 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of polymerisation conditions on the properties of polymer/clay nanocomposite hydrogels
Rinat
Nigmatullin
*,
Martin
Bencsik
and
Fengge
Gao
Nottingham Trent University, School of Science and Technology, Clifton Campus, Nottingham, NG 11 8NS, UK. E-mail: Rinat.nigmatullin@ntu.ac.uk
First published on 23rd December 2013
Abstract
Free-radical polymerisation of acrylamide derivatives in the presence of exfoliated clay platelets has recently emerged as a new technique for the synthesis of strong and tough nanocomposite hydrogels (NCHs) with a unique hybrid organic/inorganic network structure. The central intent of many research studies in the field of NCHs conducted so far was to change hydrogel properties with the introduction of various clays and variation of the clay content. Here, we demonstrate that the properties of NCHs significantly vary depending on initiating conditions used for hydrogel synthesis via in situ polymerisation in solutions of high monomer concentrations (above 1 mol L−1). A unique, complementary combination of real-time dynamic rheology and pulsed NMR/MRI has been used to study the influence of the composition of a redox initiating system on the gelation process and hydrogel properties. The molar ratio of the persulphate initiator to tertiary amine activator affects the polymerisation kinetics, morphology and mechanical properties of the hydrogels. We further show that activator-dominated systems tend to produce hydrogels with higher storage modulus and lower water intake. This trend is attributed to the increase in the cross-linking degree. From the analysis of the water state in NCH and hydrogels prepared with and without an organic cross-linker, it was concluded that clay platelets did not form covalent bonds with polymer molecules but contributed to the formation of a physical network. There is evidence of self-crosslinking of polymer chains during acrylamide polymerisation at high monomer concentration. The composition of the initiating system influences the number of formed self-crosslinks.
1 Introduction
Since the pioneering work1 by the Haraguchi group a decade ago, nanocomposite hydrogels (NCHs) have generated significant interest. This was mostly driven by a pursuit of hydrogels with improved mechanical properties. Although clay minerals and hydrophilic polymers can be processed in various different ways, Haraguchi et al.2,3 showed that hydrogels of unique structure and properties can be synthesised by the in situ polymerisation of hydrophilic monomers in a suspension of an exfoliated layered silicate. The group has proposed a structural model of NCH, which is generally accepted by the researchers in the field. It is widely agreed that clay platelets act as multifunctional cross-linking surfaces. Uniform distribution of exfoliated nanoplatelets leads to the formation of a homogeneous hybrid organic/inorganic network with clay surfaces connected to polymer chains. Such a structure gives rise to remarkably strong, tough, flexible and stretchable hydrogels. NCH materials offer a facile synthesis route compared with other types of hydrogels with improved mechanical properties such as slide-ring,4 double-network5 and tetra-PEG hydrogels.6A large number of various layered and tubular clays have been investigated for the preparation of NCH.2,7,8 Clay type has a notable effect on the mechanical properties of NCH. As anticipated, both the strength and modulus of NCH improve with the increase in the clay content. At the same time, clay concentration has little effect on the elongation at break.2 Thus most of the endeavours in the development of NCH were sought to explore different types of nanofillers and optimise their content. However, the influence of molecular characteristics and morphology of the polymer matrix on NCH properties has received little attention.
It is well established that polymer molecular weight increases with the increase of monomer concentration for the polymerisation in solutions.9,10 Moreover, significant variations in hydrogel morphology have been observed for conventional organically cross-linked hydrogels by changing initiating conditions.11 Self-crosslinking phenomenon has also been reported for the polymerisation of acrylamide derivatives in solutions with high monomer concentrations.10,12–15 All these observations suggest that molecular characteristics and morphology of the polymer matrix in NCH are susceptible to the polymerisation conditions. As a consequence, the structure and properties of NCH would depend on polymerisation conditions. This assumption is supported by the results published in a recent paper on poly(dimethyl methacrylate) (PDMA) NCH, which were synthesised using two different initiators, potassium persulfate and azobisisobutryonitrile (AIBN).16 Tensile strength increased significantly and swellability was lower for the NCH prepared by the initiation with AIBN. Additionally interference in the polymerisation process by clay can be expected in NCH synthesis. For example, the inhibition of the polymerisation has been reported in the presence of clay minerals.17–19 Thus clay may affect the NCH morphology not only as a constituent of the hybrid network but also due to its influence on the polymerisation process.
Herein, the polymerisation of acrylamide has been systematically studied under various initiating conditions in order to understand the influence of molecular characteristics and morphology of the polymer matrix on the NCH structure and properties and to gain an insight into the role of nanoplatelets in NCH. A persulphate/tertiary amine redox initiating couple was selected for this study as the most frequently used system in the NCH synthesis. Initiating conditions were varied by changing the concentrations of the components in the redox couple and their molar ratios. A real-time monitoring of the polymerisation process was conducted by non-invasive methods, oscillatory rheology and 1H NMR spin–spin relaxation. The effect of initiating conditions was investigated for acrylamide polymerisation without using any cross-linker and in systems with either an organic cross-linker or layered silicate sodium as an inorganic cross-linker. In parallel the swelling of hydrogels in water was studied. The influence of initiating conditions on the cross-linking density and morphology of hydrogels is also discussed.
2 Experimental
2.1 Hydrogel synthesis
Hydrogels were synthesised by redox initiated aqueous polymerisation of acrylamide (AA) without addition of any cross-linking agent (polyacrylamide hydrogel (AAH)) and in the presence of clay (nanocomposite hydrogel (NCH)) and N,N′-methylenebis(acrylamide) (MBA) (organically cross-linked hydrogel (ORH)) as an inorganic and organic cross-linking agent respectively. A persulphate/tertiary amine redox couple was used as the initiator system, in which the polymerisable tertiary amine, N-[3-(dimethylamino)propyl]methacrylamide (DMAPAA), was an activator. AA, MBA, DMAPAA, and potassium persulphate of purity not less than 99% were purchased from Sigma-Aldrich and used as received.Generally NCHs are synthesised by polymerisation in the systems with high monomer content namely above 1.5 mol L−1.2 In order to enable comparison of different types of hydrogels, an AA concentration of 17.0 wt% (2.4 mol L−1) was used for all hydrogels prepared in this study. At such monomer concentration even AAH is a semi-solid material. Polymerisation mixtures were prepared by mixing three types of stock solutions: monomer solution (or monomer solution in clay suspension), initiator solution and activator solution. All stock solutions were prepared using Milli-Q water (Millipore, USA) as a solvent. For the rheological and MRI in situ monitoring experiments, all formulations were prepared by mixing 4 g, 1 g and 1 g of monomer, initiator and activator stock solutions respectively. The concentrations of initiator and DMAPAA in their respective stock solutions were varied in order to achieve the required concentrations in the final formulations. Two series of experiments were carried out in order to study the influence of the composition of the redox system on the kinetics of the polymerisation and hydrogel properties. In one series, the concentration of potassium persulphate was kept constant at 3 wt% with respect to the monomer, which corresponded to approximately 0.5 wt% (20 mM) content in the whole formulation. The concentration of activator was varied from 13 to 50 mM, which resulted in the range of molar initiator/activator ratio between 0.4 and 1.5. The second series was conducted with an activator concentration fixed at 25 mM whilst concentration of potassium persulphate was changed from 5 to 60 mM. In this series the molar initiator/activator ratio was varied between 0.2 and 2.4.
All NCHs contained 0.25 wt% of sodium montmorillonite, which was supplied by Southern Clay Products Inc. (Germany) under the trademark Cloisite® Na+. For the synthesis of NCH, the monomer stock solution was prepared using 0.375 wt% aqueous clay suspension as the monomer solvent. Before the use in preparation of monomer stock solution, a 100 ml portion of clay suspension was sonicated for 3 min using an S-4000 ultrasonic processor (Misonix, USA) equipped with a 12 mm horn by applying 85% of full power amplitude. ORH contained 1 wt% of MBA with respect to AA. For the synthesis of ORH, the monomer stock solution was prepared by dissolving the required amounts of AA and MBA.
Samples in a sheet form were prepared by polymerisation in a 10 cm × 10 cm glass mould with a 1.5 mm thick silicone spacer. The final formulation was prepared as described previously by mixing 4 times larger aliquots of the stock solutions. The mixture was injected into the mould and kept in an incubator at 25 °C for 24 hours to complete the polymerisation. These samples were used in the rheological assessment, MRI and water absorption capacity experiments.
2.2 Oscillatory rheology
Rheokinetic studies of the polymerisation process were carried out using a Bohlin C-VOR rheometer (Malvern Instruments, Malvern, UK) equipped with a Peltier temperature controller. For the in situ monitoring of the polymerisation process a cone-and-plate geometry was used. In this experimental set up, a stainless steel cone with a diameter of 40 mm and a cone angle of 4° was used as the top plate. The lower plate was the stainless steel surface of the Peltier plate. During the rheological experiments, the measuring system was covered with a humidity chamber to minimise water evaporation. After adding the required amount of activator stock solution to pre-mixed portions of monomer and initiator stock solutions, the final mixture was vigorously shaken for 30 seconds. Immediately after mixing, 1.55 ml of the polymerisation mixture was placed on the bottom plate. The cone plate was brought to a 150 μm gap between the two plates. The total duration of sample loading was approximately 40 seconds. The rheological measurements were performed in strain-controlled mode. An oscillation frequency of 1 Hz and a shear strain of 0.025 were applied in the rheokinetic study. The selected deformation targeted a linear regime of oscillatory deformation. The justification of this selection will be discussed in the next section. The temperature was maintained at 25 °C throughout the measurement. After reaching steady-state readings of the storage modulus, a strain-controlled frequency sweep was performed with the change of frequency between 0.1 and 100 Hz maintaining a shear strain of 0.025 over the whole frequency range. The linear viscoelastic region was determined for fully polymerised samples prepared in the sheet form. A 40 mm diameter parallel plate was used in such experiments. Oscillatory measurements were carried out in auto-gap mode controlled by normal force, which was set at 5 N. The amplitude sweep was conducted by changing the shear strain from 0.01 to 0.55.2.3 NMR imaging
2 ml polypropylene microcentrifuge tubes were used to hold the samples in the MRI experiments. For the in situ monitoring of the polymerisation process, the mixture of required amounts of all stock solutions was vigorously shaken for 30 s and then transferred into a centrifuge tube. The centrifuge tube was completely filled with the reaction mixture. A cylindrical sample holder, which could accommodate up to 7 tubes, was used to place the samples in the MRI chamber. For the characterisation of fully polymerised hydrogels, a piece of hydrogel with a size of approximately 2.5 × 2.5 cm was cut from the hydrogel sheet, rolled into cylindrical shape and placed into a centrifuge tube. The MRI system used in this study is a 2.35 T small animal (72 mm i.d.) Bruker BIOSPEC. MR images of relaxation time T2 were obtained with the MSME (Multi Slice Multi Echo) sequence with parameters tailored differently for monitoring the polymerisation process and characterisation of fully polymerised hydrogels. The following parameters were used in polymerisation experiments: echo time, TE = 7.763 ms, number of TE images = 24, repetition time, TR = 1.2 s, imaging time = 3 h 50 min, slice thickness = 2 mm, transverse spatial resolution = 547 × 547 μm2. While fully polymerised hydrogels were imaged with TE = 10.25 ms, number of TE images = 128, TR = 4 s, imaging time = 51 min, slice thickness = 4 mm, transverse spatial resolution = 273 × 365 μm2, number of averages = 4.All MR data processing was done with in-house Matlab® software. The pixels corresponding to a specific vial were automatically selected by using the Matlab® image processing toolbox. The averaged signal outside vials was subtracted from the data, and the averaged signal in a vial was plotted against echo time for further fitting of the monoexponential function for T2 value extraction. In some experiments, each pixel found in a specific vial was fitted so as to show the T2 distribution within a specific preparation.
2.4 Equilibrium water intake
Hydrogel pieces of 4 cm diameter (produced in rheokinetic experiments or cut from the hydrogel sheet) were placed in 100 ml of Milli-Q water and kept at ambient temperature. In the first month of the swelling experiment, water was replaced on a weekly basis. Swollen hydrogels were weighed with 1 month intervals. In total, 6 months were required for hydrogels to reach their equilibrium swelling. After this, the swollen hydrogels were removed from water, tapped with filter paper to remove excess of surface water and weighed to determine the mass of the fully swollen hydrogel (mS). After that, the hydrogel was dried in an oven at 110 °C until constant mass was reached in order to determine the mass of dry polymer (mD). The equilibrium water intake (QW) was calculated by using eqn (1):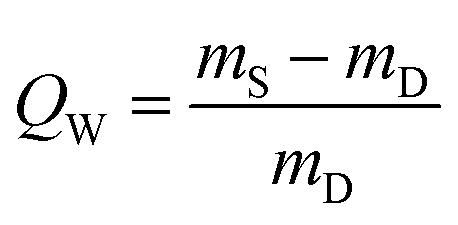 | (1) |
2.5 Tensile test
The tensile tests were performed using a single-column Zwicki Z0.5 testing machine (Zwick/Roell, Germany) under the conditions of a 5 mm min−1 loading rate. Hydrogel specimens of 10 mm width were cut from the samples polymerised in a glass mould. The sample thickness was 1.5 mm, and the sample gauge length was 30 mm.2.6 Differential scanning calorimetry
Differential scanning calorimetry (DSC) was used to monitor the melting of water crystals in frozen hydrogels. DSC thermograms were recorded using the Perkin-Elmer Pyris-Diamond instrument which was calibrated against ultrapure water. The hydrogel sample (about 20 mg) was sealed in a pierced aluminium pan and cooled to −40 °C at a scan speed of 5 °C min−1. After maintaining the sample at −40 °C for additional 5 minutes, endotherms were recorded in a temperature range between −40 and 20 °C at a heating scanning rate of 5 °C min−1 in a nitrogen atmosphere under a constant gas flow of 20 ml min−1.3 Results and discussion
3.1 Hydrogel linear viscoelastic response
Oscillatory shear rheology has become routinely used in both monitoring the gelation processes in hydrogel formation and the characterisation of physical and mechanical properties of hydrogels.20–22 In oscillatory shear rheology, material properties are probed by applying a sinusoidal shear deformation and measuring the generated stress response of the material. The rheological response of the material provides an insight into the morphological structure if the applied strains do not affect the structure and thus material deformations are reversible. With such sufficiently small deformations viscoelastic properties are independent of the amplitude of the applied strains exhibiting linear viscoelastic behaviour.During the hydrogelation a hydrogel precursor system undergoes a transformation from a liquid or viscoelastic fluid to a solid-like material. The least deformable hydrogels are obtained when polymerisation is complete. In order to ensure that the hydrogel structure remains intact during the rheological monitoring of the polymerisation process, the linear viscoelastic region was determined for completely polymerised hydrogels. If the linear viscoelastic region is defined as the strain region with shear storage modulus (G′) within 5% of its plateau value, according to Fig. 1, both AAH and NCH can tolerate significant deformations, up to 30%. Mechanical properties of hydrogels synthesised in this study are expected to vary considerably depending on composition and polymerisation conditions. Therefore in all rheological experiments a shear strain of 0.025 has been used. Such a small deformation will ensure linear viscoelastic response for all hydrogels prepared in the study.
 | ||
| Fig. 1 Normalized storage modulus (G′/G′max) as a function of strain amplitude (γ) for fully polymerised AAH and NCH. The inset shows the influence of strain amplitude on absolute values of the storage modulus. Concentrations of components of the redox couple used for hydrogel synthesis: Cinit = 6.5 mM, Cactiv = 16 mM. | ||
Both AAH and NCH are “softened” under larger deformations. For AAH, which is a binary polymer–solvent system, strain induced nonlinearity is attributed to polymer chain disentanglement with sufficiently large deformations. Although incorporating inorganic nanoplatelets into hydrogels reinforces the material as demonstrated by the higher G′ value in the inset chart of Fig. 1, the range of linear viscoelastic response is narrower for NCH compared with the corresponding AAH. Such behaviour is widely reported for particle reinforced rubbers and referred to as the Payne effect. Some recent studies23,24 on hydrogels containing nanoparticles also showed shear-induced decrease of storage modulus and narrowing of the linear viscoelastic region. Breakage of the filler network, filler deagglomeration and polymer chain debonding from the filler surface are considered as possible causes for such behaviour. Considering the low clay content in NCH studied here and possible exfoliation of clay platelets in the NCH matrix, the detachment of polymer chains from the surface of nanoplatelets is the most likely mechanism for narrowing the linear elastic region of NCH.
3.2 In situ monitoring of hydrogel formation during the polymerisation
Fig. 2 shows an example of temporal changes of the rheological parameters, G′ and G′′, together with the NMR relaxation time, T2, derived from rheological and MRI experiments, respectively, during AA polymerisation in aqueous solutions. Both methods of monitoring the polymerisation process clearly distinguish three stages in the hydrogel formation. In the early stage, there are little or very limited changes in those measured parameters. This period is followed by the sharp changes in both rheological properties and T2. Finally, all parameters converge slowly to their respective plateaus.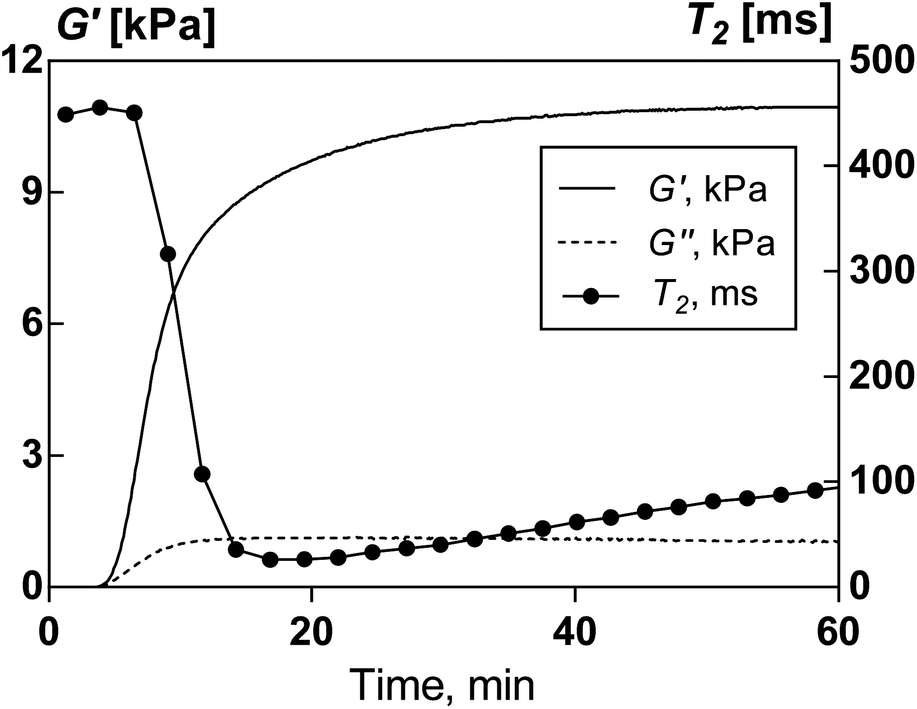 | ||
| Fig. 2 Examples of the time evolution of the storage (G′) and loss (G′′) modulus, measured in the rheokinetic studies, and T2 relaxation time, measured by 1H NMR spin–spin relaxation, during acrylamide polymerisation. | ||
Oscillatory rheology and 1H magnetic resonance explore fundamentally different material properties. Viscoelastic response reflects the material's ability to reversibly store the energy of deformation (G′) and irreversibly dissipate it (G′′), while the relaxation time T2 characterises the motional state of protons and is mostly attributed to the mobility of water. Despite fundamental differences, both viscoelastic properties and proton mobility are very sensitive to the material structure on all levels of structural organisation, i.e. molecular, supramolecular and morphological levels. As a result, structural changes during the polymerisation process lead to highly correlated patterns in the evolution of viscoelastic properties and proton relaxation time.
In the beginning of polymerisation, monomer solutions are typical Newtonian liquids with low viscosity and consequently characterised by very low G′ and G′′ values. The relaxation time is affected by the presence of dissolved or suspended substances in water (Table 1). Even at relatively low concentration (0.25 wt%), the presence of MMT causes a decrease in T2 values from 900 ms for pure water to 360 ms. AA solutions also show decreased T2 compared with pure water. The AA concentration in the systems investigated is relatively high; therefore, acrylamide contributes to the overall T2 measurements. The T2 decrease in AA solutions is a result of two independent factors: (i) intermolecular interactions between water and AA molecules and (ii) replacement of a significant fraction of water by large, less mobile AA molecules. The presence of both acrylamide and clay further decreases 1H relaxation time in mixtures before the polymerisation.
| Distilled water | 0.25 wt% clay suspension | AA solution | AA/clay suspension | |
|---|---|---|---|---|
| T 2, ms | 900 | 360 | 430 | 180 |
The initial small changes in G′, G′′ and T2 are attributed to the induction stage of the polymerisation process (Fig. 2). The induction period is usually associated with the presence of oxygen and other impurities competing with monomer molecules for free radicals. It is worth noting that T2 slightly increases during this period. Weakening of intermolecular interactions between water and AA was suggested as a cause of T2 increase at this stage.25 However, polymerisation is an exothermic process, which induces an increase in temperature of the reaction mixture. In the case of hydrogel formation, heat dissipation is hindered since stirring of the reaction mixture cannot be applied. In MRI experiments, 2 g of the polymerisation mixture was used. Separate experiments showed that the temperature of such an amount of the polymerisation mixture can reach up to 36 °C. Therefore the temperature increase in the initial stage of the reaction is likely to be responsible for the increase in T2.
The induction period is followed by a sharp increase in G′ and G′′ values and a reduction of T2. These changes correspond to sol–gel transition. G′ values swiftly overcome G′′, and the elastic modulus dominates the dissipative loss modulus during essentially the entire polymerisation process. This implies the formation of a solid-like polymer network. Linking AA monomers into macromolecules reduces their mobility and consequently the T2 signal originating from AA. Moreover, with hydrogel formation, water molecules become encaged in the polymer network and thereby less mobile. These two factors lead to the drastic decrease of T2 values, by one order of magnitude, during the sol–gel transformation.
Due to the depletion of monomer and its lowered mobility in gelled media, the reaction enters a slow kinetics stage, which is manifested as a region of slow changes of rheological parameters and T2 values before they reach a plateau, when the reaction is complete. The increase in T2 in the later stage of the polymerisation process appears counterintuitive. Such a pattern has been reported before for the formation of organic hydrogels25 and silica gels.26 Water mobility in porous networks depends on the porous structure with water molecules being more mobile in larger pores. It appears that during later stages of the polymerisation process the morphology of hydrogel undergoes some transformations accompanied by the formation of larger pores and probably the densification of polymer-rich domains. Such morphological maturation is generally detected by T2 changes even when viscoelastic properties are in a relatively steady-state.
The sigmoid pattern of the time course of G′ is convenient for fitting experimental data onto a modified three-parameter Hill equation:20,21
 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G′(θ) = G′max/2. The Hill's parameter n is related to the asymptotical slope of the curve at time θ.
:G′(θ) = G′max/2. The Hill's parameter n is related to the asymptotical slope of the curve at time θ.
3.3 Influence of initiating conditions on the kinetics of hydrogel formation
Redox-mediated polymerisation involves an initiator/activator redox couple. Therefore its kinetics has a complex response to the variations in the composition of the initiating systems, i.e. the overall concentrations of each component and their ratios. It was shown that for AA polymerisation in solutions of low and moderate concentrations (up to 1 mol L−1) the rate of polymerisation increases whilst the conversion degree decreases with the increase of the activator content.27 Moreover, the initiator/activator ratio affects the molecular weight of the formed polymer. Despite such findings and almost exclusive reliance of NCH synthesis on redox initiation, the influence of redox couple composition on NCH formation and ultimately NCH properties has not yet been studied.Fig. 3 shows the results of rheological monitoring of acrylamide polymerisation for a series of samples with different initiator/activator ratios at a fixed initiator concentration. The induction period shortens with the increase of the activator content, which suggests quicker polymerisation when the activator dominates the initiator. A similar situation occurred during the polymerisation of AA in the presence of clay.
 | ||
| Fig. 3 Effect of the composition of the redox initiation system on the evolution of storage modulus (G′) during acrylamide polymerisation in a series of syntheses with an initiator concentration of 20 mM. Initiator/activator ratio (r) increases in the direction of the arrow. r values: 0.6, 0.75, 1.0, 1.25, and 1.36. | ||
Further insight into the kinetics can be gained from the analysis of the parameters of the Hill equation. The sets of change of these parameters depending on the initiator/activator ratio are presented on Fig. 4. The results are complemented with the data on the induction period (tind), which was arbitrarily defined as time, when the storage modulus reached 3 Pa. With the increase of initiator/activator ratio, both θ and tind increase for AA polymerisation in the presence and absence of clay, which confirms faster polymerisation kinetics with the increase of relative activator concentration with respect to the initiator. At the same time, the limit values of storage modules, G′max, decrease with the increase of initiator/activator ratio. Thus the composition of the redox initiation systems affects not only the kinetics of polymerisation but also hydrogel properties. Assuming that storage modulus is dependent on molecular weight, a higher polymerisation rate for the activator-dominated redox couple led to polyacrylamide of higher molecular weight for polymerisation in the systems with high monomer content. Such a conclusion contradicts the results on polymerisation at moderate AA concentrations published by other researchers,27 which showed molecular weight decrease with the increase of activator content. This issue will be discussed in the next section.
 | ||
| Fig. 4 Influence of the composition of the redox initiation system on rheokinetic parameters: steady state storage modulus, G′max, (a); half-gelation time, θ, (b); Hills's coefficient, n, (c); induction period of the polymerisation, tind, (d) in the synthesis of AAH and NCH at the fixed initiator concentration of 20 mM. G′max, θ and n were obtained by fitting eqn (2) to storage modulus data and tind was determined as the time when the storage modulus reached 3 Pa. | ||
As can be seen from Fig. 4b and c, both θ and tind values decrease in the presence of clay. Thus the presence of clay has an accelerating effect on the dynamics of the storage modulus evolution. However, it is not possible to conclude whether clay affects the polymerisation process itself since the presence of clay changes the rheological properties of a system. In fact, the evolution of T2 has an opposite trend; the addition of clay led to slower progression of T2 values. As examples, Fig. 5 shows changes of T2 values at the initial stage of the polymerisation for compositions with small and large initiator/activator ratios. It can be seen that for both fast and slow kinetics, changes of T2 during the formation of NCH lag behind the polymerisation of corresponding mixtures without clay. Considering that acrylamide contributes to T2 measurements, the dynamics of T2 is more closely related to the kinetics of polymerisation than rheological measurements. Therefore it is more likely that clay inhibits the polymerisation process to some extent rather than promoting it. Inhibition of the polymerisation process in the presence of clay minerals has been reported previously and attributed to electron transfer from initiating or propagating radicals to the clay's Lewis acid sides.17–19 This effect is probably masked in rheological measurements due to more prominent changes of viscoelastic properties in the clay-containing system.
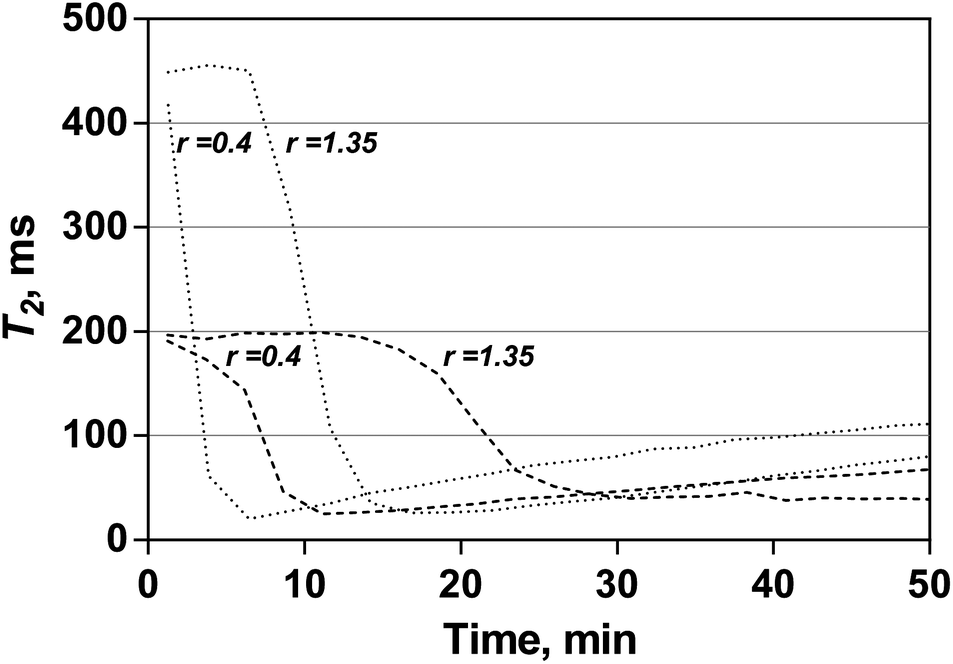 | ||
| Fig. 5 Temporal changes of T2 relaxation time for the synthesis of AAH (dotted lines) and NCH (dashed lines) at low and high initiator/activator ratios, 0.4 and 1.35 respectively. Initiator concentration – 20 mM. | ||
According to Fig. 4c, the steepness factor, n, increases with the growth of the initiator/activator ratio, as the polymerisation kinetics become slower. These are contradictive data since one should expect more shallow curves (smaller n) for slow kinetics. From Fig. 3, it appears that for slower polymerisation kinetics sigmoid curves become increasingly asymmetrical with different slopes before and after the θ-point. The Hill equation is not perfectly suitable for such asymmetric curves.28
The results discussed above demonstrate the change of polymerisation kinetics in the systems with constant initiator content. In an alternative series of experiments, the concentration of amine activator was fixed while the persulphate concentration and thereby initiator/activator ratio were changed. The concentration of initiator in this series ranged between 0.8 and 9 wt% with respect to monomer content (5 to 60 mM). Similar to the previous observation, this series confirms quicker dynamics of the G′ evolution during the NCH formation compared with AA polymerisation as shown in Fig. 6.
 | ||
| Fig. 6 Influence of the composition of the redox initiation system on rheokinetic parameters: steady state storage modulus, G′max, (a); half-gelation time, θ, (b); Hills's coefficient, n, (c); induction period of the polymerisation, tind, (d) in the synthesis of AAH and NCH at the fixed activator concentration of 25 mM. G′max, θ and n were obtained by fitting eqn (2) to storage modulus data and tind was determined as time when storage modulus reached 3 Pa. | ||
However, as revealed by Fig. 6, the polymerisation kinetics is slower in those systems with the activator-dominated redox couple, which is an opposite trend observed for the cases when the initiator concentration was fixed. The low concentration of the initiator is a dominating factor which determines the rate of polymerisation in the formulations with smaller initiator/activator ratios. In the initiator/activator range between 0.2 and 0.4 for activator concentration fixed at 25 mM, concentrations of initiator were 2 to 4 times lower than in the series with a fixed initiator concentration. When the concentration of initiator is low, even an excess of the activator cannot generate a high rate of free radical formation. Another startling difference between these two series is that in the experiments with fixed activator concentration, the steady-state G′ values decrease with the increase of the polymerisation rate (Fig 6a) whereas G′max increases for the faster reaction when the initiator concentration is fixed (Fig. 4a). This reflects the complexity of the polymerisation processes initiated by redox couples. Sulphate anion radicals, hydroxyl radicals and alkylaminomethyl radicals have been confirmed as initiating radicals for persulphate/tertiary amine couples.27 Moreover, the primary free radicals derived from amine parent molecules can participate in chain transfer due to the reactions with sulphate and hydroxyl radicals, as well as in further transformations leading to other amine-containing radicals. Therefore the total amounts of different free radicals have a complex response to changes of overall and relative concentrations of the components in the redox system.
3.4 Influence of the kinetics on the hydrogel structure and properties
Results discussed in the previous section demonstrated that steady-state values of storage modulus of hydrogels of the same composition, in terms of polymer and clay contents, can be varied significantly depending on initiating conditions. Thus the hydrogel structure formed is determined by the initiating conditions. Numerous publications were generated in an attempt to improve the properties of NCH by changing the type and content of nanofillers.2,3,7 However, it was essentially never discussed that the hydrogel structure and consequently properties can defer in the same systems synthesised under different conditions. To the best of our knowledge only one study investigated the influence of initiating conditions on NCH properties.16 In this paper, persulphate and organic initiator, azobisisobutyronitrile, were used for the preparation of NCH. It was shown that the mechanical properties of NCH synthesised by the polymerisation in the presence of an organic initiator were superior compared with the NCH polymerised via persulphate initiation.Generally, initiation is a well-studied topic in polymer chemistry. Initiation conditions determine the rate of generation of free radicals and their concentration. In the formation of linear polymers, higher concentrations of free radicals result in polymers of lower molecular weight due to free radical disproportion and recombination, which leads to chain termination. The influence of initiating conditions on the structure of cross-linked polymer structures is not so predictable considering that a polymer network is formed by monomers of different relative activities, monomer(s) and cross-linking monomer(s). Although the NCH does not include an organic cross-linker, clay is a potentially reactive component and can have an influence on the polymer molecular weight.
In the two series of hydrogels synthesised with variation of the initiating conditions, two opposite trends in changes of the steady-state values of storage modulus were observed; G′max values can either increase (Fig. 4) or decrease (Fig. 6) with the increase in polymerisation rate. Therefore such results cannot be interpreted simply in terms of polymer molecular weight. The theory of rubber elasticity is widely applied to hydrogels relating material storage modulus to cross-linking density and polymer chain length (molecular weight) between two junctions.20,21,29 Although the adequacy of quantitative description from this theory for hydrogel containing nanoplatelets is not clear, for materials with the same content of inorganic filler there is an obvious correlation between cross-linking density and storage modulus. Thus the dependence of ultimate storage modulus on initiating conditions implies that cross-linking density varied in response to changes in the composition of the redox initiator couple.
On the other hand, molecular weight between crosslinks is related to hydrogel equilibrium swelling. In swelling experiments, even some of the AAHs investigated in this study preserved shape and did not disintegrate in 6 months, maintaining the same swelling capacity during the last two months. Even those AAH samples which disintegrated cannot be classified as soluble materials since significant parts of the hydrogel were present in relatively small insoluble particles. As expected, all NCH were swellable but insoluble materials. It can be seen from Fig. 7 that the water intake capacity of hydrogels significantly varies with the change in the composition of the initiator system. The figure presents the only available data for AAH. Equilibrium swelling of NCH can vary as much as three times depending on the initiating conditions. The swelling of AAH is higher compared with that of the corresponding NCH. Comparison of the results on mechanical properties (Fig. 4 and 6) and swelling capacity (Fig. 7) indicates that trends in change of swelling capacity are in line with the variation of G′max values with the change in initiating conditions. Thus swelling experiments support the suggestion that initiating conditions influence the cross-linking density of the hydrogels.
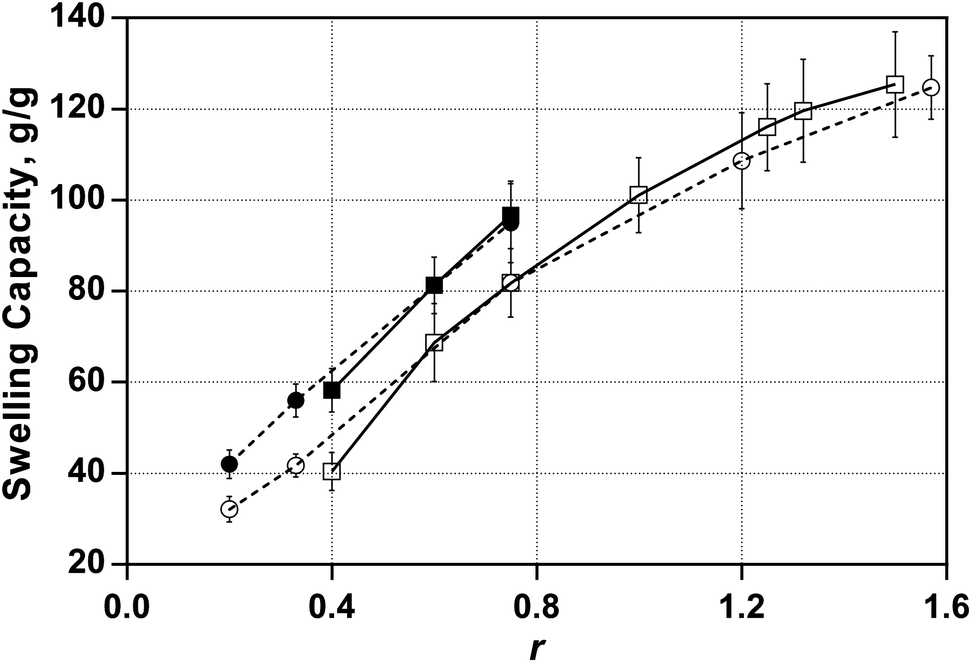 | ||
| Fig. 7 Influence of the composition of the redox initiation system on hydrogel swelling capacity for hydrogels synthesised in two series with the fixed initiator concentration of 20 mM (square symbols) and fixed activator concentration of 25 mM (circle symbols). Open symbols represent data for NCH and closed ones for AAH. | ||
The insolubility of AAH synthesised in this study implies that even in the absence of any cross-linking agent some covalent cross-links can be formed as a result of redox-initiated AA polymerisation in the solutions with relatively high concentrations. Self-crosslinking effect has been reported for the polymerisation of substituted-amide monomers such as N-isopropylamide,12,13,30N,N-dimethylacrylamide,15 and vinylformide.14 This phenomenon was observed for the polymerisation in the solutions with high monomer content. It is generally accepted that self-crosslinking occurs due to radical transfer to polymer chain, which results firstly in polymer branching and ultimately in cross-linking. Amide-pendant groups and tertiary carbon in the polymer backbone are considered as probable sites for chain transfer. The latter one can be potentially responsible for self-crosslinking of polyacrylamide. As a matter of fact, branching at high monomer conversion and concentration is a common phenomenon in free radical polymerisation including acrylamide polymerisation.31 Therefore self-crosslinking of acrylamide is a plausible event in the redox-initiated acrylamide polymerisation at high monomer concentrations, especially when polymerisation proceeds to high monomer conversion which is typical for the synthesis of nanocomposite hydrogels. These conditions are common in the preparation of nanocomposite hydrogels. Acrylamide branching is more pronounced at high temperatures and initiator concentrations31 and, therefore, sensitive to initiating conditions. This supports the view that variations in storage modulus and swelling degree with the changes in composition of redox system can be caused by altering the density of covalent self-crosslinking.
However, hydrogel properties are determined not only by permanent covalent cross-links but also by the network of physical cross-links. Actually, the hydrogel network can be entirely formed by physical cross-links. For the hydrogels with the same compositions, material properties are controlled by their morphological structures. The porous structure and the structure of polymer-rich domains are among the factors which determine material properties. Some insights into the morphological structure can be gained by studying the water state in hydrogels. A set of representative curves of T2 evolution (Fig. 8) have been selected to demonstrate three types of T2 temporal changes, which apparently correspond to three types of hydrogel morphological structures. Experiments with AAH and NCH were complemented with the data for ORH in an attempt to elucidate the contribution of rigid permanent network formation to the hydrogel morphology.
 | ||
| Fig. 8 Three types of T2 evolution curves in the synthesis of AAH (dashed lines), ORH (solid lines) and NCH (dotted lines). Initiator concentration – 20 mM. Initiator to activator molar ratio: a − 0.4; b − 0.8; c − 1.6. | ||
All curves show a characteristic sharp drop of T2-values during gelation reaching a minimal value. After this period, T2-values either increase sharply (Fig. 8c), or moderately (Fig 8b), or only slightly deviate from the minimal values (Fig 8a) during the “maturation” of the hydrogel structure. We may assume that hydrogels with minimal T2-values are uniform materials with small evenly distributed regions of polymer or polymer/clay deficient domains. Such a structure provides efficient water interactions with the polymer or polymer/clay network. In contrast, in hydrogels giving high T2-values, most of the water is in a state close to the free unbound water. This implies that the morphology of such a hydrogel is dominated by large polymer-deficient domains (large pores) which contain free water. Depending on the initiating conditions various morphological structures can be achieved with corresponding T2-values between the lower and upper limits.
The influence of initiating conditions on the formation of hydrogels of different morphologies and consequently on the state of the water in hydrogels was also confirmed by DSC. Fig. 9 illustrates DSC normalised endotherms for some of the hydrogels, whose T2 evolution profiles are presented in Fig. 8a and c. All thermograms are characterised by a large endothermic peak with a maxima above 0 °C and a melting event at temperatures below 0 °C. The latter does not produce a distinctive peak. The higher temperature peak corresponds to the melting of water in a state close to the bulk water, while the lower temperature melting event is attributed to the phase transition of freezing bound water. As can be seen from Fig. 9, the endotherm for AAH with a smaller steady-state T2 value (Fig. 8a) shifts towards lower temperatures compared with AAH showing a larger steady-state T2 value (Fig. 8c). This indicates the decrease in hydrogel pore size and stronger association of water with the polymer matrix leading to lower mobility of water molecules. Also further thermogram shifts to lower temperatures were observed for endotherms of NCH that is completely in line with the T2 trend; smaller T2 values were obtained for NCH compared with the corresponding AAH. Thus T2 values reflect the state of water, which is defined by the hydrogel morphology.
 | ||
| Fig. 9 DSC endotherms normalised to sample weight for hydrogels synthesised using an initiator concentration of 20 mM. AAH: initiator to activator molar ratio of 0.4 (dotted line), initiator to activator molar ratio of 1.6 (solid line). NCH: initiator to activator molar ratio of 0.4 (dashed line). | ||
Examples presented in Fig. 8 demonstrate that ultimate T2 values for hydrogels prepared under any specific composition of the initiating system always follow the same order: ORH > AAH > NCH. Thus rigid networks formed by permanent cross-links promote the formation of large domains filled with free water. However, although NCHs (according to the measurements of storage modulus and swelling capacity) have higher cross-linking densities, T2 values are smaller than the values for corresponding AAH. On one hand good dispersion of clay platelets in the hydrogel, perhaps even their exfoliation, contributes to the restriction of water mobility. On the other hand, clay in NCHs can lead to more uniform distribution of polymer chains across the hydrogel volume and thereby to the improvement of water binding by the polymer network.
Importantly variations in the redox couple composition lead to orchestrated changes in T2 values for three types of hydrogels. For example, for initiating conditions presented in Fig 8a even ORH had a T2 value around 200 ms while for AAH and NCH the values were approximately 160 and 125 ms respectively. However, the composition of the initiating system presented in Fig 8c resulted in ORH with a T2 around 800 ms and correspondingly significantly higher T2 for AAH and NCH than for hydrogels shown in Fig. 8a. Therefore initiating conditions predetermine the morphological structure of the hydrogel. Moreover, despite some morphological changes due to the presence of either an inorganic or organic cross-linking agent, it appears that the formation of the acrylamide network is the dominating factor in the morphology of ORH and NCH.
The mechanism of the formation of NCH with layered silicates is still a subject of discussion.15,32 It is generally agreed that silicate platelets play the role of multifunctional cross-linking surfaces. However, it is not clear whether binding of polymer chains to the platelet surface is covalent or due to purely physical interactions. The results obtained in this study demonstrate that despite the storage modules of NCH being higher than the corresponding AAH, changes in initiating conditions led to the same patterns of storage modulus variations for hydrogels of both types. If growing chains formed covalent bonds, the number of such bonds and consequently the cross-linking degree would depend on initiating conditions. Therefore a significant deviation of the G′max trend for NCH would occur compared with the trend for AAH. No steepening has been observed for NCH curves compared with AAH (Fig. 4a and 6a). This strongly suggests that covalent bonds do not form between silicate and polymer chains. The properties of NCH are changed due to physical interactions between clay platelets and macromolecules. A recently reported self-healing ability of NCH33 also supports this view. It was shown that NCH returned to its initial shape and more importantly was restored its mechanical strength when two cut off pieces were kept together in contact. Only non-covalent bonds can be renewed under such conditions.
The strongest argument suggesting covalent bonding was lack of success in making NCHs by direct mixing of polymer solutions with a clay suspension. However, it becomes apparent that the in situ hydrogel synthesis by the polymerisation at high monomer concentrations results in polymers of ultrahigh molecular weight. Moreover such polymers most likely have some degree of branching and perhaps self-crosslinking. The swelling test of AAH confirms that self-crosslinking is possible in the polymerisation of unsubstituted acrylamide. Studies from other groups demonstrated even more pronounced self-crosslinking tendency for amide-substituted monomers.12–15,30 Such molecular structures of the polymer can be uniquely produced only by in situ polymerisation.
Our study suggests that the extent of self-crosslinking can be changed depending on the initiating conditions. Therefore the properties of NCH are dependent on synthesis conditions. In fact, recently Hu et al.34 synthesised fully degradable poly(N-isopropylacrylamide) hydrogels by avoiding self-crosslinking via the selection of initiating conditions. Interestingly Abdurrahmanoglu et al.35 reported higher cross-linking degrees for NCH of more hydrophobic poly(N-isopropyl acrylamide) and poly(N,N-dimethyacrylamide) compared with more hydrophilic poly(acrylamide). The authors attributed the increase in cross-linking density to better physical interaction of the former two polymers with clay platelets. However, this rather more efficient self-crosslinking gave rise to the formation of NCH with increased total cross-linking density.
It is worth mentioning that G′max values obtained in rheokinetic experiments can be barely considered as accurate data for fully polymerised hydrogels. First of all, despite making attempts to minimise water evaporation for those experiments lasting more than 1 hour, some loss of water can be expected. Moreover, the volume of reaction mixture decreases due to hydrogel shrinkage and the degree of shrinkage varies depending on material properties. Shrinkage will affect the precision of oscillatory rheology since the contact area of the sample with the surface of the measuring system can change. Also it is possible that polymerisation proceeds even when the rheometer does not detect changes in storage modulus. T2 stabilised at much longer times than the storage modulus. Therefore we characterised selected samples of fully polymerised hydrogels, which were prepared in a sheet shape by keeping in the mould for 24 hours at 25 °C. Two initiating conditions were chosen: initiator and activator concentrations of 6 and 16 mM (ratio 0.4), and 20 and 20 mM (ratio 1.0). In the rheology experiments, these conditions resulted in NCH with G′max 16.5 and 12.3, while G′max of AAH were 13.2 and 11.0 kPa respectively (Table 2).
| G ′max, kPa | Q W, g g−1 | Elastic modulus, kPa | Tensile strength, kPa | Elongation at break, % | |
|---|---|---|---|---|---|
| [Initiator] = 6 mM, [activator] = 16 mM | |||||
| AAH | 12.3 | 38.0 ± 1.9 | 26.3 ± 0.7 | 36.5 ± 2.5 | 590 ± 10 |
| NCH | 16.5 | 28.7 ± 1.5 | 31.5 ± 0.5 | 45.0 ± 1.6 | 605 ± 10 |
| [Initiator] = 20 mM, [activator] = 20 mM | |||||
| AAH | 11.0 | — | 17.8 ± 0.6 | 29.7 ± 0.9 | 485 ± 15 |
| NCH | 13.2 | 101.1 ± 8.3 | 19.3 ± 0.8 | 32.4 ± 1.2 | 610 ± 10 |
As shown in Table 2, tensile measurements of fully polymerised hydrogels are in line with rheokinetic measurements. Initiating conditions, which resulted in hydrogels of higher G′max values, give rise to higher tensile strength and elastic modulus for fully polymerised samples. As expected, NCH showed improved strength and modulus compared with the corresponding AAH. NCHs were also more ductile with extension at break above 600%. Such fully polymerised hydrogels were further characterised in MRI experiments. According to Fig. 10, similar to in situ MRI monitoring, T2-values for fully polymerised hydrogels varied significantly depending on initiating conditions. Both AAH and NCH with lower G′max have larger T2-values, which reflects the influence of initiating conditions on the morphology and mechanical properties of the hydrogels. Interestingly AAH showed a significant distribution in relaxation times (low peak height and large peak width). In contrast, T2 values of the NCH have a much narrow distribution. This implies that the morphological structure of the NCH is more uniform than that of AAH, which contributes to the improvement of the mechanical properties of the NCH.
 | ||
| Fig. 10 Pixel T2 distribution for fully polymerised AAH and NCH synthesised under two initiating conditions: hydrogels represented by dotted lines were synthesised at initiator and activator concentrations of 6 and 16 mM (r = 0.375), respectively; hydrogels represented by dashed lines were synthesised at an initiator and activator concentration of 20 mM (r = 1). | ||
4 Conclusions
The process of hydrogel formation in free-radical AA polymerisation was monitored in situ by non-invasive methods using oscillatory rheology and 1H magnetic resonance imaging. Polymerisation was carried out in the solutions of high monomer concentration, which is a typical condition in the synthesis of nanocomposite hydrogels containing layered silicates as multifunctional cross-linking surfaces. The study focussed on the influence of initiating conditions on the polymerisation kinetics and the properties of hydrogels. Initiating conditions were varied by changing concentrations of components of a redox couple. The experiments were conducted at fixed concentrations of the initiator or activator and the variable initiator/activator molar ratio. At the fixed initiator concentration (20 mM) faster polymerisation was observed with the increase of the content of the activator (decrease of the initiator/activator ratio), while the polymerisation accelerated with the increase of the ratio in the experiments with fixed activator concentration (25 mM). However, in both series the ultimate storage modulus of hydrogels was higher in activator-dominated systems. Such observations cannot be attributed to the change in polymer molecular weight since it usually drops with the increase of the rate of polymerisation. Equilibrium water adsorption capacity correlated with the ultimate values of storage modulus; hydrogels with lower storage modulus had swollen to higher degrees. Thus initiating conditions influenced the hydrogel cross-linking degree.1H MRI showed that T2 relaxation time, which was used as a characteristic of water mobility, significantly changed in response to variations in the composition of the initiating system. Depending on the composition of the initiator system, T2 values changed from approximately 150 ms to the values almost corresponding to T2 of bulk water. This indicates the variations in interactions between water and network constituents. Thus initiating conditions play an important role in determining the morphological structure of the hydrogel.
It was shown that for hydrogels prepared with the same compositions of the initiating system, T2 increases in the following order: NCH < AAH < ORH. It was concluded that the rigid network of ORH formed by covalent cross-links led to the segregation of the polymer into polymer-rich domains accompanied by the formation of large polymer-deficient areas (pores). The later accommodate unbound water with the mobility close to the one of free water. However, despite having a higher cross-linking degree than AAH, NCH showed lower water mobility. This suggested that clay platelets did not form covalent bonds with polymer molecules but contributed to the formation of the physical network. Swelling experiments demonstrated that at some initiating conditions AAH preserved the shape and did not disintegrate in 6 months. Even those AAHs, which did disintegrate, could not be considered as soluble because of the presence of small pieces of insoluble hydrogels. The self-crosslinking effect has been reported in other studies for amide-substituted monomers during polymerisation in solutions of high monomer concentrations. It appears that acrylamide also formed a self-crosslinked network and cross-linking density of such a network depends on the initiating conditions. Thus self-crosslinking can be an important factor in the formation of the unique morphological structure of NCH prepared by in situ polymerisation.
References
- K. Haraguchi and T. Takehisa, Adv Mater., 2002, 14, 1120–1124 CrossRef CAS.
- K. Haraguchi, Curr. Opin. Solid State Mater. Sci., 2007, 11, 47–54 CrossRef CAS PubMed.
- K. Haraguchi, Macromol. Symp., 2007, 256, 120–130 CrossRef CAS.
- K. Ito, Polym. J., 2007, 39, 489–499 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- T. Sakai, React. Funct. Polym., 2013, 73, 898–903 CrossRef CAS PubMed.
- K. Kabiri, H. Omidian, M. J. Zohuriaan-Mehr and S. Doroudiani, Polym. Compos., 2011, 32, 277–289 CrossRef CAS.
- P. Schexnailder and G. Schmidt, Colloid Polym. Sci., 2009, 287, 1–11 CAS.
- A. Giz, H. Çatalgil-Giz, A. Alb, J. Brousseau and W. F. Reed, Macromolecules, 2001, 34, 1180–1191 CrossRef CAS.
- K. Haraguchi, Y. Xu and G. Li, Macromol. Symp., 2011, 306–307, 33–48 CrossRef CAS.
- H. Tamagawa, S. Popovic and M. Taya, Polymer, 2000, 41, 7201–7207 CrossRef CAS.
- J. Gao and B. J. Frisken, Langmuir, 2003, 19, 5212–5216 CrossRef CAS.
- J. Gao and B. J. Frisken, Langmuir, 2003, 19, 5217–5222 CrossRef CAS.
- L. Gu, S. Zhu, A. N. Hrymak and R. H. Pelton, Macromol. Rapid Commun., 2001, 22, 212–214 CrossRef CAS.
- L. Carlsson, S. Rose, D. Hourdet and A. Marcellan, Soft Matter, 2010, 6, 3619–3631 RSC.
- Y. Wang, J. Ma, S. Yang and J. Xu, Colloids Surf., A, 2011, 390, 20–24 CrossRef CAS PubMed.
- D. H. Solomon and J. D. Swift, J. Appl. Polym. Sci., 1967, 11, 2567–2575 CrossRef CAS.
- D. H. Solomon, Clays Clay Miner., 1968, 16, 31–39 Search PubMed.
- P. Bera and S. K. Saha, Polymer, 1998, 39, 1461–1469 CrossRef CAS.
- O. Okay and W. Oppermann, Macromolecules, 2007, 40, 3378–3387 CrossRef CAS.
- D. Calvet, J. Y. Wong and S. Giasson, Macromolecules, 2004, 37, 7762–7771 CrossRef CAS.
- J. Kalfus, N. Singh and A. J. Lesser, Polymer, 2012, 53, 2544–2547 CrossRef CAS PubMed.
- A. S. Sarvestani and E. Jabbari, in Rubber Nanocomposites, ed. T. Sabu and S. Ranimol, John Wiley & Sons, Ltd, 2010, pp. 331–351 Search PubMed.
- M. Liu, W. Li, J. Rong and C. Zhou, Colloid Polym. Sci., 2012, 290, 895–905 CAS.
- H. Tanaka, K. Fukumori and T. Nishi, J. Chem. Phys., 1988, 89, 3363–3372 CrossRef CAS PubMed.
- W. H. Dokter, H. F. van Garderen, T. P. M. Beelen, J. W. de Haan, L. J. M. van de Ven and R. A. van Santen, Colloids Surf., A, 1993, 72, 165–171 CrossRef CAS.
- X. D. Feng, X. Q. Guo and K. Y. Qiu, Makromol. Chem., 1988, 189, 77–83 CrossRef CAS.
- J. Giraldo, N. M. Vivas, E. Vila and A. Badia, Pharmacol. Ther., 2002, 95, 21–45 CrossRef CAS.
- K. S. Anseth, C. N. Bowman and L. Brannon-Peppas, Biomaterials, 1996, 17, 1647–1657 CrossRef CAS.
- Y. Xu, G. Li and K. Haraguchi, Macromol. Chem. Phys., 2010, 211, 977–987 CrossRef CAS.
- W. Kulicke, R. Kniewske and J. Klein, Prog. Polym. Sci., 1982, 8, 373–468 CrossRef CAS.
- Y. Wu, M. Xia, Q. Fan, Y. Zhang, H. Yu and M. Zhu, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 263–266 CrossRef CAS.
- K. Haraguchi, K. Uyama and H. Tanimoto, Macromol. Rapid Commun., 2011, 32, 1253–1258 CrossRef CAS PubMed.
- X. Hu, Z. Tong and L. A. Lyon, Langmuir, 2011, 27, 4142–4148 CrossRef CAS PubMed.
- S. Abdurrahmanoglu, V. Can and O. Okay, J. Appl. Polym. Sci., 2008, 109, 3714–3724 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |