 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gel architectures and their complexity
Walter
Richtering
a and
Brian R.
Saunders
*b
aInstitute of Physical Chemistry, RWTH Aachen University, Landoltweg 2, D-52056 Aachen, Germany.
bBiomaterials Science Research Group, Manchester Materials Science Centre, School of Materials, The University of Manchester, Grosvenor Street, Manchester, M13 9PL, UK. E-mail: brian.saunders@manchester.ac.uk
First published on 3rd March 2014
Abstract
Gels have made the transition from brittle materials with few potential applications to high performance systems with mechanical properties approaching that of rubber. They have a wide variety of structures and provide the opportunity to tailor these structures to achieve well-controlled properties over a range of length scales. In this review we consider and compare the structures and properties of a range of gels that have been studied in recent years. In comparing these gels we highlight the importance of key structural parameters in defining gel mechanical properties. It is hoped that this article will provide authors who discover new gels a resource that will easily enable them to determine the differences of their new gels to existing gels.
 Walter Richtering | Walter Richtering studied chemistry at the universities of Bochum and Freiburg and obtained his PhD with Prof. Burchard at the Hermann-Staudinger Institute of Macromolecular Chemistry in Freiburg as a Fellow of the Graduiertenkolleg Polymerwissenschaften in 1990. Afterwards he was a post-doc as a Feodor-Lynen Fellow of the Alexander von Humboldt-Foundation at the University of Massachusetts. He was appointed as Professor for Physical Chemistry at Kiel University in 2000 and to the chair in Physical Chemistry at RWTH Aachen University in 2003. He received the Raphael-Eduard-Liesegang award of the Kolloid-Gesellschaft and is the editor of Colloid & Polymer Science. He is the coordinator of the Collaborative Research Centre (SFB 985) “Functional microgels and microgel systems” funded by the Deutsche Forschungsgemeinschaft. |
 Brian R. Saunders | Brian R. Saunders obtained his PhD at Monash University and worked as a post-doc with Prof. Brian Vincent at the University of Bristol between 1994 and 1997. He is a Professor of Polymer and Colloid Chemistry at the School of Materials, University of Manchester. His research career at Manchester began in 2002 and involves the application of polymer and colloid chemistry principles to healthcare and solar energy research. A highlight of his healthcare research is a new method for constructing gels using pre-assembled sub-micrometer microgel particles. He is co-founder and scientific director of a University of Manchester spin-out, Gelexir Healthcare Ltd. |
Introduction
Gels continue to attract much interest in the literature.1–3 There is a clear transition from simple, approximately homogeneous gels towards more complex and architecturally sophisticated gels. The introduction of specific morphologies across different length scales has resulted in gels with mechanical properties that begin to approach that of synthetic rubbers.4 Furthermore, increasingly sophisticated intra- and inter-molecular interactions that have wide ranges of tunability have become common. With this explosion of structural and interaction complexity there has also been a plethora of new terms introduced for these new gels. The purpose of this review is to consider some of the most recent examples of structurally complex gels.In this review, which concentrates on synthetic gels, we assess the structures and properties of a number of new gels that have emerged and compare them to previous gels. More general reviews on hydrogels with emphasis on their biomaterial applications have recently been published elsewhere.5,6 An aim of this focussed review is to show that crosslinking and assembly are crucial for gel properties and that control of these processes can provide excellent mechanical properties. The manuscript concludes with a recommendation that authors seeking to publish new gels should clearly compare and contrast their systems to existing gels and then critically assess whether a new name/terminology is actually required.
Hydrogel fundamentals
Polymer gels consist of networks of polymer chains that are either swollen in water (hydrogels), swollen in oil (lipogels) or air (aerogels).7 The term ‘gel’ is often used to encompass both hydrogels and lipogels. Hydrogels absorb and retain large amounts of water. As for the polymer networks, they comprise crosslinked polymer chains which swell but do not dissolve in a good solvent. The crosslinks may be physical or chemical. Chemically crosslinked gels have covalent linkages between different chains (usually not reversible). However, dynamic covalent bonds are reversible and this has resulted in self-healing hydrogels.8 Physically crosslinked gels have physical (reversible) interactions between different chains that prevent dissolution. These are often due to electrostatic attraction, hydrogen bonding or hydrophobic interactions (below). Another class of physical gels are those from concentrated colloid dispersions9 – but they are beyond the scope of the present discussion.The most important parameters to characterise swollen networks are10 the polymer volume fraction (ϕ), number-density of elastically effective chains (νe) and the correlation length or distance between two adjacent crosslinks (ξ). The value of ϕ provides a measure of volume swelling ratio (1/ϕ) and global porosity (1 − ϕ). In addition, Mc(ave) is the mean molar mass of the elastically effective chains. The latter controls swelling, elasticity and ductility of gels. The value of ξ provides a measure of space available (local porosity) between macromolecular chains. The ability to control values of ϕ, Mc(ave) and ξ both at the local and global level enables the design of hydrogels with bespoke mechanical, diffusive and swelling properties.
The following equation applies for the shear modulus (G) of a gel.7
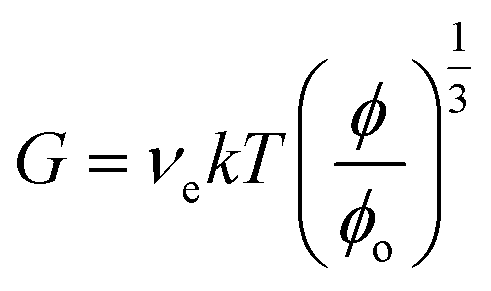 | (1) |
For the above equation ϕo is the as-prepared polymer volume fraction. In the case of a Poisson ratio of 0.5 (which is often assumed7,11), the Young's modulus follows from:
| E = 3G | (2) |
Eqn (1) assumes that each elastically effective chain contributes kT to the energy required to stretch the chain. The equation shows that G for a gel decreases with swelling. However, if the swelling state is the same as preparation then ϕ = ϕo and eqn (1) simplifies further.
The length scales for conventional hydrogels are defined by key parameters such as ξ and these are more or less uniformly distributed throughout conventional hydrogels. Of course, they are likely to be polydisperse. However, for more complex gels, inter penetrating networks (IPNs), semi-IPNs, doubly crosslinked (DX) microgels and double network (DN) gels, they are less well-defined. In fact, for the last three there may be two different ξ values. This has interesting implications for attempts to mimic natural hydrogels, such as articular cartilage where a range of hierarchical structures with different inherent ξ values are present. We return to this topic below when the more complex gels are discussed.
Hydrogels with low structural complexity
An important consequence of conventional hydrogel preparation is that the local values for νe differ substantially within the hydrogel. In other words, the Mc(ave) and ξ values are strongly polydisperse. The broad distribution of elastically effective chain lengths results in stress concentration in regions of lowest νe when placed under strain causing localised failure and macroscopic rupture.7 The gels undergo multiple fracture events which occur at the weakest points. This process is responsible for the inherent brittleness of conventional gels. The inherent brittleness of conventional gels is enhanced when they are swollen because the elastically effective chains are already extended prior to any further strain being applied.The key general difference between physical gels and chemical gels is reversibility; i.e., the time period over which the crosslink disassembles compared to the experimental time period. For physical gels these two times are comparable. For chemical gels the former is orders of magnitude larger than the latter. Conventional chemical gels are well known and the science governing their properties is mature. However, there is an emerging area of dynamic hydrogels, especially in the biomaterial context. Burdick et al. highlighted that the dynamic complexity of hydrogels is an emerging new concept whereby the hydrogel's structure and properties should evolve with time in response to user-defined triggers or cellular behaviour.1
The bond dissociation energy for a C–C covalent bond is ca. 347 kJ mol−1 (140 kT (ref. 12)). The interactions that underpin the different types of physical gels available2 are ionic interactions, hydrogen bonding (8–35 kJ mol−1 (ref. 13)) and London dispersion type hydrophobic interactions (4–8 kJ mol−1 (ref. 13)). Another indirect method for physical gel formation involves crystallisation of gels.14 Because physical gels involve reversible crosslinking, they have the ability to reform and consequently exhibit self-healing. This has been considered in detail by Sun et al.,12 where the effect of bond strength on self-healing was assessed. The attractive interactions are additive and this is why physical gels can be readily prepared using cooperative ionic, hydrogen bonding or London dispersion interactions.
Chemical gels can also approach physical gels in their ability to self-heal if they contain covalent bonds that are not permanent. Dynamic covalent bonds8,15 are those that have lower than usual bond dissociation energies or bonds that can be reversibly cleaved using a small change in environmental conditions. An example is RS–SR where the bond can be reversibly cleaved readily by reduction, thiol–disulfide exchange16 or light. It is through the use of dynamic covalent bonds (for chemical gels) or cooperative attractive interactions (for physical gels) that the intriguing implications of personalised implants that can be encompassed by the dynamic hydrogel concept may be realised.
To overcome the inherent brittleness of conventional gels resulting from polydisperse Mc values, a hydrogel was created using pairs of macromonomers with four pegylated arms bearing mutually reactive end functional groups. The arms were of equal length for each macromonomer. These tetra-PEG gels had much lower polydisperse Mc values and were assumed to occupy a tetrahedral geometry17 (see Fig. 1). For these gels the local and global νe values were much closer. This resulted in a more uniform stress distribution and cooperative response, which distributed the stress over a much larger number of elastic chains. The result was increased yield strains at break and, hence, improved ductility. This was elegantly demonstrated with tetra-PEG hydrogels prepared using PEGs with different PEG lengths in the arms.18 Those gels had a controlled polydisperse elastically effective chain length distribution and were less ductile than more uniform tetra-PEG gels.
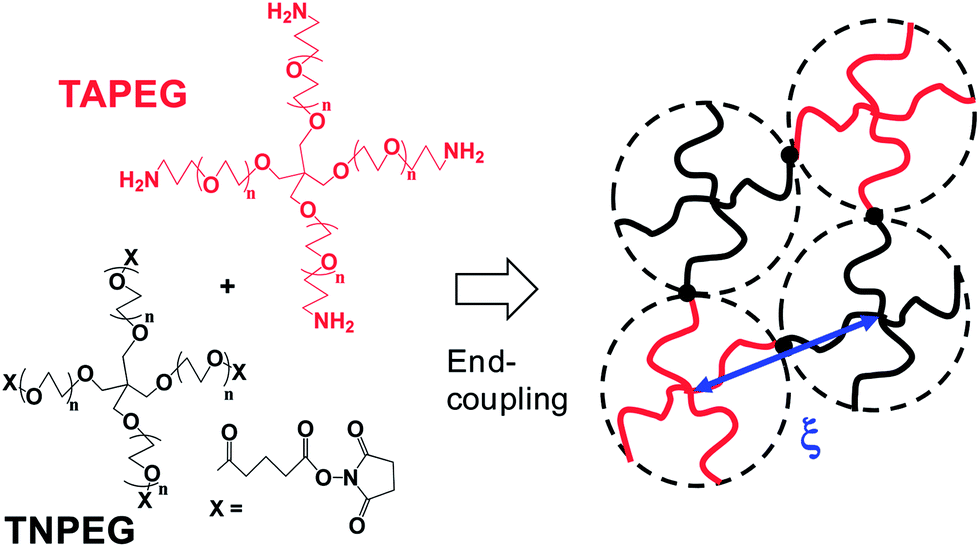 | ||
| Fig. 1 Preparation and structure of tetra-PEG gels. TAPEG and TNPEG are tetra-amine terminated PEG and tetra-NHS-glutarate terminated PEG, respectively. The figure is adapted from ref. 7 and 17. A tetra-PEG gel was formed with a diamond-like architecture due to end-coupling between complementary end groups. | ||
Inter-penetrating and semi-interpenetrating networks
IPNs are gels comprised of two polymers for which both have crosslinked structures19 but are not joined together. The polymers that comprise IPNs have orthogonal crosslinking. An example of an IPN is poly(N-isopropylacrylamide) (PNP)–chitosan where each network was independently crosslinked using orthogonal chemistry.19 An alternative approach for IPN formation is to prepare the first crosslinked gel (e.g., poly(dimethylacrylamide), PDMAAm) and then prepare the second gel (e.g., crosslinked poly(acrylic) acid, PAA) within the first.20 In this interesting work the polymers formed inter-polymer complexes (due to electrostatic attraction). This approach was also used for PNP/PMAA IPNs (PMAA is poly(methylacrylic acid)).21 In that work it was shown that the properties of each network were maintained within the IPN.Semi-IPNs are gels consisting of two polymers where one of them is linear and not crosslinked. The polymer network and linear polymer exist independently and are not covalently connected within the gel. Typically, a hydrogel is prepared from a solution of monomer and crosslinker (initiator and accelerator) in the presence of a linear polymer. Lee et al. investigated a semi-IPN of PNP and chitosan.19 Their gels showed both temperature and pH-responsive behaviours. In the case of semi-IPNs the value for Mc(ave) will originate only from the network. PAAm/PNP semi-IPNs have also been reported where the PAAm (poly(acrylamide)) network was crosslinked and PNP was linear. Those semi-IPNs showed enhanced elasticities compared to the control PAAm hydrogels.22 PNP/PAA semi-IPNs have also been reported.23 Although the volume change of PNP/PAA was similar to that of a control PNP hydrogel,23 the injectability and transparency of the systems was altered.
Double network hydrogels
Gong established gels with outstanding toughness that are termed double network (DN) gels.24 They were considered as a new class of hydrogels and contained a neutral crosslinked polymer (second network) which contained a relatively large Mc(ave) value and was formed within a crosslinked polyelectrolyte (first network) with a much lower Mc(ave) value. The optimised DN hydrogel structures contained about 90% water, had an elastic modulus of 0.1–1.0 MPa, failure compressive strengths of 20–60 MPa and high toughness values (100–1000 J m−2). However, a consequence of the mechanism responsible for these impressive mechanical properties of DN hydrogels is that they have negligible fatigue resistance.Gong identified key requirements for DN network formation.3 The first network must be rigid and brittle (e.g., polyelectrolyte); whilst the second network must be soft and ductile (e.g., a non-ionic polymer). The molar concentration of monomer for the second network should be 20–30 times that for the first network. The first network should be relatively highly crosslinked compared to the second, i.e., νe(1st network) ≫ νe(2nd network).
Fig. 2 shows an example of the extraordinary toughness of DN hydrogels.3 The toughness of a material can be measured from the area under the stress–strain curve. DN hydrogels have exceptionally high toughness.3 Close examination of elongation of DN gels (and comparison of the mechanical properties with the parent first network) has led to the conclusion that the first network fragments under strain, which results in energy dissipation.3 After this process has been completed it is the second, more lightly crosslinked, ductile network that allows the gel to deform to high strains without macroscopic failure. This mechanism is supported by the large hysteresis observed in stress–strain curves for DN hydrogels.25
 | ||
| Fig. 2 Image of a PAMPS/PAAm DN gel. Image taken from ref. 3. | ||
Fig. 3 depicts the mechanism that is proposed to be operative for DN gels. As mentioned above, the molar concentration of the second (neutral) network should be much higher than that of the first (polyelectrolyte) network for a DN gel. The PAMPS network fractured to clusters when the stress exceeded a critical value. The PAMPS clusters that were produced as a result of this process acted as sliding crosslinkers for the PAAm network. As a consequence, the DN gel became soft after the process and this was associated with necking.
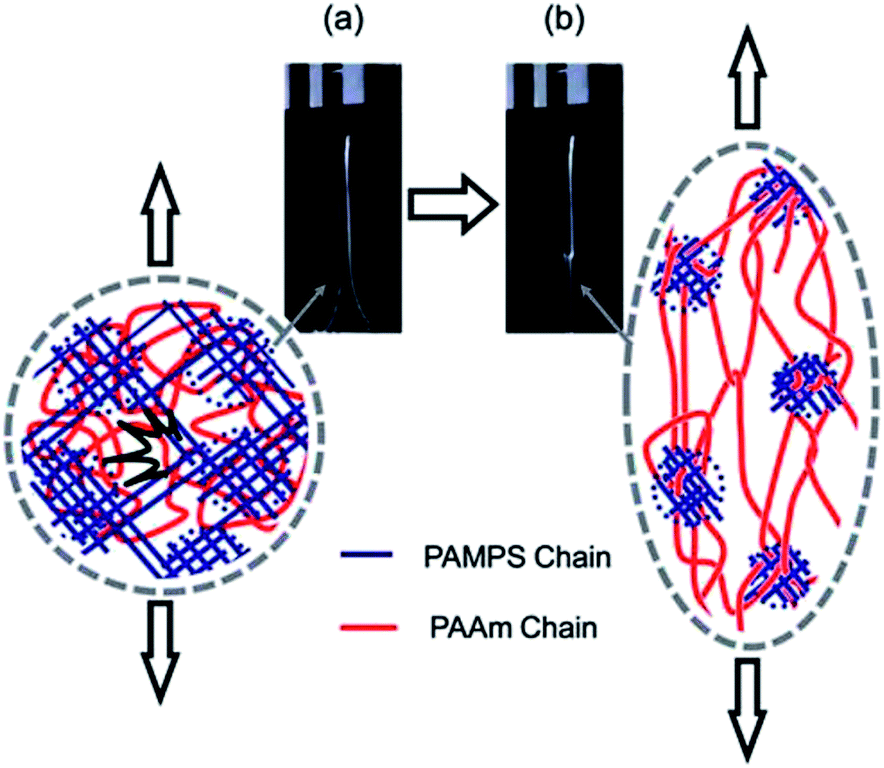 | ||
| Fig. 3 Depiction of the network structure of a DN gel before (a) and after (b) necking. PAMPS and PAAm are poly(2-acrylamido-2-methyl-1-propanesulfonic acid) and poly(acrylamide), respectively. Image taken from ref. 3. | ||
An important clarification to the mechanism of mechanical property improvement for DN hydrogels was reported by Nakajima et al.26 They examined hydrogels prepared using the conventional DN method and also those where great care was taken to prevent survival of pendant vinyl groups from the first network. DN hydrogels prepared by the conventional method were found to be connected through the reaction of vinyl groups from the second network via residual pendant vinyl groups on the first network. This provided a source of non-independent crosslinking for the second network. The stress applied to the DN network was transferred from the first network to the second network via the interconnected structure. They also prepared “truly independent” networks and found that toughening could occur provided that the second network contained some added crosslinks. It would appear to the present authors that the truly independent DN gels from that work are a subset of IPNs. This is because those networks were independent. Accordingly, each part of the gel should retain its own intrinsic character. It is further suggested that the connected DN hydrogels (conventional DN hydrogels) are equivalent to a hydrogel reinforced by a second network. The DN concept has been applied to a hydrogel based on the biopolymer chondroitin sulfate. That study supports the view that a brittle/ductile combination of networks is key to obtaining the DN effect.27
Nanocomposite hydrogels
In 2002 Haraguchi and Takehisa28 introduced nanocomposite (NC) hydrogels. Examples of syntheses involved NP28 or dimethylacrylamide,29N,N,N′,N′-tetramethylethylenediamine (TEMED), ammonium persulfate (APS) and laponite. Interestingly, no crosslinker was added. Their NC hydrogels had low turbidity and remarkably high ductilities (yield strains > 1000%). Thin cylinders could be stretched and knots tied in them without macroscopic fracture. Laponite is a synthetic hectorite clay with a diameter and thickness of about 20 and 1 nm, respectively.30 The platelets were exfoliated and adsorbed APS. The platelets acted as crosslinking points and this was in part attributed to the presence of TEMED.28 The NC hydrogels were termed rubbery polymeric hydrogels4 because they showed extraordinarily large and reversible extensions. This enabled remarkably large scale samples to be prepared that were transparent (Fig. 4). | ||
| Fig. 4 NC hydrogels of various shapes: (a) shows a thin film (200 μm thick), (b) a sheet (30 mm thick), (c) a hollow tube, and (d) a regular array of pillars. Images are reproduced from ref. 31 with kind permission from Springer Science and Business Media. | ||
The reason the NC hydrogels had superior ductility is that νe was greatly decreased compared to conventional hydrogels and was also spatially controlled. Laponite acted as a multifunctional crosslinking agent. The resulting flexible chains adopted nearly random conformations between the laponite plates. The authors claimed to have established independent control over νe and Mc(ave), whereas these two parameters are normally inversely related. The key factors that favoured success of the NC hydrogels (low turbidity and high ductility) were the small size and large surface area of laponite and strong interaction between the initiator and laponite.
A range of other polymers, silicates and other particles have been combined to form NC hydrogels and these were reviewed earlier.32,33 Haraguchi's NC hydrogels are unequalled in terms of their high, reversible, strain and low turbidity. However, an obstacle for their potential biomaterial application is that they have to be prepared in situ using free-radical polymerisation of water-soluble monomers, which may damage cells due to monomer–cell interactions. This potential obstacle also applies to many conventional hydrogels as well as IPNs, semi-IPNs and DN hydrogels.
Hydrogels containing microgels
A different type of hydrogel involves those containing microgel particles. Microgel particles are crosslinked polymer colloids that swell in a good solvent or when the pH approaches the pKa of the polymer subchains if these comprise polyacids or polybases.34 The microgels introduce additional length scales to the structure of the hydrogel: (i) the size of the microgel particle, (ii) the ξ value of the microgel network, and (iii) the length of the dangling chains at the microgel periphery. While the size of the entire particle introduces a large length scale that typically is not present in gels, the other two factors ((ii) and (iii)) are generally comparable to the respective values present in gels. The structural characteristics of the particle periphery are especially relevant for very small swellable particles (i.e. nanogels) because of the higher surface area to volume ratio (cf. microgels). The crosslink density and length of dangling chains affect any inter-penetration of (and entanglements between) microgels and polymer chains that surrounding them. Small angle neutron scattering (SANS) provides access to these length scales and the gel structure, especially as contrast variation is possible by selective deuteration of one component.35,36Soft nanocomposite hydrogels have recently been reported that consist of thermoresponsive PNP based microgels entrapped or covalently linked to a non-thermally responsive carbohydrate-based hydrogel.37 Those systems were investigated from the viewpoint of controlling drug release kinetics. Bencherif38 studied hyaluronic acid–GMA (HA–GMA) hydrogels containing covalently incorporated EO-based microgels prepared by inverse emulsion polymerisation. They used redox cleavable crosslinkers within microgels to enable reductant-triggered release of additives. The microgels were present at low concentrations and did not affect the hydrogel composite's mechanical properties.
Jha et al.39 also used HA–GMA and in this case prepared GMA-functionalised HA microgels. They prepared hydrogels containing microgel particles covalently linked to the hydrogel matrix. Their data show that the covalently linked microgel particles did not significantly increase the modulus of the hydrogel composites compared to the pure hydrogel. Jha et al. and Bencheriff et al. termed their systems nanostructured hydrogels.
Jia et al.40 used the complementary amine/aldehyde reactions to prepare HA-based microgels and then linked residual functional groups on the surface together via additional functionalised HA. Their microgel particles were covalently linked to the hydrogel matrix. Their systems were studied in the context of vocal fold regeneration. They termed their systems doubly crosslinked networks because there were two levels of crosslinking: (1) the intra-microgel crosslinking and (2) secondary network inter-connecting the microgels. The mechanical properties (e.g., elasticity) were tuneable using the composite hydrogel composition.
For two of the systems above38,39 there were locally distinct νe values (intra and inter-microgel) and these were tuneable. However, for these three systems it was mostly the case that a mechanical property benefit did not occur. It is possibly because there was not always a major difference in the νe values for the microgel and hydrogel components.
Meid et al.41 studied composite hydrogels containing microgels. In that study the microgels were not covalently linked to the hydrogel phase and were well dispersed within that phase (Fig. 5). They reported a modulus increase at high temperature (>VPTT) but not at low temperature (<VPTT). At higher temperature the microgel particles behaved as hard sphere fillers.
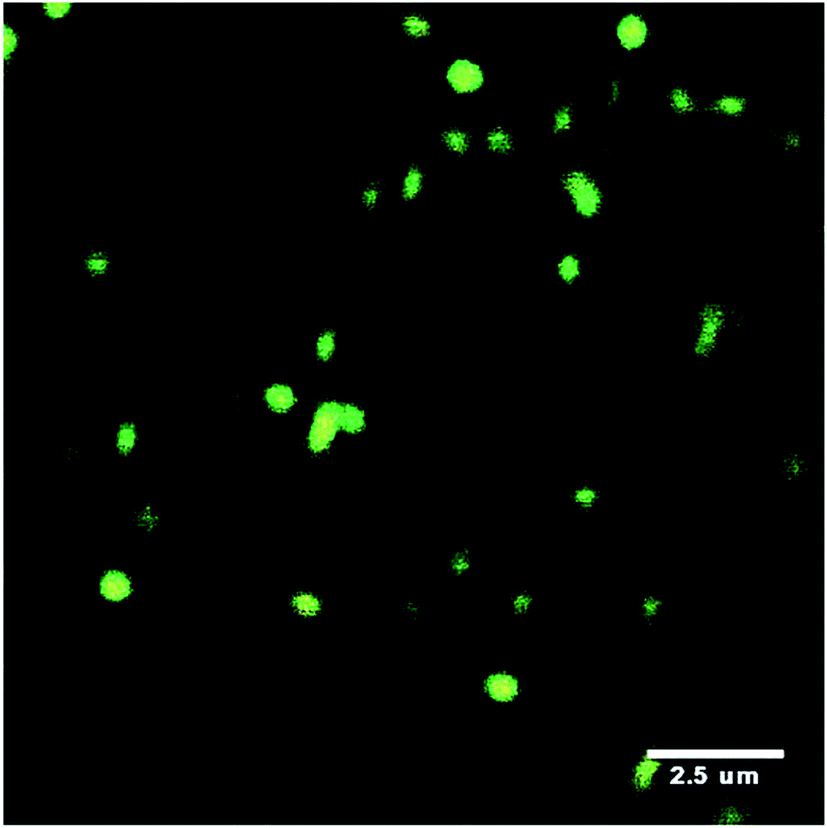 | ||
| Fig. 5 Confocal scanning microscopy image of labelled PNP microgels embedded within a hydrogel matrix. Image taken from ref. 41. | ||
In a different approach Supasuteekul et al.42 reported N,N′-methylenebis(acrylamide) (MBAAm) hydrogels containing GMA-functionalised PEA/MAA microgel particles. The difference between this work and that discussed above is that the hydrogel was formed within a physical gel of microgel particles. The microgel particles overlapped with each other. The hydrogel composites had much higher modulus values compared to the parent hydrogels. This differs from the above examples and probably arose from (a) the relatively high concentration of microgel particles used and (b) that the MBAAm phase inter-penetrated the microgel phase. This system had two inter-penetrating networks (one from microgel and one from MBAAm). If residual vinyl groups were present within the microgels then inter-network crosslinking also occurred.
Doubly crosslinked microgels and composites
Hu et al. were the first to construct materials from inter-linked microgels.43 Their materials had interesting photonic properties. Later, Cho et al.44 constructed hydrogels from aggregated PNP microgels. The aggregated morphology persisted within the hydrogels and this resulted in accelerated volume swelling/collapse kinetics. In 2011 it was shown at Manchester that hydrogels could be prepared by crosslinking concentrated microgel glasses using free-radical chemistry.45 The microgel particles were GMA-functionalised and these groups were sufficiently close in the swollen state to enable formation of a permanent gel. Importantly, there is no additional monomer or hydrogel required for DX microgels. It is the inter-linking of neighbouring microgels in the physical gel state that produces a hydrogel. Because a second round of crosslinking was required to form the gels they were termed doubly crosslinked microgels.45 These macroscopic hydrogels are modular and constructed from pre-formed colloidal sized gel particles. DX microgels have particular advantages from the viewpoint of using the fluid-to-gel transition to give an injectable gel for load support of degenerated soft tissue and this has been demonstrated.46 From the biomaterials perspective they have the advantage of enabling formation of a hydrogel in vivo from systems that are mostly pre-formed. This minimises the chemistry that must be done in the body and avoids the use of conventional monomers. DX microgels also have good mechanical properties and transparency (Fig. 6).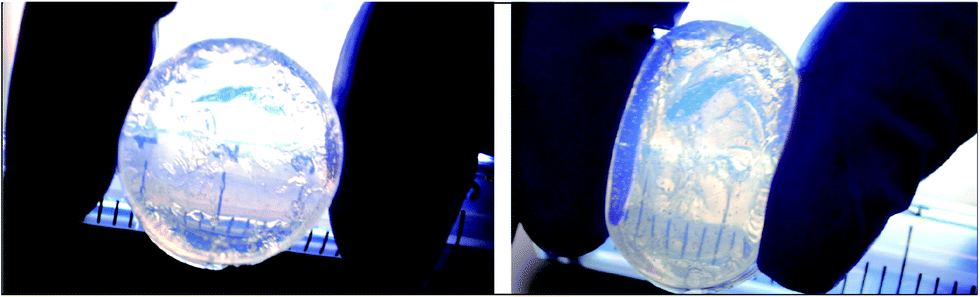 | ||
| Fig. 6 Digital photographs of a PMMA-MAA-EGD DX microgel before and after compression. | ||
The DX microgels have also been swollen with a non-ionic monomer which was subsequently crosslinked (lightly) to form DN DX microgels.47 These new types of DN hydrogels showed improved elasticity and ductility. Recently, DX microgels have been prepared using cationic poly(vinylamine) microgels and those systems were shown to be injectable.48 That work showed that the DX microgel formation concept was generally applicable. The requirements being vinyl functionalised microgel particles and inter-penetration (or contact) of the peripheries of neighbouring particles in the physically gelled state prior to free-radical covalent crosslinking. It is noted that definitive proof for inter-penetration of microgels has not yet been reported in the context of DX microgels. An alternative interpretation is that inter-linking occurs between the peripheral pendant vinyl groups of neighbouring microgel particles. This aspect should become clear with further research in this area.
DX microgels can also be considered as nanostructured hydrogels. However, it may be that the microgels retain their initial identities within the hydrogels. This was most apparent in the DX PVAM microgels.49 For DX microgels there are two νe regions. The first is within the core of the microgels and this should be the same as the “as prepared” value. The second is in the region that inter-connects the microgels and this value may be lower or higher than the former νe value depending on the GMA functionalisation content in the peripheries.
Hydrogel terminology and structures: is a systematic approach warranted?
When the gel literature is reviewed it is clear that there are a wide range of terms used to describe different types of systems. A question we considered was whether it is sensible to propose a common naming terminology for all the gels? One problem for that approach is that the gel complexity has become such that a systematic terminology soon becomes long and unwieldy. For example NC hydrogels could be considered as covalently-inter linked clay particle gels. DN hydrogels could be considered as covalently-inter linked IPNs. Topological details that in principle are altered by the presence of few connections between two components are often very difficult to be detected or excluded by experimental techniques. Thus, it would seem that a pragmatic approach of using the terms already in common usage should prevail. Furthermore, it is often not straightforward to experimentally distinguish between the influences of network structure and chain interactions (e.g., electrostatic or H-bonding). It is suggested that workers who believe they have produced a new type of gel should closely consider the similarities and differences of their gels to those shown in Fig. 7. The field would benefit if they would then start from the existing terms and decide whether or not their gel is truly novel and whether a new term is required.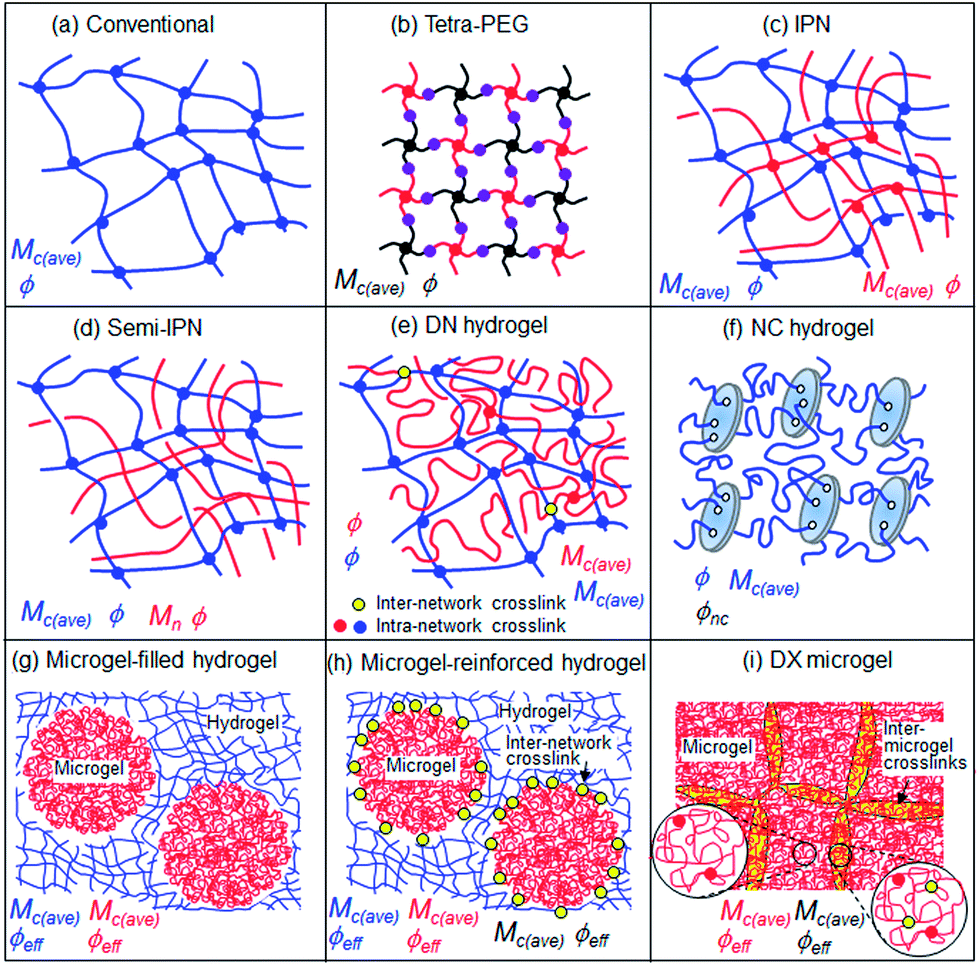 | ||
| Fig. 7 Depiction of the structures of well-studied hydrogels. The important compositional and structural parameters that control the properties are shown. | ||
Fig. 7(f)–(i) illustrate the increased complexity of hydrogels when they contain colloidal scale additives. The hydrogels containing microgel particles are of particular interest because they contain a pre-formed network. In the case where there is inter-network crosslinking (Fig. 7(h)) then the value for Mc(ave) should decrease at the interface. Three different local Mc(ave) values are expected which means the mechanical properties (elasticity and ductility) should vary greatly across the hydrogel at the colloidal scale. In the case of the DX microgels (Fig. 7(i)) there is a region of inter-microgel crosslinking which corresponds to the overlap region near to the interface. In this region there should be a lower Mc(ave) value than in the microgel interior and hence the DX microgel mechanical properties are believed to be affected by the effective volume fraction of this phase as well as that not directly involved in inter-linking (i.e., the interior of the particles). The differences in local Mc(ave) values should also affect the local mobility of dissolved molecules inside DX microgels.50 The latter property is relevant for many applications, e.g., in drug release and sensing.
Conclusions and future perspective
This review has considered a representative selection of new gels that have been reported recently. There is clearly an ongoing trend towards gel design with increased structural complexity and this has resulted in much improved mechanical properties. Although it may be difficult to imagine gels becoming more structurally complex than some of those depicted in Fig. 7 this is certain to occur. Future hydrogels are likely to contain increasingly well-controlled morphologies, with a move toward local compositional gradients. These could also include well-controlled compartmentalised hydrogels with length scales with controlled architectures over the nanometre and micrometre range. Fine control over the assembly of these structures would inevitably lead to complex hydrogels for biomaterial applications that mimic the morphologies of architecturally complex load-bearing soft tissue, such as articular cartilage. However, much is yet to be done for this goal. For the gels considered here, the ones with the best mechanical properties (NC hydrogels and DN hydrogels) had preparation methods that were not well suited for in vivo formation via minimally invasive injection. It is clear that a major part of hydrogel research for years to come will be driven by the potential practical application of structurally complex hydrogels for regenerative medicine and this will benefit greatly from minimally invasive approaches that cause assembly of user-defined architectures. Accordingly, it can be suggested that more emphasis be placed on dispersions of pre-formed gel particles that can be inter-linked in vivo with minimal chemistry.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft for support within SFB 985 and also the EPSRC for funding.References
- J. A. Burdick and W. L. Murphy, Nat. Commun., 2012, 3, 1269 CrossRef PubMed.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2012, 64, 223 CrossRef PubMed.
- J. P. Gong, Soft Matter, 2010, 6, 2583 RSC.
- K. Haraguchi, T. Takehisa and S. Fan, Macromolecules, 2002, 35, 10162 CrossRef CAS.
- N. Annabi, A. Tamayol, J. A. Uquillas, M. Akbari, L. E. Bertassoni, C. Cha, G. Camci-Unal, M. R. Dokmeci, N. A. Peppas and A. Khademhosseini, Adv. Mater., 2014, 26, 85 CrossRef CAS PubMed.
- J. Thiele, Y. Ma, S. M. C. Bruekers, S. Ma and W. T. S. Huck, Adv. Mater., 2014, 26, 125 CrossRef CAS PubMed.
- T. Matsunaga, T. Sakai, Y. Akagi, U.-I. Chung and M. Shibayama, Macromolecules, 2009, 42, 1344 CrossRef CAS.
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jian and Y. Chen, ACS Macro Lett., 2012, 1, 275 CrossRef CAS.
- P. N. Pusey and W. van Megan, Nature, 1986, 320, 340 CrossRef CAS.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345 CrossRef CAS PubMed.
- U. Gulyuz and O. Okay, Soft Matter, 2013, 9, 10287 RSC.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932 CrossRef CAS PubMed.
- D. E. Packham, Handbook of Adhesion, John Wiley and Sons, 2nd edn, 2005 Search PubMed.
- J. L. Holloway, A. M. Lowman and G. R. Palmese, Soft Matter, 2013, 9, 826 RSC.
- J. A. Syrett, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 978 RSC.
- J. F. Quinn, A. P. R. Johnston, G. K. Such, A. N. Zeliken and F. Caruso, Chem. Soc. Rev., 2007, 36, 707 RSC.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U.-I. Chung, Macromolecules, 2008, 41, 5379 CrossRef CAS.
- S. Kondo, H. Sakurai, U.-I. Chung and T. Sakai, Macromolecules, 2013, 46, 7027 CrossRef CAS.
- W.-F. Lee and Y.-J. Chen, J. Appl. Polym. Sci., 2001, 82, 2487 CrossRef CAS PubMed.
- T. Aoki, M. Kawashima, H. Katono, K. Sanui, N. Ogata, T. Okano and Y. Sakurai, Macromolecules, 1994, 27, 947 CrossRef CAS.
- J. Zhang and N. A. Peppas, Macromolecules, 2000, 33, 102 CrossRef CAS.
- E. C. Muniz and G. Geuskens, Macromolecules, 2001, 34, 4480 CrossRef CAS.
- R. A. Stile and K. E. Healy, Biomacromolecules, 2002, 3, 591 CrossRef CAS PubMed.
- J. P. Gong, Y. Kastuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155 CrossRef CAS PubMed.
- R. E. Webber, C. Creton, H. R. Brown and J. P. Gong, Macromolecules, 2007, 40, 2919 CrossRef CAS.
- T. Nakajima, H. Furukawa, Y. Tanaka, T. Kurokawa, Y. Osada and J. P. Gong, Macromolecules, 2009, 42, 2184 CrossRef CAS.
- T. Suekama, J. Hu, T. Kurokawa, J. P. Gong and S. H. Gehrke, ACS Macro Lett., 2013, 2, 137 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120 CrossRef CAS.
- K. Haraguchi, R. Farnworth, A. Ohbayashi and T. Takehisa, Macromolecules, 2003, 36, 5732 CrossRef CAS.
- E. Balnois, S. Durand-Vidal and P. Levitz, Langmuir, 2003, 19, 6633 CrossRef CAS.
- K. Haraguchi, Colloid Polym. Sci., 2011, 289, 455 CAS.
- C. W. Peak, J. J. Wilker and G. Schmidt, Colloid Polym. Sci., 2013, 291, 2031 CAS.
- P. Schexnailder and G. Schmidt, Colloid Polym. Sci., 2009, 287, 1 CAS.
- B. R. Saunders, N. Laajam, E. Daly, S. Teow, X. Hu and R. Stepto, Adv. Colloid Interface Sci., 2009, 147, 251–262 CrossRef PubMed.
- J. Meid, T. Friedrich, B. Tieke, P. Lindner and W. Richtering, Phys. Chem. Chem. Phys., 2011, 13, 3039 RSC.
- J. Musch, S. Schneider, P. Lindner and W. Richtering, J. Phys. Chem. B, 2008, 112, 6309 CrossRef CAS PubMed.
- D. Sivakumaran, D. Maitland, T. Oszustowicz and T. Hoare, J. Colloid Interface Sci., 2013, 392, 422 CrossRef CAS PubMed.
- S. A. Bencherif, D. J. Siegwart, A. Srinsvasan, F. Horkay, J. O. Hollinger, N. R. Washburn and K. Matyjaszewski, Biomaterials, 2009, 30, 5270 CrossRef CAS PubMed.
- A. K. Jha, S. M. Manisha, M. C. Farach-Carson, R. L. Duncan and X. Jia, Soft Matter, 2010, 6, 5045 RSC.
- X. Jia, Y. Yeo, R. J. Clifton, T. Jiao, D. S. Kohane, J. B. Kobler, S. M. Zeitels and R. Langer, Biomacromolecules, 2006, 7, 3336 CrossRef CAS PubMed.
- J. Meid, F. Dierkes, J. Cui, R. Messing, A. J. Crosby, A. Schmidt and W. Richtering, Soft Matter, 2012, 8, 4254 RSC.
- C. Supasuteekul, A. H. Milani, J. M. Saunders, S. Lally, T. Freemont and B. R. Saunders, Soft Matter, 2012, 8, 7234 RSC.
- Z. Hu, X. Lu and C. Wang, Adv. Mater., 2000, 12, 1173–1176 CrossRef CAS.
- E. C. Cho, J.-Y. Kim, A. Fernandez-Barbero and D. A. Weitz, Nano Lett., 2008, 8, 168 CrossRef CAS PubMed.
- R. Liu, A. H. Milani, T. J. Freemont and B. R. Saunders, Soft Matter, 2011, 7, 4696 RSC.
- A. H. Milani, T. J. Freemont, J. A. Hoyland, D. J. Adlam and B. R. Saunders, Biomacromolecules, 2012, 13, 2793 CrossRef CAS PubMed.
- T. Lane, J. L. Holloway, A. H. Milani, J. M. Saunders, A. J. Freemont and B. R. Saunders, Soft Matter, 2013, 9, 7934 RSC.
- S. Thaiboonrod, A. H. Milani and B. R. Saunders, J. Mater. Chem. B, 2014, 2, 110 RSC.
- S. Thaiboonrod, F. Cellesi, R. V. Ulijn and B. R. Saunders, Langmuir, 2012, 28, 5227 CrossRef CAS PubMed.
- S. Lehmann, S. Seiffert and W. Richtering, J. Am. Chem. Soc., 2012, 134, 15963 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |