. after a possible polarization transfer. If a polarization transfer occurred during tm, a crosspeak will appear in the correlation spectrum arising from a spatial proximity between the nuclei with the respective chemical shifts. However, due to complex dependence of the cross-correlation on the distance between the nuclei and their dynamics, the NOESY-experiment only yields qualitative information on possibly transient contacts.
For the NMR measurements the samples were prepared directly within the NMR rotor by loosely filling it with the sample mixture (DMPC and polymer), adding 50 wt% of D2O afterwards and letting it equilibrate at a temperature above the main phase transition of the lipid in order to obtain a homogeneous suspension. All NMR experiments were carried out on a Bruker Avance III 400 spectrometer with resonance frequencies of 400.03 MHz for protons, using a double-resonance 4 mm probe at a rotation frequency of 4 kHz. The 90° proton pulse was 3 μs, the mixing time was set to 200 ms and a total of 512 t1 increments were acquired with 16 scans for each increment and a relaxation delay of 1 s. For best spectral resolution, the samples were investigated in the Lα phase and hence heated to a temperature of 40 °C.
Results and discussion
DSC: influence of polymer binding on the transition temperatures of lipid membranes
Using DSC measurements the influence of the polymers on the gel to liquid crystalline (Lβ′ → Lα) phase transition of lipid membranes composed of phosphatidylcholines can be monitored. This transition is a cooperative first order transition and gives a peak in the Cp = f(T) thermogram, where the integral of the peak corresponds to the phase transition enthalpy and the width is a measure for the cooperativity (the narrower, the more cooperative). The maximum of the DSC trace is the main transition temperature (Tm) of the lipid membrane.T m is sensitive to interactions on the membrane surface and/or insertion of hydrophobic substances into the hydrophobic part of the membrane. Tm increases when the interaction leads to a screening of headgroup charges, a displacement of hydration water from the hydrophilic membrane layer,27,28 or hydrogen bond formation between the headgroups.29,30 When hydrophobic molecules or moieties insert into the hydrophobic layer of the membrane Tm decreases due to a perturbation of the chain packing.29–32
The copolymers under investigation, being amphiphilic (GP) or triphilic (F-GP), have the potential to interact with the hydrophilic headgroup layer via the PGMA blocks and with the hydrophobic acyl chain layer via the PPO block.8,33 Due to their hydrophobicity also the perfluoro segments have the tendency to partition into the hydrophobic membrane layer. However, their lipophobic nature might lead to a segregation of inserted fluorinated moieties within the membrane. An influence on Tm would depend on the nature and the balance of the interactions between the lipids and the copolymers. Thus, the change in Tm is a marker for the type of interaction that occurs in this system and about the influence of end-cap fluorination of the copolymers.
To decipher the possible interactions we performed a number of experiments under systematic variation of (i) the type of polymer, (ii) the added polymer concentration, (iii) the hydrophobic thickness of the membrane and (iv) the nature of the lipid headgroup.
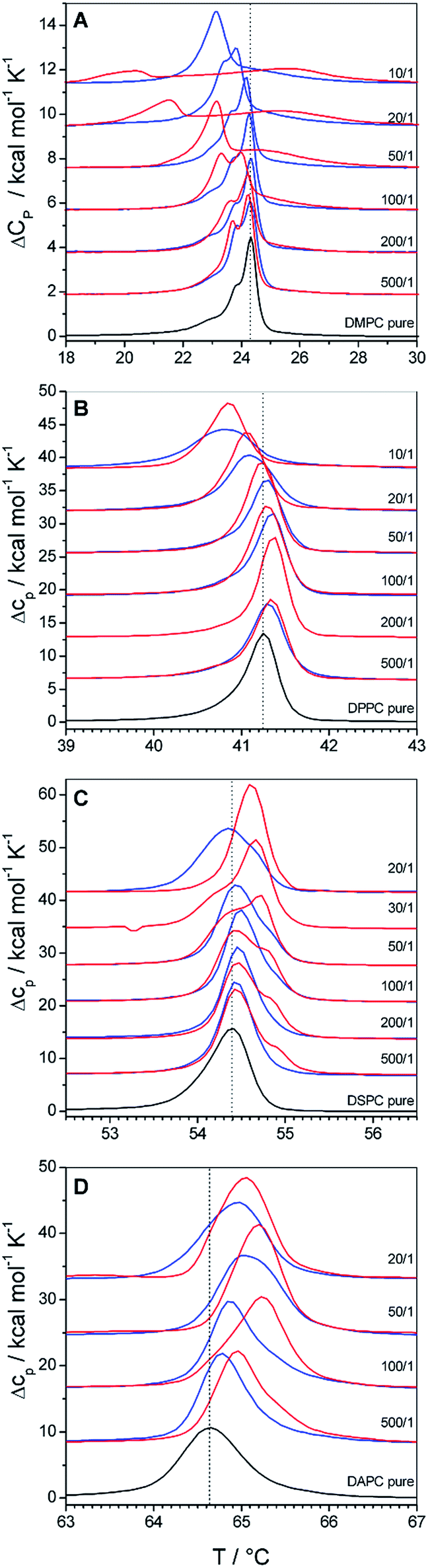 | ||
| Fig. 2 DSC thermograms of polymer bound lipid vesicles prepared from (A) DMPC, (B) DPPC, (C) DSPC and (D) DAPC. Black traces represent the phase transition of the pure lipid vesicles, blue traces their mixtures with GP and red traces their mixtures with F-GP. The lipid/polymer molar mixing ratio is given in the figure. Dotted lines are drawn at Tm of the pure lipid membranes. | ||
Fig. 2A summarizes the DSC scans of the second heating of DMPC–copolymer mixtures. Already a very low polymer content of 0.2 mol% leads to a shift of Tm to lower values (ΔTm < 0). This shift gets systematically larger as the copolymer content increases, leading to a 1 °C downshift at a GP content of 10% and even a 4 °C downshift for the same F-GP content. From Fig. 2A it becomes evident that F-GP binding (red curves) has a stronger influence on the DMPC phase transition than GP-binding (blue curves). The shift of Tm to lower temperature indicates the insertion of the copolymers into the hydrophobic part of the lipid bilayer. Probably, the hydrophobic PPO block inserts into the acyl chain region of the membrane perturbing the acyl chain packing, as it was also postulated for the related poloxamers.8 The fact that F-GP decreases Tm more than GP shows that not only the PPO block but also the terminal perfluoroalkyl chains insert into the membrane. This confirms that the hydrophobic nature of the perfluoroalkyl chains dominates their lipophobic nature. For transitions of F-GP–DMPC mixtures with low F-GP content no single peaks are observed but additional minor transition components emerge on the low temperature side of the main peak. With higher F-GP content, a broad transition at higher temperature develops which is clearly separated from the transition at lower temperature. This biphasic transition is an indication for a partial phase separation into ordered domains of different compositions. Possibly, there are polymer rich regions in the membrane where the acyl chain melting is retarded either by strong headgroup interactions or by an order that is conferred by inserted, rigid fluorocarbon chains.
Whatever the exact reason, it must be due to the presence of the fluorinated moieties in the system, as it is not observed for the non-fluorinated triblock copolymer (GP).
In Fig. 2B we show the influence of copolymer binding on the phase transition of DPPC vesicles. Even though DMPC and DPPC are homologous phospholipids which differ only in the length of their acyl chains, the influence of copolymer binding on their phase transitions is surprisingly different: at low polymer contents (0.2 to 1 mol%) Tm of DPPC is shifted to higher temperatures (ΔTm > 0). Only at higher polymer content (>1 mol%) Tm decreases as it was observed for DMPC membranes. The ability of the polymers to induce both, an upshift and a downshift of Tm shows that the insertion of the hydrophobic polymer block and the fluorinated chain in the case of F-GP is not the only mode of interaction. The increase of Tm indicates that also polar interactions occur between the lipid headgroups and the hydrophilic moieties of the copolymers, i.e. the PGMA blocks. The overall effect on Tm is a result of the delicate balance of hydrophobic and polar interactions, which occur concurrently but influence the phase transition in an opposite manner. The compensation of both counteracting effects explains that the changes in Tm are relatively small in absolute values. A similar balance of Tm increasing and Tm decreasing effects was observed before for binding of polyelectrolytes to lipid membranes.39
The balance of hydrophobic effects and polar interactions is concentration dependent. At low polymer contents polar interactions dominate the influence on Tm and over-compensate the effect of perturbation of chain packing by insertion of the hydrophobic polymer block into the acyl chain region. At higher polymer content it is well imaginable that the hydrophilic polymer blocks are stretched into the surrounding aqueous solution because of crowding effects. As a consequence, the polar interactions between the PGMA block and the bilayer surface are reduced leading to a decrease in Tm due to perturbations of chain packing. A concentration dependent change in binding mechanism was also predicted by molecular dynamics simulations for poloxamer binding to lipid membranes8 and a concentration dependent change in the sign of ΔTm was also shown for poloxamer binding to DPPC vesicles.9 The difference in the influence of GP and F-GP binding on Tm of DPPC membranes seems to be small. However, slightly narrower transitions are observed for mixtures with F-GP.
A further increase in the hydrophobic thickness of the membrane leads to the transitions depicted in Fig. 2C for DSPC (18 C-atoms per acyl chain) and Fig. 2D for DAPC (20 C-atoms per acyl chain). Copolymer addition to these thicker membranes does not lead to a Tm decrease and ΔTm stays positive at all concentrations and for both copolymers. The increase is more pronounced for DAPC than for DSPC and more pronounced for addition of F-GP compared to GP. For both types of membranes and for both types of polymers the concentration dependence that leads to a maximum in ΔTm at intermediate polymer contents is still visible. This shows that both, insertion into the hydrophobic membrane layer and hydrophilic interactions in the headgroup layer still compete in their effect on Tm. However, for these thicker membranes the polar interactions always dominate the effect on Tm, leading to an over-all stabilization of the membranes gel phases. The increase in the lipids acyl chain length increases the van-der-Waals interactions in the hydrophobic membrane layer, which is reflected by the increasing transition temperatures of the pure membranes (black traces). The perturbation of the acyl chain order which is exerted by the insertion of PPO-blocks and perfluorocarbon chains is the less pronounced the more stable this layer is. Thus, the effect of PPO/perfluorocarbon insertion on Tm decreases with increasing bilayer thickness. Simultaneously, the effect on the headgroup layer is always the same as it does not depend on the bilayer thickness. In combination, this leads to the observed increasing influence of the polar headgroup interaction on Tm with increasing bilayer thickness.
The comparison of the effects of GP and F-GP on DAPC and DSPC shows again that both copolymers have qualitatively the same effects on Tm but that the addition of perfluorinated chains leads to an increase in its magnitude. The addition of perfluorinated segments increases on the one hand the downshift of Tm (for DMPC) and on the other hand the upshift of Tm (for DAPC and DSPC). This indicates that the addition of the two perfluorinated moieties to the ends of each polymer chain leads to an increase in the apparent binding constant, thus amplifying both hydrophobic and polar interactions at the membrane surface. The perfluoroalkyl chains thus function as additional membrane anchors due to their high hydrophobicity.
Fig. 3 summarizes the effects of GP and F-GP binding on Tm of the different examined PC membranes. The following systematic trends can be extracted:
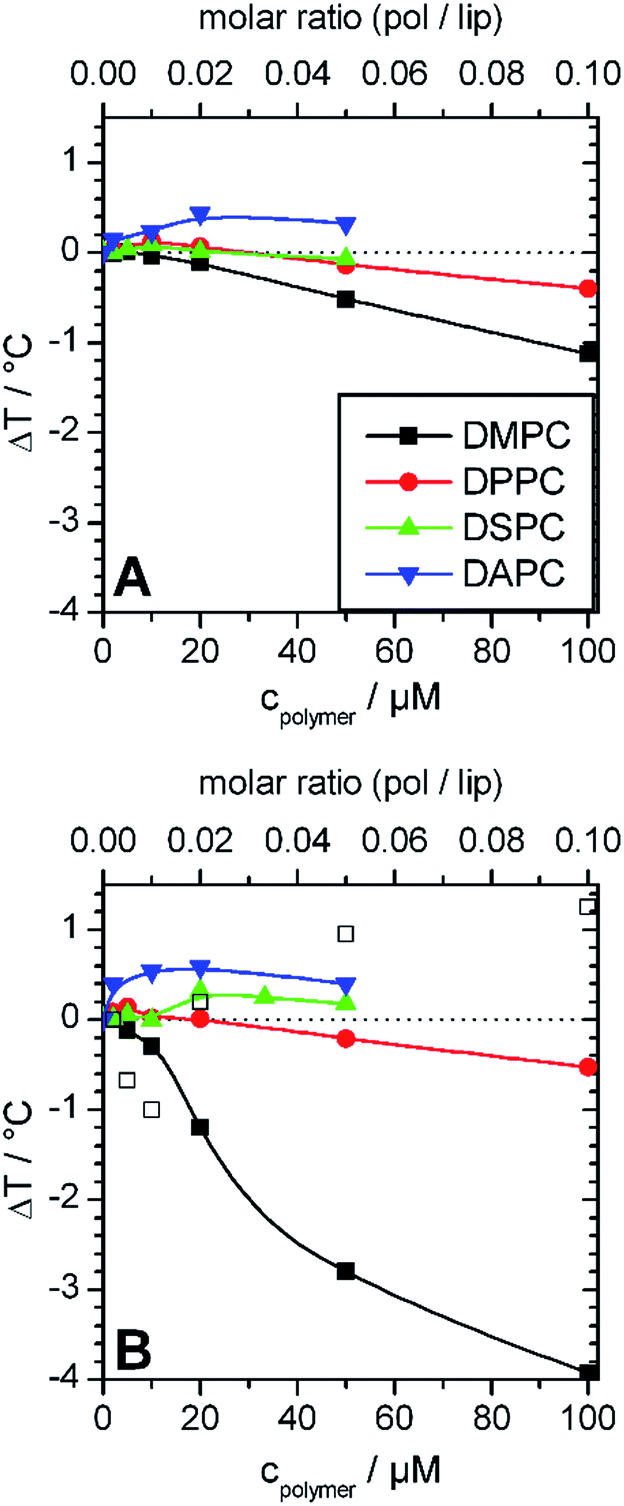 | ||
| Fig. 3 Change of main transition temperatures of the lipid membranes prepared from DMPC (black), DPPC (red) DSPC (green), DAPC (blue) after addition of different amounts of (A) GP and (B) F-GP. The lipid concentration was always 1 mmol L−1. In (B) major (solid symbol) and minor (open symbol) transition components are shown for DMPC, where major refers to the transition component with the maximal cp value and minor to shoulders or transition peaks with lower cp values (see Fig. 2A). Copolymer/lipid molar ratios are presented on the top x-axes. The inserted lines are drawn to guide the eyes. | ||
i. Stabilization (ΔTm > 0) and destabilization (ΔTm < 0) of the membrane are possible. The overall effect is a superposition of both.
ii. The effect on Tm is concentration dependent, with a maximum of ΔTm at intermediate concentrations (about 1 mol% polymer).
iii. Sign and absolute value of ΔTm depend on the hydrophobic thickness of the membrane. The higher the membrane thickness, the less important is the effect of perturbation of acyl chain packing and the more positive is ΔTm.
iv. Addition of perfluorinated moieties at the chain ends of the polymers leads to an amplification of all effects.
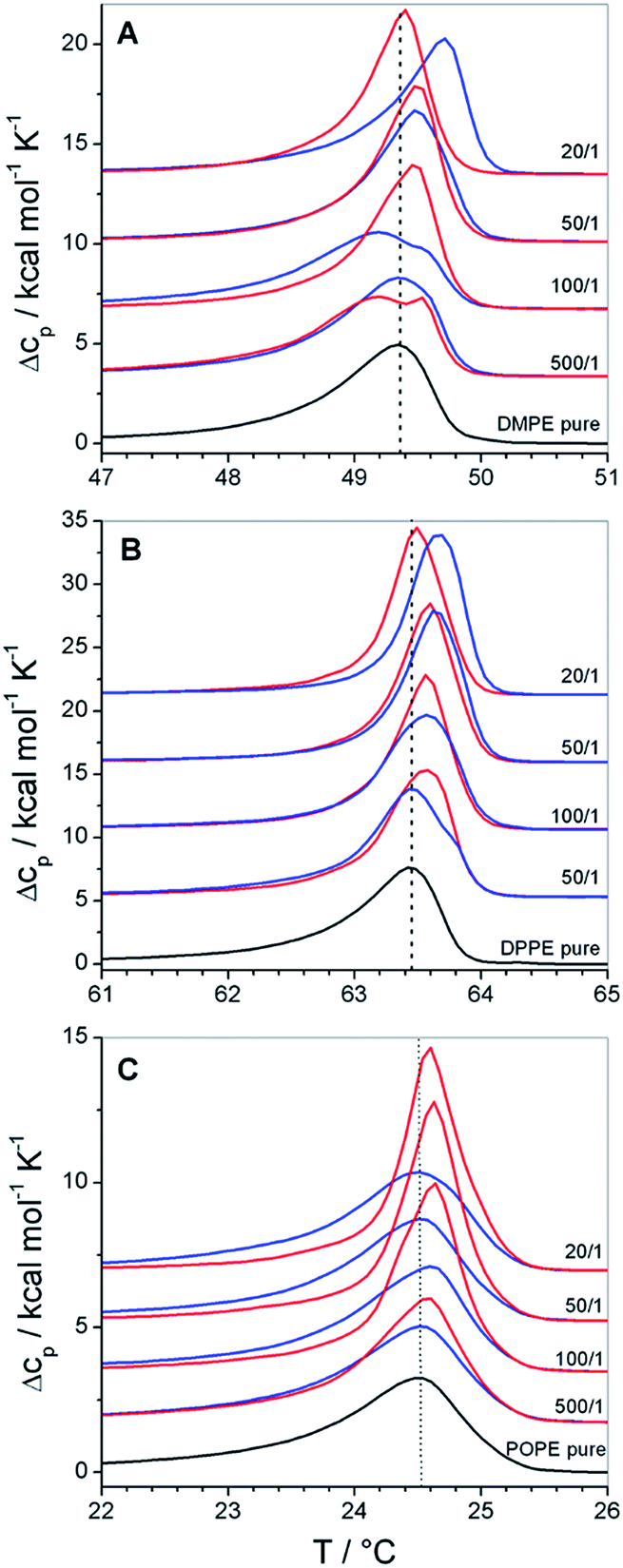 | ||
| Fig. 4 DSC thermograms of polymer bound lipid vesicles prepared from (A) DMPE, (B) DPPE and (C) POPE. Black traces represent the phase transition of the pure lipid vesicles, blue traces their mixtures with GP and red traces their mixtures with F-GP. The lipid/polymer molar mixing ratio is given in the figure. Dotted lines are drawn at Tm of the pure lipid membranes. The lipid concentration is always 1 mmol L−1. | ||
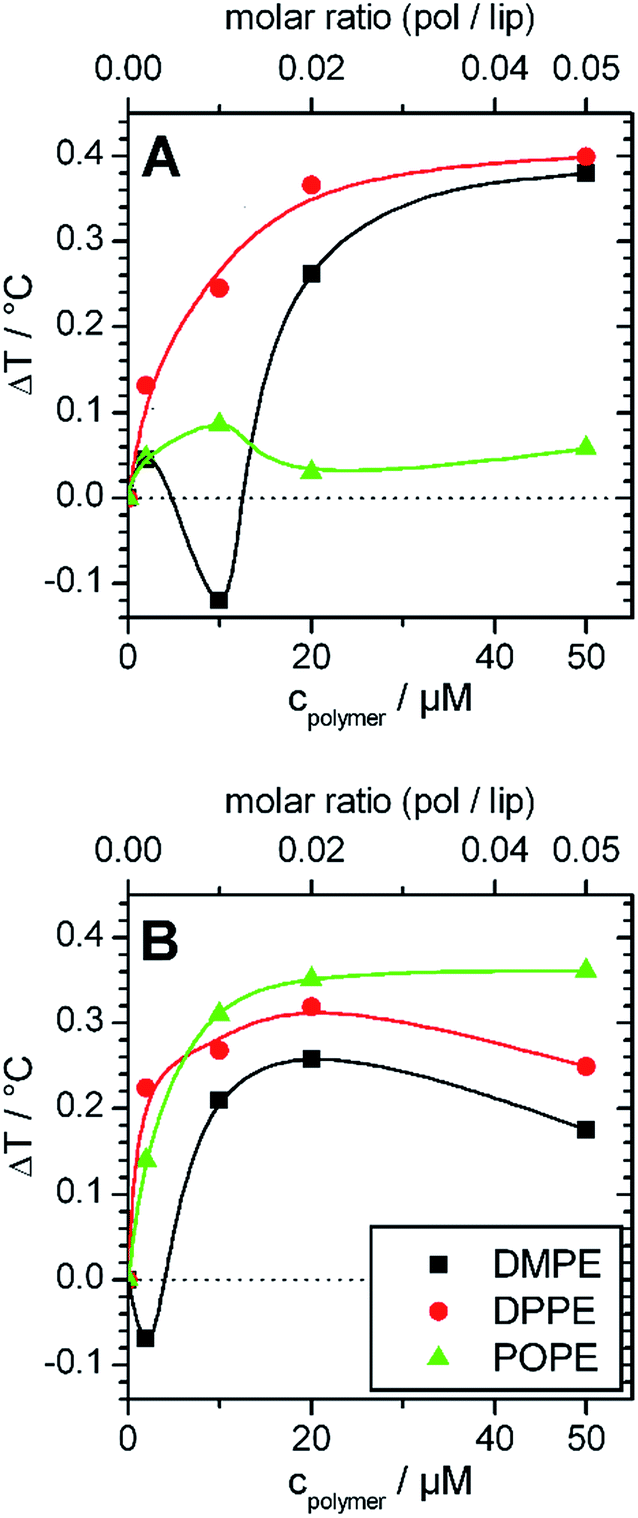 | ||
| Fig. 5 Change of main transition temperatures of lipid membranes prepared from DMPE (black), DPPE (red) and POPE (green) after addition of different amounts of (A) GP and (B) F-GP. The lipid concentration is always 1 mmol L−1. The temperature where half of the transition enthalpy was consumed was interpreted as transition temperature. The inserted lines are drawn to guide the eyes. | ||
These effects are small, but become interesting in comparison with the corresponding data of copolymer/PC membrane aggregates, were the Tm shift was negative (see Fig. 2A, 2B and 3). The only difference between DMPC and DMPE as well as between DPPC and DPPE is the chemical structure of the headgroup. Thus, the different influence on Tm must be due to polar interactions between the copolymers and the headgroups. Therefore, the presented results are a direct proof for the existence of interactions between the PGMA blocks of the polymer and the lipid headgroups. The fact that the polymer binding induces a more positive Tm shift for PE membranes than for PC membranes of equal chain length indicates that polar interactions between the polymers and the PE headgroup are more dominant. This is due to the ability of PE to act as both, as hydrogen bond donor and acceptor.40 High propensities for hydrogen binding are also known for PGMA.14,18 Thus, a contribution of hydrogen bonds between the PGMA blocks of the polymers and the PE headgroup to the polar interactions can be assumed.
Moreover, it can be seen that POPE/copolymer transitions which are depicted in Fig. 4C are always unimodal and highly cooperative. It means that the bimodal transitions which were detected for the DMPC–F-GP mixtures in the same temperature range are not due to any change in the aggregation state of the copolymer.
NMR: localization of the PPO block
From DSC measurements at different conditions we concluded that the hydrophobic parts of the polymer insert into the lipid bilayer. To be able to observe more directly the localization the copolymer within the lipid membrane, we performed solid state magic angle spinning (MAS) NMR spectroscopy.Taking advantage of the high-resolution conditions provided by MAS solid-state NMR, the (temporary) spatial proximity of nuclei can be elucidated by means of the 2D-NOESY experiment.47 This experiment can give answers to the question whether there is at least a temporary close proximity between the protons of the lipid molecules and those of the polymer molecules. This would allow to conclude on the depth of penetration of the polymer molecules into the lipid bilayer.
All experiments were performed on DMPC membranes in the Lα-phase, because gel phase membranes do not yield a sufficient spectral resolution for a 2D-NOESY analysis. In Fig. 6 the proton NMR spectrum of the mixture of DMPC and F-GP (5 mol%) recorded at a spinning speed of 4 kHz is shown. Due to many different proton types in the sample and the narrow chemical shift range, most of the polymer resonances overlay with resonances of the lipid. The only isolated resonance from the polymer is the signal of the PPO methyl group at a chemical shift value of approximately 1.1 ppm. Thus, information on the proximity of the PPO methyl group to nuclei of the lipid molecules can be obtained from crosspeaks between this polymer resonance and lipid resonances.
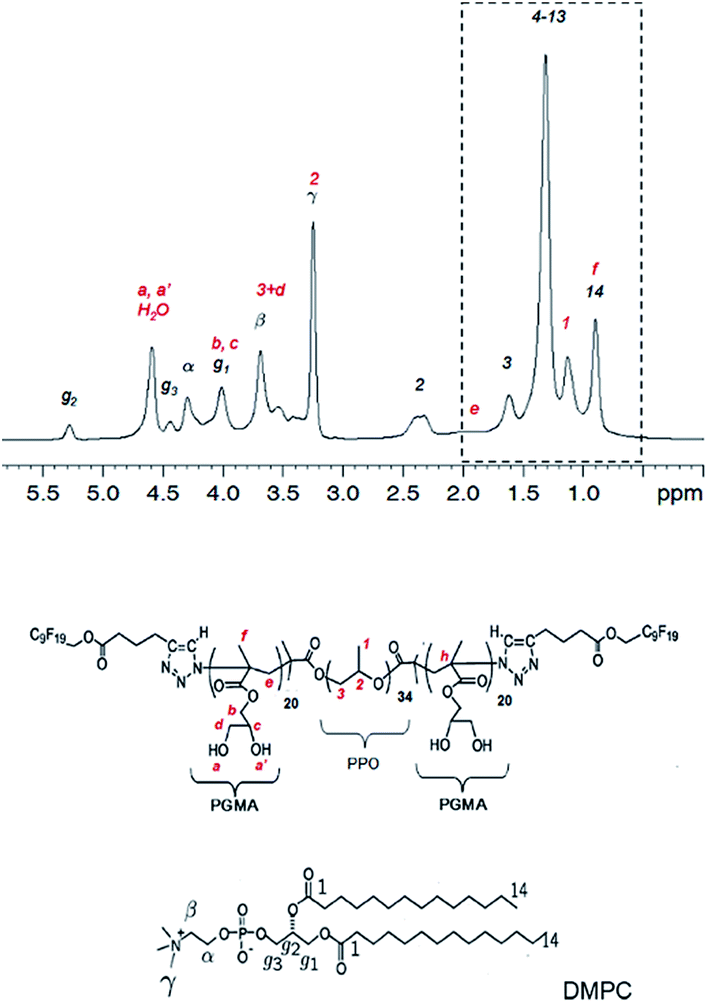 | ||
| Fig. 6 1H NMR spectrum of the mixture of DMPC with F-GP (5 mol%) and structures of the molecules including assignment of the 1H resonances. Peaks labelled in red originate from the polymer and peaks labelled in black originate from lipid resonances. The region of the spectrum inside the dashed box is evaluated in a 2D-NOESY experiment (see Fig. 7). | ||
In Fig. 7 a close-up of the region of interest of the full 2D-NOESY spectrum is depicted for the mixture of DMPC with both polymers (Fig. 7A: GP, Fig. 7B: F-GP). In both spectra an off-diagonal signal is clearly detectable for the PPO methyl group and the (CH2)4-13 signal of the lipid acyl chains. To illustrate this in more detail, a slice through the PPO–methyl signal at approximately 1.1 ppm in the ω1 – dimension is shown in each of the spectra. The strong peak corresponds to the PPO–methyl resonance on the diagonal. To the left of this peak a small signal shows up at the chemical shift of the methylene groups of the lipid acyl chain (ca. 1.3 ppm), revealing that the corresponding nuclei exchanged magnetization during the mixing period. These crosspeaks confirm the insertion and mixing of the polymer molecules into the lipid bilayer. The crosspeaks are only of relatively small intensity, which is likely due to only weak and rather transient contacts in the fluid bilayers. Whether the polarization transfer occurs via an actual cross-relaxation (NOE) effect caused by rapidly fluctuating dipole–dipole couplings, or via spin diffusion caused by weak residual quasi-static couplings is not clear.48,49 At any rate, the observed temporarily close molecular contacts between the hydrophobic groups of the lipid (acyl chains) and the hydrophobic block of the polymers (PPO) leads to the conclusion that the polymer is not only adsorbed onto the surface but is indeed inserted into the lipid membrane to a significant degree. This finding confirms the conclusions drawn from the DSC experiments.
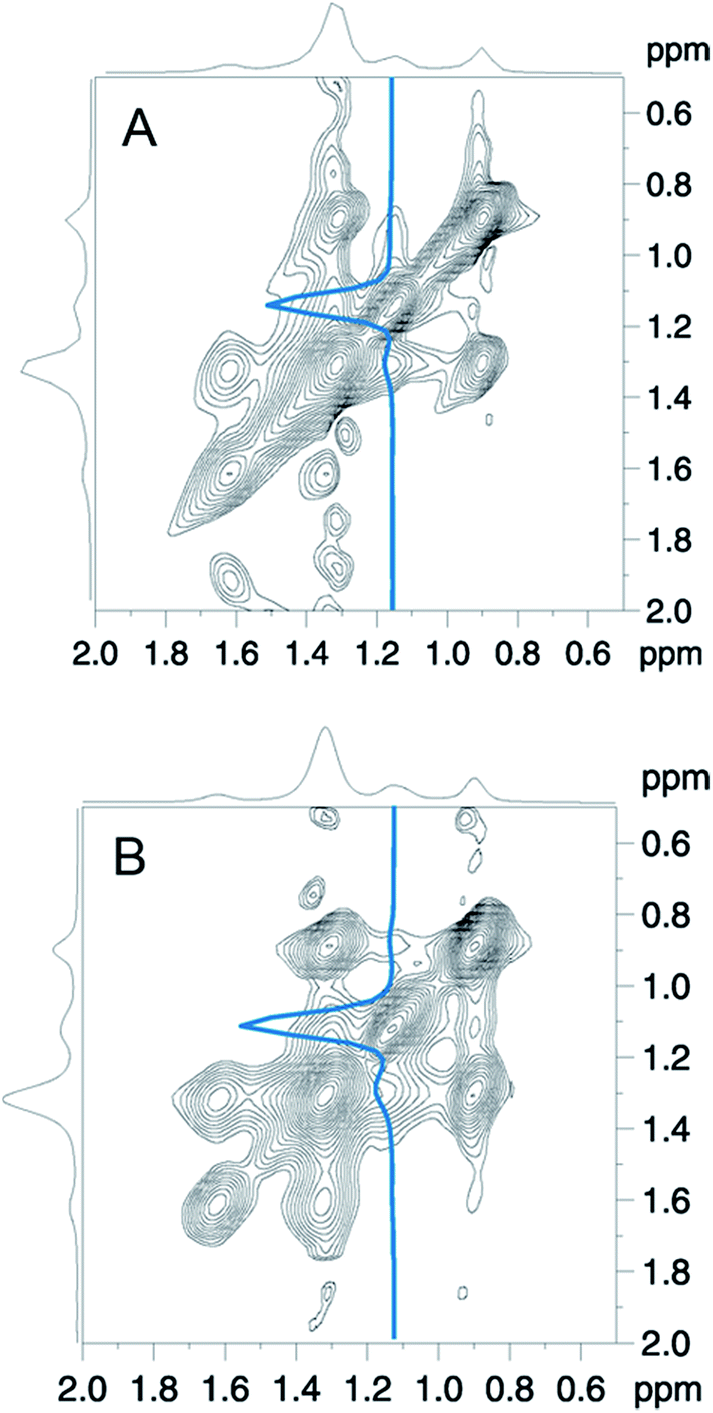 | ||
| Fig. 7 2D-NOESY-spectra of DMPC mixed with (A) GP and (B) F-GP polymer (both 5 mol%) measured at 40 °C. The base level of the counter plots is set to 5% of the highest intensity. The blue lines represent slices at the position of the PPO–methyl resonance in the ω1 – dimension. | ||
The fraction of inserted polymer can also be estimated from a detailed study of residual dipole–dipole couplings within the PPO-chains as arising from pronounced anisotropic dynamics, as will be reported shortly.
CLSM: membrane integrity and pore formation
After having evidence that both, GP and F-GP insert into the hydrophobic part of the lipid bilayer the question arises to which extent the membrane integrity is perturbed by this insertion. The questions whether the barrier function of the membrane is maintained, whether the copolymer insertion leads to pore formation or membrane defects, and whether the polymer can translocate through the membrane into the inner volume of the vesicles are addressed by two types of fluorescence microscopy experiments performed with giant unilamellar vesicles (GUVs). The GUVs were formed from POPC by electro-swelling. As the phase transition temperature of POPC is at −3 °C (ref. 50) and all experiments were performed at room temperature the results allow conclusions on the interactions of the polymers with fluid membranes. | ||
| Fig. 8 Confocal laser scanning microscopy images of solutions containing the water soluble dye Alexa 488 and upper row: POPC GUVs; middle row: POPC GUVs + GP (8 mmoL L−1) and lower row: POPC GUVs + F-GP (7.1 mmoL L−1) at different time intervals after injecting the GUVs to the other components. The indicated times are times after the beginning of the microscopy. There is a small lag time between the GUV/polymer mixing and the begin of the microscopy of approximately 10 s. | ||
The middle row of Fig. 8 represents a time series of images taken after the GUVs have been injected into a GP–dye mixture. To be able to identify the same GUV in the different images, selected GUVs are marked by a number. A low fluorescence intensity can be detected in the lumen of most, but not all, of the GUVs after mixing. The intensity is always lower inside the GUVs than outside, indicating that complete mixing of the two volumes did not occur. Moreover, there is no evolution of fluorescence intensity within the observation time of 10–15 minutes, i.e. no dye influx was observed after the lag time of the experiment. This suggests that some transient pores or defects formed upon mixing of the GUVs with the polymer solution within the lag time, which then healed after an equilibrium was reached.
The same experiment performed with the fluorinated copolymer (F-GP) yields the microscopic images depicted in the bottom row of Fig. 8. Some of the GUVs are already filled with dye when the first image was taken (e.g. GUV 4), which means that there is a rapid dye influx after mixing. Other GUVs fill within the time scale of the experiment (e.g. GUV 1 and GUV 3). This provides clear evidence that F-GP adsorption and insertion leads to the formation of stable hydrophilic pores or defects, which allow the water soluble dye to diffuse into the interior volume of the GUVs. The images show that the liposomes stay intact and do not disintegrate or form other structures after being injected into the polymer solution. The images also show that the permeation of the dye is quite inhomogeneous, i.e. dye-filled GUVs coexist with GUVs that still maintain their barrier function over the time scale of the experiment. This is very similar to what was observed for some pore forming peptides (PFPs) interacting with GUVs51 and indicates that the leakage mechanism is “heterogeneous graded”.52 Some of the vesicles might also be permeabilized by the “all or none” mechanism (GUV 4).
For quantification of dye influx we integrated the fluorescence intensity of the lumen of some selected GUVs (marked by numbers in Fig. 8) at different times and normalized it to the average intensity of the respective image. The resulting normalized intensities of some GUVs of the control sample are shown in Fig. 9A. The results for GP and F-GP containing GUV suspensions are shown Fig. 9B and C, respectively. The images of the GUVs of the control sample shown in Fig. 9A confirm that the normalized intensity values are lower than 1 and do not change with time indicating that no dye influx occurs. In Fig. 9B it can be seen that the fluorescence intensity within the selected GP-bound GUVs is higher than in the control but does not increase within the timescale of the experiment (15 min). In contrast, Fig. 9C shows that the fluorescence intensity inside GUVs mixed with F-GP increases with time. However, the kinetics of dye influx is different for the individual GUVs. The content of GUV 4 already completely mixed with the bulk solution at the beginning of the image recording. This indicates that pores of different size and lifetimes are formed upon F-GP binding to POPC vesicles and that the sample is not in a thermodynamic equilibrium at the end of the experiment.
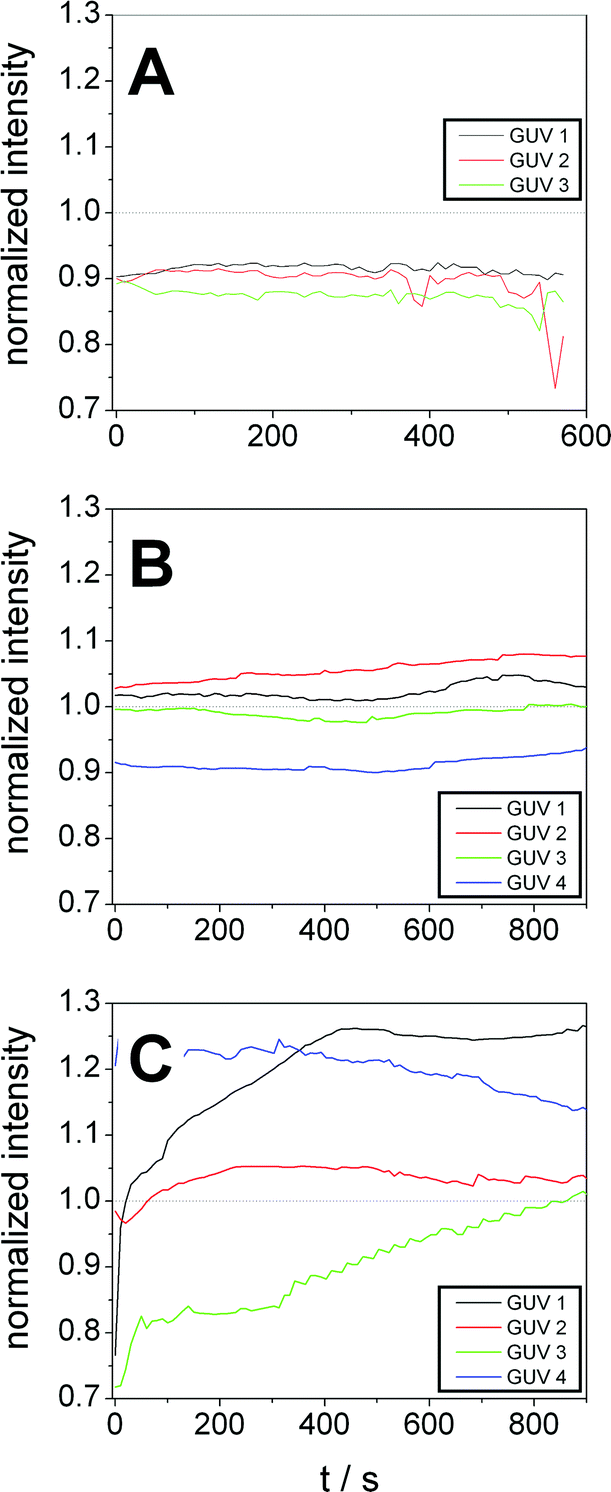 | ||
| Fig. 9 Fluorescence intensities within selected POPC GUVs after addition to a dye solution containing (A) no additive, (B) GP (8 mmol L−1) and (C) F-GP (7.1 mmol L−1) normalized to the average fluorescence intensity of the complete image. The numbering of the GUVs corresponds to the numbering in Fig. 8. | ||
The different dye influx kinetics of GP and F-GP containing samples reveals a clear influence of the perfluoroalkyl moieties. In the case of GP binding, the permeabilization is only minute and transient and is followed by resealing of the membrane once the equilibrium is reached. In the case of F-GP binding pores and/or defects are more stable and heterogeneous. An equilibrium is not reached within the time of the experiments. This might be due to a more complex binding mechanism of the triphilic polymer as compared to the amphiphilic one. In case of F-GP binding not only polar and hydrophobic forces have to be balanced, but also the tendency of the perfluoroalkyl chains to segregate, has to be taken into account. This additional driving force leads to a higher number of possible intermediates in the different steps of the interaction, as micellization/demicellization, adsorption, insertion and possibly translocation. The tendency of the inserted fluorinated chains to segregate within the membrane might play a role in the formation of membrane pores or defects.
 | ||
| Fig. 10 Confocal laser scanning microscopy images of POPC GUVs labelled with BODIPY (green) after addition of TMR labelled GP (0.9 mmol L−1) (left column) and F-GP (0.8 mmol L−1) (right column). (a and e) Superposition of TMR and BODIPY fluorescence; the encircled vesicles are shown in (b and f) in a higher magnification. (c and g) TMR and (d and h) BODIPY channel of the vesicle shown in (b) and (f), respectively. | ||
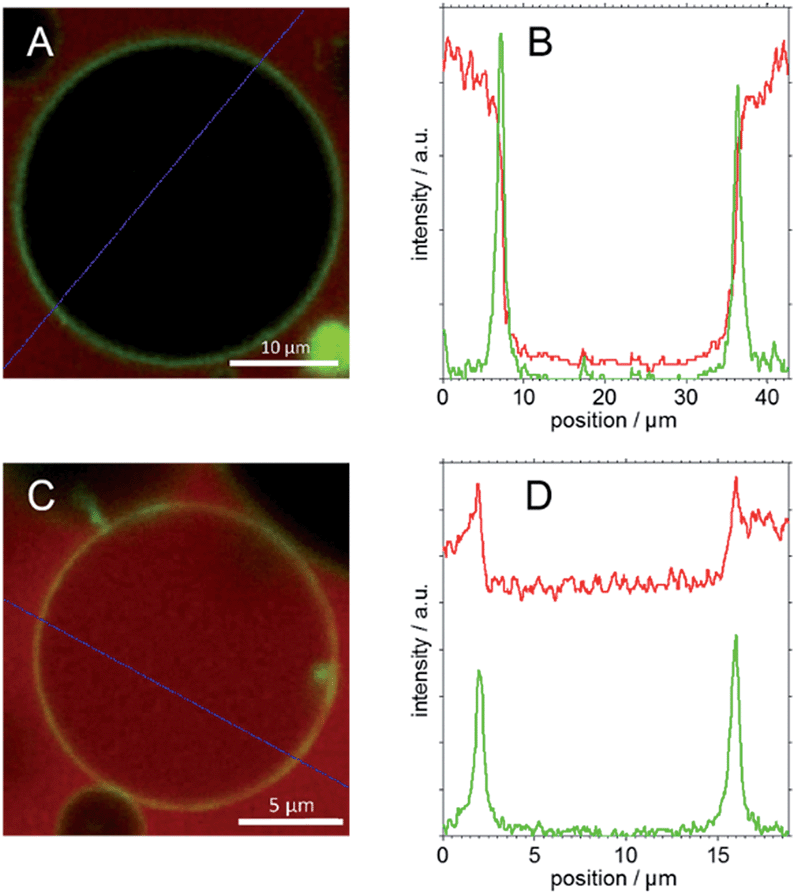 | ||
| Fig. 11 CLSM images of BODIPY labelled GUVs in (A) TMR labelled GP solution and (C) TMR labelled F-GP solution and relative intensity values of red (TMR) and green (BODIPY) channels (B and D) along the blue lines drawn in (A and C), respectively. | ||
The first column of Fig. 10 represents images of GP–GUV mixtures recorded about 5 min after mixing. Fig. 10a and b show an overlay of the TMR and BODIPY channels in different magnifications; Fig. 10c and d show the TMR and the BODIPY channel, respectively. Already in the TMR channel and the overview (Fig. 10a) it can be seen that there is no TMR fluorescence intensity in the interior volume of the GUVs. In the overlay of TMR and BODIPY channel (Fig. 10b) it can clearly be seen that the POPC membrane (green fluorescence) constitutes a diffusion barrier for the GP copolymer (red fluorescence). Fig. 11C shows the intensity values of the TMR and the BODIPY channel along a cut through the GUV image presented in Fig. 10b and 11A. The peaks in the green channel indicate the position of the membrane. The intensity of the red channel drops nearly to zero in between the two peaks, i.e. in the interior of the GUV. That indicates that GP does not translocate through the membrane. Furthermore, it is concluded that there is no surface excess of GP at the site of the membrane. These findings corroborate the results of a study of similar amphiphilic triblock copolymers with DLPC GUVs,53 where polymer adsorption to the vesicle surface could only be observed at much higher polymer concentration and no polymer was found in the aqueous interior of the GUVs.
Very different results are obtained when the same experiment is repeated with the block copolymer F-GP (right column in Fig. 10). In Fig. 10e–g TMR fluorescence can be observed within the inner volume of the selected GUV. This indicates that the complete copolymer F-GP with its hydrophilic, hydrophobic and fluorophilic moieties is able to translocate through the lipid bilayer. Fig. 10e represents an overview of a small volume containing several GUVs. It shows that 5 min after mixing the distribution of the TMR fluorescence in the GUV suspension is inhomogeneous. Many of the GUVs are filled with F-GP, whereas numerous others show no TMR fluorescence within their lumen. This is similar to what was observed for the dye-influx experiments, discussed above and points towards a heterogeneous graded permeabilization.52 In the BODIPY channel (Fig. 10h) we see an almost equal distribution of membrane staining dye, indicating that the GUV stays intact and does not disintegrate upon pore formation and copolymer translocation. Fig. 10f also shows that the highest TMR fluorescence intensity is co-localized with the fluorescence of the membrane staining BODIPY, i.e. the site of the membrane.
This becomes even clearer in Fig. 11D, where the intensity values along a line through the image presented in Fig. 10f and 11C are plotted. In between the peaks of the green channel the red fluorescence decreases only slightly with respect to the intensity before the first and after the second peak. Moreover, the intensity of the red channel is maximal at the position of the membrane, i.e. at the position of the highest BODIPY florescence. This indicates that (i) F-GP translocates through the vesicle membrane and (ii) the highest concentration of F-GP is found at the GUV membrane, i.e. strong adsorption and/or insertion of F-GP leads to an enrichment of the fluorinated copolymer at the membrane. This is clearly different to what was observed for GP and reveals a clear effect of the perfluoroalkyl end groups, which apparently increases the interaction with the lipid membrane.
The exact mechanism of F-GP translocation cannot be derived from the data presented here. The question remains, whether the polymer is freely diffusing through pre-formed pores or defects or whether it translocates by a sequence of adsorption, percolation8 and desorption at the other side of the bilayer. As both, translocation and pore formation occur concomitantly the former pathway seems to be probable. Moreover, it is unlikely that the hydrophilic PGMA blocks of the copolymers may pass the hydrophobic barrier of the membrane without using any defects.
The fluorescence experiments indicate clear differences in the action of F-GP and GP and thus reveal the effect of the copolymer perfluoro end groups. The addition of perfluoro moieties leads to a stronger adsorption to POPC membranes and to the formation of membrane defects that allow the diffusion of water soluble probes as well as the translocation of the complete triphilic block copolymer. Moreover, the finding of an enhanced binding of F-GP corroborates the conclusion we have drawn from the DSC experiments.
Conclusion
The interaction of new symmetric amphi- and triphilic block copolymers with lipid membranes has been investigated. The amphiphilic block copolymers consist of a hydrophobic PPO middle block to which hydrophilic PGMA blocks are attached on either side. The triphilic block copolymer is derived from the former one by terminally attaching perfluoroalkyl chains to the PGMA blocks. These perfluoro segments are both hydrophobic and lipophobic and thus have a tendency to phase segregate in water as well as in a hydrocarbon matrix. DSC was used to study the phase transition temperature Tm of lipid membranes upon interaction with the polymers. Tm is a thermodynamic parameter that reflects the stability of the gel- and liquid crystalline phases of the membrane and therefore allows conclusions about the nature of the interactions. An extensive set of DSC data was acquired under systematic variation of the polymer concentration, the chain length of the phospholipids, the nature of their headgroups, and the addition of perfluoro moieties to the copolymer. The resulting variations in Tm are small but rather systematic. The following observations were made:i. Decrease and increase in Tm are possible upon polymer binding, depending on the chosen conditions. The actual value of Tm is a result of a delicate balance of counteracting effects.
ii. The shift in Tm (ΔTm) after polymer binding increases with the hydrophobic thickness of the bilayer. It is negative for the thinner DMPC and positive for the thicker DAPC membranes.
iii. ΔTm is higher for PE headgroups than for PC headgroups, i.e. it increases with the capacity of the lipid headgroups to donate hydrogen bonds.
iv. ΔTm is maximal at intermediate copolymer concentrations (1 to 2 mol%).
v. All effects are amplified by the addition of perfluoro end groups to the copolymer.
vi. Perfluoro moieties induce the tendency for phase separation within the membrane.
These findings lead to the conclusion that the binding mechanism of the polymers consists of an adsorption of the hydrophilic PGMA blocks to the headgroup layer and a concomitant insertion of the PPO blocks and perfluorinated chains into the hydrophobic part of the lipid bilayer. Clear evidence for both processes is given by the chain length and headgroup dependency of Tm. A contribution of H-bonding to the headgroup interactions is probable, as the interactions increase with the availability and accessibility of H-donors and -acceptors. It is worth noting that a positive ΔTm was rarely reported for poloxamer binding to lipid membranes, which usually leads to a decrease in Tm.7,9,21,22 Therefore, we think that the positive shift of Tm that we observe systematically upon binding of the PGMA containing block copolymers is due to the higher propensity of the GMA side groups to donate and to accept H-bonds. The addition of perfluoro segments to the copolymer leads to a stronger binding, the perfluoroalkyl chains acting as lipid anchors. Additionally, they induce a tendency for segregation within the bilayer.
Magic angle spinning NMR spectroscopy was applied to directly prove the insertion of the PPO blocks into the hydrocarbon membrane layer. On a sub-molecular level we could show the close proximity between PPO methyl groups and the lipid acyl chains of DMPC. This intimate contact could be shown for both fluorinated and non-fluorinated copolymer and provides unambiguous evidence for the copolymer insertion.
On a larger length scale we investigated the influence of polymer adsorption and insertion on the integrity of the membrane. By fluorescence microscopy on giant unilamellar POPE vesicles (GUVs) we could show that binding of the copolymer with perfluoro moieties leads to the formation of membrane defects that allow the influx of water soluble dyes as well as the translocation of the complete triphilic block copolymer through the bilayer. In contrast, the binding of the non-fluorinated triblock copolymer does not or only marginally disturb the barrier function of the membrane. Only very moderate dye influx and only rare events of polymer translocation could be observed. Moreover co-localization experiments with labelled polymer and stained GUVs showed an enrichment of F-GP at the lipid membrane, indicating a strong adsorption, which could not be observed for GP.
All experiments lead to a consistent picture of the interaction of the copolymers with lipid membranes and reveal the role perfluoro segments. In comparison to the extensively studied (yet not completely understood) poloxamers the introduction of the GMA side chains and the perfluoro anchors lead to new modes of interaction and new structuring elements. The further study of these amphi- or triphilic molecules is promising, as the fine tuning of hydrogen bonding interactions, hydrophobic interactions and the segregation tendency of fluorinated moieties may lead to the controlled design of target structures, such as defined pores or channels.
Acknowledgements
The authors thank Dr Zheng Li for the synthesis of the polymers, D. Glatte for help with the CLSM measurements, and the Deutsche Forschungsgemeinschaft (DFG) for financial support (FOR 1145).References
- J. Y. Wang, J. M. Chin, J. D. Marks and K. Y. C. Lee, Langmuir, 2010, 26, 12953–12961 CrossRef CAS PubMed.
- V. Y. Erukova, O. O. Krylova, Y. N. Antonenko and N. S. Melik-Nubarov, Biochim. Biophys. Acta, 2000, 1468, 73–86 CrossRef CAS.
- S. A. Maskarinec, G. H. Wu and K. Y. C. Lee, in Cell Injury: Mechanisms, Responses, and Repair, ed. R. C. Lee, F. Despa and K. J. Hamann, 2005, vol. 1066, pp. 310–320 Search PubMed.
- R. C. Lee, L. P. River, F. S. Pan, L. Ji and R. L. Wollmann, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 4524–4528 CrossRef CAS.
- F. A. Merchant, W. H. Holmes, M. Capelli-Schellpfeffer, R. C. Lee and M. Toner, J. Surg. Res., 1998, 74, 131–140 CrossRef CAS PubMed.
- C. Y. Cheng, J. Y. Wang, R. Kausik, K. Y. C. Lee and S. Han, Biomacromolecules, 2012, 13, 2624–2633 CrossRef CAS PubMed.
- M. A. Firestone, A. C. Wolf and S. Seifert, Biomacromolecules, 2003, 4, 1539–1549 CrossRef CAS PubMed.
- S. Hezaveh, S. Samanta, A. De Nicola, G. Milano and D. Roccatano, J. Phys. Chem. B, 2012, 116, 14333–14345 CrossRef CAS PubMed.
- G. Pembouong, N. Morellet, T. Kral, M. Hof, D. Scherman, M. F. Bureau and N. Mignet, J. Controlled Release, 2011, 151, 57–64 CrossRef CAS PubMed.
- E. Amado, A. Blume and J. Kressler, React. Funct. Polym., 2009, 69, 450–456 CrossRef CAS PubMed.
- E. Amado, C. Augsten, K. Mäder, A. Blume and J. Kressler, Macromolecules, 2006, 39, 9486–9496 CrossRef CAS.
- T. Demina, I. Grozdova, O. Krylova, A. Zhirnov, V. Istratov, H. Frey, H. Kautz and N. Melik-Nubarov, Biochemistry, 2005, 44, 4042–4054 CrossRef CAS PubMed.
- E. Amado, A. Blume and J. Kressler, Langmuir, 2010, 26, 5507–5519 CrossRef CAS PubMed.
- E. Amado, A. Kerth, A. Blume and J. Kressler, Langmuir, 2008, 24, 10041–10053 CrossRef CAS PubMed.
- S. O. Kyeremateng, E. Amado, A. Blume and J. Kressler, Macromol. Rapid Commun., 2008, 29, 1140–1146 CrossRef CAS.
- M. P. Krafft and J. G. Riess, Biochimie, 1998, 80, 489–514 CrossRef CAS.
- E. Amado and J. Kressler, Soft Matter, 2011, 7, 7144–7149 RSC.
- S. O. Kyeremateng, K. Busse, J. Kohlbrecher and J. Kressler, Macromolecules, 2011, 44, 583–593 CrossRef CAS.
- S. O. Kyeremateng, T. Henze, K. Busse and J. Kressler, Macromolecules, 2010, 43, 2502–2511 CrossRef CAS.
- E. Feitosa and F. M. Winnik, Langmuir, 2010, 26, 17852–17857 CrossRef CAS PubMed.
- J. D. Castile, K. M. G. Taylor and G. Buckton, Int. J. Pharm., 2001, 221, 197–209 CrossRef CAS.
- Y. Y. Chieng and S. B. Chen, J. Phys. Chem. B, 2009, 113, 14934–14942 CrossRef CAS PubMed.
- W. M. Yau, W. C. Wimley, K. Gawrisch and S. H. White, Biochemistry, 1998, 37, 14713–14718 CrossRef CAS PubMed.
- L. L. Holte and K. Gawrisch, Biochemistry, 1997, 36, 4669–4674 CrossRef CAS PubMed.
- D. Huster, K. Arnold and K. Gawrisch, J. Phys. Chem. B, 1999, 103, 243–251 CrossRef CAS.
- J. Jeener, B. H. Meier, P. Bachmann and R. R. Ernst, J. Chem. Phys., 1979, 71, 4546–4553 CrossRef CAS PubMed.
- G. Cevc, A. Watts and D. Marsh, FEBS Lett., 1980, 120, 267–270 CrossRef CAS.
- A. Watts, K. Harlos, W. Maschke and D. Marsh, Biochim. Biophys. Acta, 1978, 510, 63–74 CrossRef CAS.
- D. Papahadjopoulos, M. Moscarello, E. H. Eylar and T. Isac, Biochim. Biophys. Acta, 1975, 401, 317–335 CrossRef CAS.
- C. Schwieger and A. Blume, Eur. Biophys. J., 2007, 36, 437–450 CrossRef CAS PubMed.
- V. P. Ivanova, I. M. Makarov, T. E. Schaffer and T. Heimburg, Biophys. J., 2003, 84, 2427–2439 CrossRef CAS.
- C. h. Huang and S. Li, Biochim. Biophys. Acta, 1999, 1422, 273–307 CrossRef CAS.
- E. Amado, A. Kerth, A. Blume and J. Kressler, Soft Matter, 2009, 5, 669–675 RSC.
- J. F. Nagle and S. Tristram-Nagle, Biochim. Biophys. Acta, 2000, 1469, 159–195 CrossRef CAS.
- N. Kučerka, Y. Liu, N. Chu, H. I. Petrache, S. Tristram-Nagle and J. F. Nagle, Biophys. J., 2005, 88, 2626–2637 CrossRef PubMed.
- M. Garavaglia, S. Dopinto, M. Ritter, J. Furst, S. Saino, F. Guizzardi, M. Jakab, C. Bazzini, V. Vezzoli, S. Dossena, S. Rodighiero, C. Sironi, G. Botta, G. Meyer, R. M. Henderson and M. Paulmichl, Cell. Physiol. Biochem., 2004, 14, 231–240 CrossRef CAS PubMed.
- M. Caffrey and J. Hogan, Chem. Phys. Lipids, 1992, 61, 1–109 CrossRef CAS.
- R. Lewis, N. Mak and R. N. McElhaney, Biochemistry, 1987, 26, 6118–6126 CrossRef CAS.
- C. Schwieger and A. Blume, Biomacromolecules, 2009, 10, 2152–2161 CrossRef CAS PubMed.
- M. Dyck, P. Krüger and M. Losche, Phys. Chem. Chem. Phys., 2005, 7, 150–156 RSC.
- A. Blume, Biochemistry, 1980, 19, 4908–4913 CrossRef CAS.
- P. b. Hitchcoc, R. Mason, K. M. Thomas and G. G. Shipley, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3036–3040 CrossRef.
- R. Koynova and M. Caffrey, Chem. Phys. Lipids, 1994, 69, 1–34 CrossRef CAS.
- M. Save, J. V. M. Weaver, S. P. Armes and P. McKenna, Macromolecules, 2002, 35, 1152–1159 CrossRef CAS.
- K. Mortensen, J. Phys.: Condens. Matter, 1996, 8, A103–A124 CrossRef CAS.
- Y.-l. Su, J. Wang and H.-z. Liu, Macromolecules, 2002, 35, 6426–6431 CrossRef CAS.
- J. Forbes, C. Husted and E. Oldfield, J. Am. Chem. Soc., 1988, 110, 1059–1065 CrossRef CAS.
- D. Neuhaus, in Encyclopedia of Magnetic Resonance, Vol. 5, ed. D. M. Grant and R. K. Harris, J. Wiley & Sons, Chichester, 1996, p. 3290 Search PubMed.
- K. Schmidt-Rohr and H. W. Spiess, Multidimensional Solid-State NMR and Polymers, Academic Press Ltd, London, 1994 Search PubMed.
- K. L. Koster, M. S. Webb, G. Bryant and D. V. Lynch, Biochim. Biophys. Acta, 1994, 1193, 143–150 CrossRef CAS.
- B. Apellániz, J. L. Nieva, P. Schwille and A. J. García-Sáez, Biophys. J., 2010, 99, 3619–3628 CrossRef PubMed.
- H. Patel, C. Tscheka and H. Heerklotz, Soft Matter, 2009, 5, 2849–2851 RSC.
- R. Schöps, E. Amado, S. S. Müller, H. Frey and J. Kressler, Faraday Discuss., 2013, 166, 303–315 RSC.
| This journal is © The Royal Society of Chemistry 2014 |