 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microarrays and single molecules: an exciting combination
Stefan
Howorka
*ab and
Jan
Hesse
b
aDepartment of Chemistry, Institute for Structural and Molecular Biology, University College London, London WC1H 0AJ, UK. E-mail: s.howorka@ucl.ac.uk
bCenter for Advanced Bioanalysis GmbH, A-4020 Linz, Austria
First published on 9th January 2014
Abstract
Biomolecules positioned at interfaces have spawned many applications in bioanalysis, biophysics, and cell biology. This Highlight describes recent developments in the research areas of protein and DNA arrays, and single-molecule sensing. We cover the ultrasensitive scanning of conventional microarrays as well as the generation of arrays composed of individual molecules. The combination of these tools has improved the detection limits and the dynamic range of microarray analysis, helped develop powerful single-molecule sequencing approaches, and offered biophysical examination with high throughput and molecular detail. The topic of this Highlight integrates several disciplines and is written for interested chemists, biophysicists and nanotechnologists.
Introduction
Analyzing biomolecular interactions with microarrays has helped usher in the genomic and proteomic area and revolutionized the biosciences. Initially DNA-chips,1,2 and later, arrays of proteins,3 peptides,4 and carbohydrates5 have become popular research tools. They condense biomolecular assays into a parallel and highly miniaturized format with the added benefit of reduced sample and reagent consumption.Of similar importance, analyzing biomolecules at the single-molecule level has been the driving force towards an improved understanding of how proteins and DNA function. Single-molecule detection can uncover unique sub-populations within the observed biomolecule species or trace temporal variations in their bioactivity, which can be masked in conventional ensemble assays.6
This Highlight explores the exciting interface between biomolecular arrays and single-molecule sensing. Two topics will be covered in separate chapters: the first is about the analysis of conventional microarrays with ultrasensitive methods. The read-out at the single-molecule level is of relevance as it offers scientific advantages. For example, mRNA and proteins can be directly measured at very low concentrations without signal amplification that may lead to distortions.
The second topic is the generation and use of single-molecule arrays. These are composed of individual proteins or nucleic acids that are positioned at defined sites on substrate surfaces. Similar to their conventional counterparts, single-molecule arrays can be analyzed to yield high sensitivity. As a further benefit, their high packing densities can increase the throughput of assays such as sequencing-by-synthesis of singulated strands.7 The known position of isolated molecules can furthermore facilitate their biophysical and structural examination without interference from other nearby molecules.8–12 Finally, patterns of single biomolecules can be exploited in cell biology as tools to mimic cell surfaces and thereby study cell adhesion and signaling processes.13–18 Hence, while the second topic overlaps with the first in the ultrasensitive read-out of arrays, it goes beyond this classical remit. Rather, it includes biophysical and cell biological studies which are of equal importance.
A concluding chapter will identify current trends in single-molecule array analysis and potential future areas of development and research.
Single-molecule readout of microarrays and advanced ultrasensitive detection techniques
This chapter covers the sensing of microarrays with ultra-sensitive methods and provides a brief survey of relevant advanced methods to detect individual protein or DNA molecules. Not all methods have yet been applied for array scanning. However, some have been used in the related context of creating single-molecule arrays.22 This indicates the dynamic and overlapping nature of research into arrays and single-molecule sensing.In one of the first examples of the ultrasensitive readout of conventional mRNA microarrays,21 wide-field fluorescence microscopy was used.23 Scanning of microarray slides with areas of several cm2 was achieve with a new fast read-out mode.29 In addition, slides with optimized microarray coatings were used to attain highly specific analyte binding and minimize unspecific binding. Under these optimized conditions, the sensitivity was in the femtomolar range and the dynamic range was 4.7 logs (Fig. 1).21 The high-speed scanning of slides was also applied to monitor the expression of cancer cell subpopulations24 and to characterize the variety of DNA molecules with large genomic and tandem repeat mutations.25,26 As a limitation, wide-field fluorescence microscopy can only detect individual molecules when they are spaced beyond the diffraction limit at low surface densities.
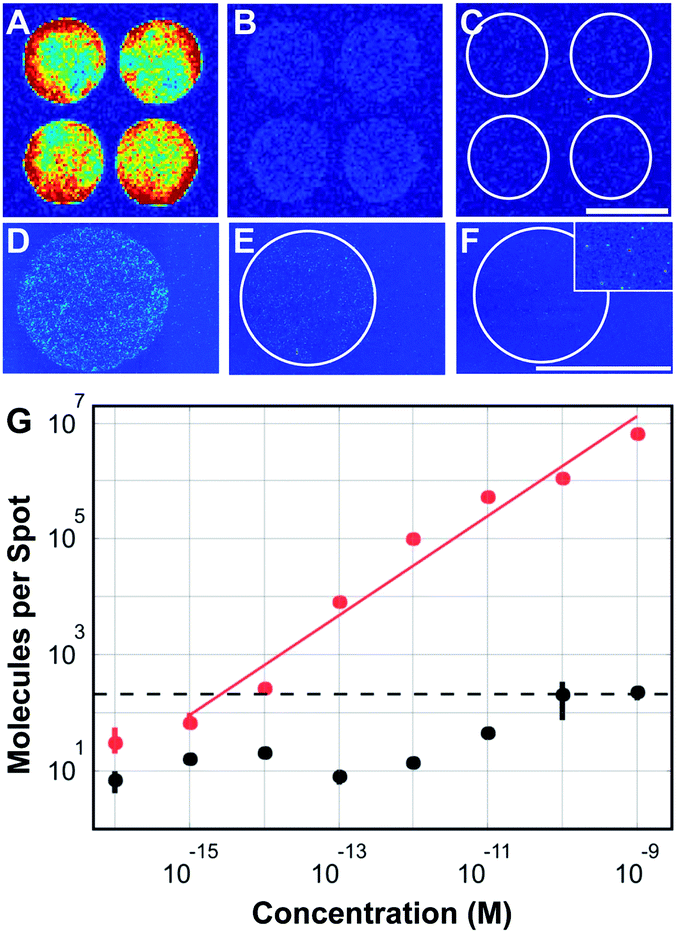 | ||
| Fig. 1 Ultransensitive read-out of nucleic acid microarrays. (A–F) Fluorescence scans of DNA microarrays spotted on (A–C) commercial substrates (SuperAldehyde, Telechem Inc) and on (D–F) custom-coated aldehyde-glass coverslips19 acquired using (A–C) a ScanArray4000 reader and (D–F) a single-molecule reader.20 Microarrays were hybridized with different concentrations of Cy5-labeled target-oligonucleotide: (A) 100 pM, (B) 10 pM, (C) 1 pM, (D) 100 fM, (E) 10 fM, (F) 1 fM. The inset in (F) shows single molecule signals at 5-fold higher magnification. Scale bars: 100 μm. (G) Number of specifically hybridized (red) and unspecifically adsorbed (black) oligonucleotide molecules per spot vs. sample concentration. The surface density increases linearly over six orders of magnitude; unspecific binding limits the dynamic range to 4.7 logs. Assuming a minimum signal-to-noise ratio of 10 as the criterion for reliable analysis (dashed line), a detection limit of c = 1.3 fM can be specified. Figure adapted from (ref. 21). | ||
To resolve individual molecules at high bulk concentrations, zero-mode waveguides have been developed.27 In these analysis platforms, glass substrates are coated with 100 nm thin metallic masks featuring nanoscopic holes with diameters smaller than the wavelength of light. The resulting small excitation volume of a few zeptoliters helps illuminate single molecules within bulk concentrations of up to 20 μM. Zero-mode waveguides are used for a sequencing-by-synthesis approach which tracks the polymerase-mediated incorporation of fluorescence-labeled nucleotides present at high concentrations.28
Other routes based on conventional light microscopy can achieve nm-resolution for single molecules spaced below the diffraction limit.29 The first approach of these super-resolution methods, stimulated emission depletion (STED) microscopy,30 restricts the excitation volume to a diameter of around 100 nm, analogous to zero-mode waveguides. In STED, the restriction is achieved with a pair of laser pulses. The first focused excitation laser pulse is followed by a STED pulse for efficient de-excitation of the previously excited fluorophores, resulting in a residual excited volume of sub-wavelength diameter.31,32 In combination with raster scanning, super-resolution images can be obtained for the diffusion of individual lipid and protein molecules within the plasma membrane of living cells.33 The concept of STED has also been used for generating arrays of binding sites that can be decorated with single protein molecules,22 as discussed in the next chapter.
The other super-resolution microscopy methods rely on the sequential activation of photoswitchable fluorophores and subsequent single-molecule imaging. In photoactivated localization microscopy (PALM)34,35 fluorescent dyes are activated using a laser pulse. The illumination time and intensity is chosen to limit the density of activated fluorophores to a sufficiently small number which can be detected with optical, diffraction-limited imaging systems.36 Utilizing PALM for time-resolved super-resolution microscopy was made possible by an engineered fluorescent protein which is sufficiently photostable to enable localization with high precision yet amenable to efficient photo-deactivation to achieve a high contrast between the on and off state.
Stochastic optical reconstruction mircoscopy (STORM)37 is the second method of generating a sparse set of active fluorophores. As the probe, a pair of Cy3 and Cy5 fluorophores is used. Cy5, excited using a red laser, is imaged till it switches to its dark state. Exposing the Cy3–Cy5 pair to a green laser converts Cy5 back to its fluorescent state. The process can be repeated hundreds of times before irreversible photobleaching occurs. Recently, STORM has been utilized for measuring mRNA levels within intact cells in a parallel manner.38 Utilizing the resolution power of STORM in combination with 7 super-resolution fluorophore pairs for spectral barcoding, 32 genes in individual S. cerevisiae cells could be quantified. The concept of STORM has been extended to 3D-imaging39 to monitor the dynamics of transferrin and clathrin-coated pits in live cells.40 A simplified version of STORM called direct STORM (dSTORM) utilizes conventional cyanine dyes (Cy5, Alexa647).41 The reversible switching between dark and fluorescent state is achieved by illumination with 647 nm (turn dark) and 514 nm (turn fluorescent) laser light, respectively. Using dSTORM, the dynamics of protein complexes within cells has been imaged with 20 nm resolution and movements with 3 nm s−1 could be captured.42
The last technique for high-resolution fluorescence detection is NSOM.43,44 Here, visible light from an optically coated fiber emanates from an aperture of a few tens of nm to illuminate a small volume. Raster scanning with the probe allows imaging of micrometer-sized samples with high sub-wavelength resolution and single fluorophore sensitivity45 as shown for DNA46 and membrane-diffusing lipids.47
Information on other optical single-molecule detection assays in bioanalytical applications as well as advantages and limitations of corresponding ultrasensitive detection techniques are provided in several reviews.48,49
The non-optical scanning probe approach of atomic force microscopy (AFM) is the most popular method for analyzing the shape, dimensions, and intramolecular and intermolecular forces of individual molecules.50 Among the many AFM variants and modes, three are most relevant for biopolymers. For molecular recognition, surface-bound biomolecules can be detected by cognate receptor molecules tethered to the AFM tip.51 This imaging can be combined with the regular topographic mode.52 Another related variant termed force spectroscopy, does not provide visual information in terms of lateral scans but studies the force-dependent interactions between individual surface-bound molecules and a receptor-coated cantilever53,54 or within individual biological molecules55 to elucidate energetic, dynamic, and structural aspects of molecular processes such as DNA unzipping56 or protein unfolding.57,58 Finally, the slow-scanning read-out of serial AFM is being overcome by high-speed atomic force microscopy. This mode allows direct visualization of dynamic structural changes and dynamic processes of functioning biological molecules in physiological solutions and at high spatiotemporal resolution.59,60
Single-molecule arrays
This chapter describes the generation and different applications of single-molecule arrays. What constitutes these regular assemblies? In keeping with the format of conventional microarrays, single-molecule arrays can be understood as spatially defined and usually regular alignments of individually discernible molecules on planar surfaces. Indeed, single-molecule arrays represent the endpoint of a trajectory which has started with DNA and protein microarrays and has led to nanoarrays featuring a small number of molecules per spot. It is, however, also clear that the concept of planar assemblies has to be expanded into 3D to include nanotopographical surfaces and arrays of nanoparticles, beads and lipid vesicles which carry or contain individual biomolecules.How, and how easily, can single-molecule arrays be generated? Their fabrication can be challenging due to the experimental difficulty in handling and positioning individual biomolecules. Several established nanopatterning methods61–64 can be adapted to achieve this aim. In general, arrays of biopolymers can be obtained following one of the two routes of nanopatterning, namely top-down or bottom-up.63,64
Top-down
In the top-down route, nanopatterns are made by physical or physico-chemical methods employing macroscopic tools. The large variety of top-down approaches can traditionally be further sub-categorized depending on whether the patterning is conducted in a serial fashion using e.g. an AFM cantilever or in a parallel fashion with a macroscale stamp. But this distinction has been blurred with the arrival of highly parallel AFM cantilever arrays.65Given this Highlight's focus on the positioning of individual molecules, a different criterion is used. The distinction is made between the direct and indirect deposition. In the direct route, target biopolymers are transferred from a nanoscale carrier onto an otherwise unstructured surface. By comparison, in the indirect route molecules specifically adsorb from solution onto nanoscale sites of a pre-structured substrate.
Representative methods for the first, direct approach use a biomolecule-filled nanopipette which ejects individual or small numbers of biopolymer molecules.66 Other nanoscale carriers are “dip-pen” AFM cantilevers which are “inked” with biopolymers67 to transfer them onto the substrate upon writing. Deposition may also be achieved in a more parallel fashion with arrays of AFM cantilevers or via contact printing with nanostructured stamps68,69 which may optionally be assembled from microscale particles.70
For indirect patterning, biomolecules bind to chemically or electrostatically adhesive nanopatterns. Patterning with a chemical nanopatch was, for example, achieved with serial AFM-based nanografting where the cantilever scratches a hole into a surface-passivating layer to expose an adhesive nanopatch onto which individual biomolecules can bind.10,71,72 Similarly, the AFM-based dip-pen nanolithography can generate nanopatterns of organic capture molecules for the immobilization of target biomolecules.73–75 The adhesive nanoislands can also be generated with complementary parallel methods including classical procedures and variants of e-beam and UV lithography,76–82 nanoimprint lithography,83 and the above mentioned nanocontact printing.84
Several of the methods have been used to generate single-molecule arrays. A few reports are highlighted here. In the direct deposition route, individual nucleic acid molecules are positioned onto a substrate by taking advantage of the precision of AFM (Fig. 2A). Using a copy-and-paste approach based on reversible hybridization, DNA oligomers were picked up from a depot area on a chip by means of a complementary DNA strand which had been tethered to an AFM tip.67,85 The picked-up strands were then transferred to and deposited on a target area by formation of a more stable duplex with a substrate-immobilized capture strand. The approach helped create simple geometrical structures assembled from the single-molecule units. To confirm that individual molecules had been transferred, each cut-and-paste transfer event was characterized by single-molecule force spectroscopy and single-molecule fluorescence microscopy.
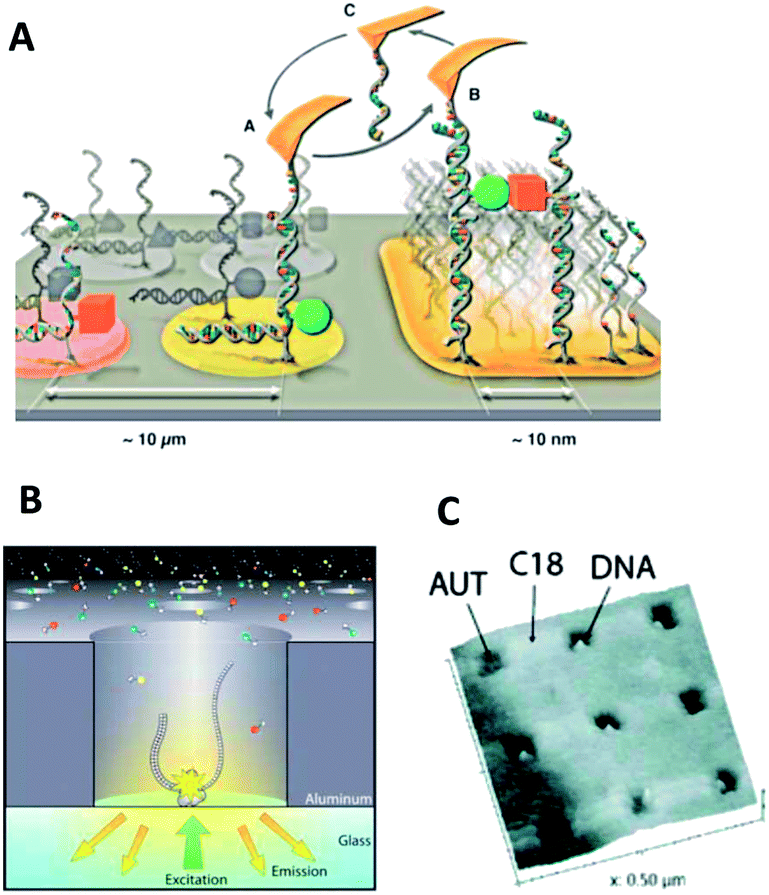 | ||
| Fig. 2 Top-down approaches for the patterning of single molecules on surfaces. (A) Copy-paste of DNA strands with a capture-DNA-modified AFM cantilever.60 (B) Zero-mode waveguides carrying a single DNA strand within a nanocavity.28 (C) AFM image of a nanografted surface pattern carrying individual DNA duplexes within the scratched holes.10 | ||
In comparison to the rather time-consuming nature of the direct deposition route, the complementary indirect route can bind in highly parallel fashion, molecules to millions of pre-defined sites on a nanopatterned substrate. Key to achieving the desired stoichiometry of one molecule per nanosite is to limit the dimensions of the latter to those of the former. Hence, arraying large biopolymers with a diameter of around 100 nm is easier than those of 5 nm because substrate patterns with feature sizes of 100 nm are more readily fabricated. In one example of the indirect deposition of large particles, individual single stranded DNA molecules resembling nanoballs of 300 nm diameter were immobilized onto lithographically generated nanodisks of equal diameter.7 The nanoballs were formed due to the engineered presence of self-complementary sequence elements which caused DNA to fold back onto itself. Electrostatic interaction caused the negatively charged nucleic acid nanoball to bind to the organically coated and positively charged surface nanopatches. The arraying of DNA was carried out to facilitate single-molecule sequencing-by-synthesis. Similar to related assays, the template-directed incorporation of fluorescent nucleotides into the nascent DNA was monitored. The new aspect was that each DNA strand contained concatenated copies of the same DNA sequence, thereby increasing the signal-to-noise ratio as multiple identical molecular fluorescence reactions took place per nanospot. Positioning DNA nanoballs in the array format had the added benefit of increasing their surface density resulting in a higher through-put in read-out compared to random, less dense biomolecule assemblies.
DNA strands were also immobilized onto size-matched nanopatterns in another sequencing approach based on zero-mode waveguides. In these analysis platforms, a glass slide is coated with a 100 nm-thick aluminum layer which contains an array of holes 70 nm across which is similar to the size of the analyzed DNA duplexes (Fig. 2B). For sequence-by-synthesis, a DNA polymerase is first immobilized at the bottom of the nanocavities to capture. In a second step, individual DNA strands at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry per well.28 The zero-mode waveguides limit the fluorescence excitation volume to a few zeptoliters to enable the use of μM concentrations of fluorescence-labeled deoxyribonucleotides. These high concentrations are required to drive enzymatic polymerization.
:1 stoichiometry per well.28 The zero-mode waveguides limit the fluorescence excitation volume to a few zeptoliters to enable the use of μM concentrations of fluorescence-labeled deoxyribonucleotides. These high concentrations are required to drive enzymatic polymerization.
Smaller biopolymers have been arrayed by decreasing the size of the nanopatterns, as illustrated for a double stranded DNA of 100 base pairs or around 30 nm length. In this case, the indirect deposition method of AFM nanografting was employed.10 A film of octadecanethiol was first physisorbed onto an atomically flat gold substrate to create a non-adsorptive layer. Patches of this film around 50 nm in size were subsequently scratched away with the AFM cantilever to expose the underlying adhesive gold surface. Incubating the holes with a solution of thiolated DNA duplexes led to the binding of individual molecules (Fig. 2C). To avoid immobilization of multiple strands, an excess of positively charged alkanethiol was added to coat the remaining areas of bare gold. The formed positive patch also electrostatically stabilized the binding of the DNA duplex to the surface. The DNA duplexes were visualized by AFM. In further studies, the individualized DNA strands were observed within the controlled nanoscale environment thereby helping to decouple the intrinsic properties from the heterogeneity caused by the local environment.9,10
Compared to relatively large DNA duplexes, the immobilization of individual smaller protein molecules requires the generation of nanospots which approach or match the size of protein targets. In one example, a few to individual antibody molecules of 10–15 nm size were immobilized on small polymeric nanopatches of 60–70 nm. Arrays of the nanofeatures were fabricated by photo-polymerizing acrylic precursors with a two-photon STED technique22 which was originally developed for high-resolution fluorescence microscopy. The adsorbed antibodies remained bioactive as shown by probing their interaction with the cognate antigen which was located within bilayer membranes.
In another study, the immobilization sites were sub-ten nm gold nano-particles. The small particles were generated by scanning probe block copolymer lithography.86 In this specific form of dip-pen lithography, polymers chelated with metal ions are deposited on a chip and later annealed and reduced to form the metal particles. Proteins were immobilized on top of gold crystal scaffolds via an intermediary alkylthiol monolayer. The number of immobilized proteins was found to depend on the particles' size; when the nanospheres' size approached the dimensions of a protein molecule, each gold scaffold supported a single protein. This was demonstrated with both gold nanoparticle and quantum dot labeling coupled with transmission electron microscopy imaging experiments. The immobilized proteins remained bioactive, as shown by enzymatic assays and antigen–antibody binding experiments.
The concept of immobilizing biomolecules on spherical templates has also been exploited with larger microscale beads. Unlike the previous method of immobilization on top of the nanoscaffold, individual DNA strands were beneath the microspheres as they function as a molecular tether to the underlying nanopatterned substrate prepared by contact printing.87 The interfacial position was central in achieving a 1:1 stoichiometry because, following Poisson distribution, only one to a few DNA strands on the curved bead surface were physically proximal to the planar substrate. Hence, the density of biomolecules on the bead surface is an important parameter to achieve single-molecule arrays.88 The particular DNA-bead array was used to measure the variation in the effective length of the DNA tether. The experimental observable was the Brownian motion of the beads. This helped to uncover biophysical changes to the DNA structure which were induced by interaction with proteins. The advantage of the single-molecule array was an increased data output for the parallel force spectroscopy as opposed to conventional time-consuming serial assays.
Apart from the immobilization on surfaces of physically hard planar or curved substrates, arrays have also been obtained with soft spherical bilayer vesicles or water-in-oil droplets. Encapsulation in aqueous environments can help maintain the biomolecules' activity or structural integrity which may otherwise suffer by immobilization on inorganic substrates.89 The fragile fluid compartments require, however, careful patterning approaches. In one study, lipid bilayer vesicles have been assembled on microstructured substrates via multiple DNA duplexes formed by oligonucleotide anchors.90 With water-in-oil droplets, the assembly was shown to take place on hydrophilic-hydrophobic micropatterned surfaces91 which were optionally linked to fiber optic arrays for direct optical readout.92,93 When applied to encapsulated enzymes, the atto- to femtoliter nanoreactors enabled single-molecule kinetics studies in a highly parallel format.93 The signal of individual enzymes was amplified by fluorogenic substrates.91–93
It is noted that in the above mentioned micro-sized droplets and vesicles, single molecules are no longer present at a 1:1 stoichiometry but Poisson-distributed because the nanocontainers are larger than the proteins which removes the size-dependent restriction of other physically hard nanopatterns such as zero-mode waveguides. In another variant droplet array, water-in-oil vesicles are mixed with phospholipids to form a lipid-monolayer lining the water–oil interface. When droplets are in physical contact, a bilayer is formed.94,95 Individual protein channels can insert at the interface bilayer leading to electrically detectable channel networks.96
Lipid bilayers in the planar rather than vesicular form have also been exploited for single-molecule studies. By their nature, planar bilayer arrays are ideal to study membrane proteins including ion channels and nanopores with microscopic techniques. Planar arrays have been created by layering membranes on microstructured solid substrates and,97–101 more recently, by spanning them across water-filled cavities.102–104 The latter format is scientifically attractive as the absence of any underlying solid support enables the integral membrane protein to freely diffuse in the bilayer thereby mimicking their natural biophysical behavior. As an additional benefit, powerful single-channel current analysis can examine ion channels and nanopores provided the bilayer is formed over cavities containing individual microelectrodes.102–104 These chip-based nanopore microarrays are important as they enable the parallel high-resolution electrical analysis of channels as well as analytes which pass through individual protein nanopores. In future, the bilayer arrays will likely play an important role in the sequencing of single DNA strands by nanopore recording.105,106 In this respect, arrays of top-down fabricated solid-state nanopores are also mentioned107–109 even though they are not yet able to distinguish individual bases.
Bottom-up
The top-down route towards single-molecule arrays is complemented by bottom-up self-assembled lattices. Top-down methods are excellent for periodicities from the microscale down to around 50 nm, but the range from 50 nm to 1 nm is accessible via bottom-up routes. In general, self-assembly is a remarkable process and can lead to the formation of planar arrays on interfaces as shown for nucleotides,110 small organic aromatic molecules,111 organic polymers,13 and inorganic particles.112 Natural and engineered proteins113–115 and DNA nanostructures116–119 also form lattices but do not require solid supports because the biopolymers' sophisticated structure enables a lock-and-key-like interaction of the subunits during self-assembly.Among the most prominent proteins capable of self-assembling into planar lattices are bacterial S-layers. These protein arrays constitute the outermost cell-wall coat of many bacteria and all archaea and form, in most cases, by self-assembly of a single protein species.120 Relevant for patterning purposes, protein subunits are arranged in different lattice symmetries (oblique, square or hexagonal) and are separated by inter-subunit distances which vary between 2 and 20 nm depending on the S-layer type.113,121,122 The proteins also self-assemble in vitro into microscale products.123,124 The structures of different lattices have been visualized with AFM and electron microscopy at nanoscale resolution, and, more recently, with X-ray crystallography to atomic resolution (Fig. 3A).125 The availability of the detailed molecular picture will facilitate rational engineering of S-layers but is also an achievement in itself. In particular, the recent X-ray study has overcome the inherent difficulty126,127 of obtaining 3D lattices from protein that naturally assembles into 2D lattices.
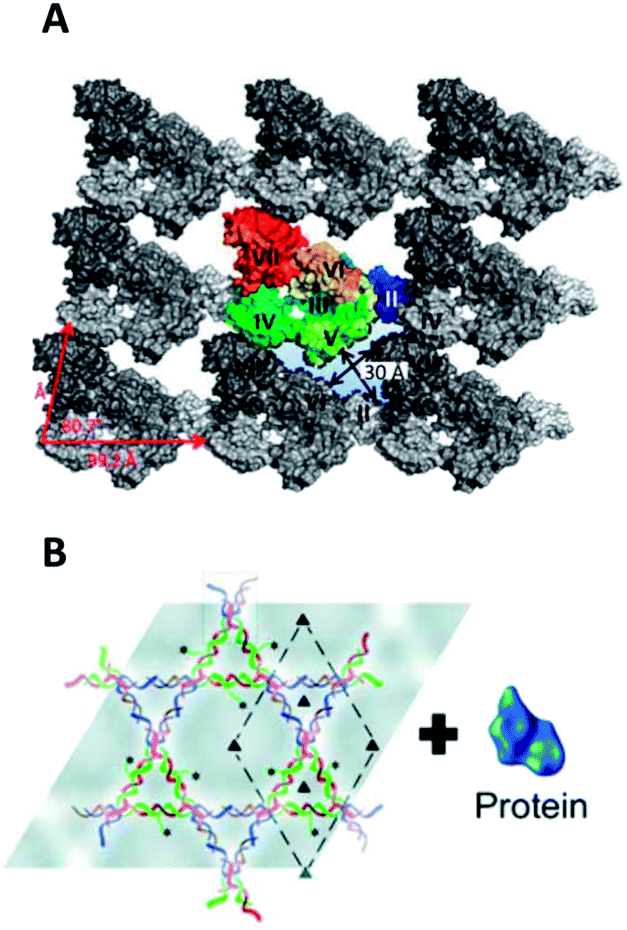 | ||
| Fig. 3 Bottom-up biopolymer assemblies. (A) Molecular model of the S-layer lattice from Geobacillus stearothermophilus obtained by fitting the X-ray structure of an assembly-blocked protein into cryo-EM of the native lattice.125 (B) DNA nanoaffinity templates create dense, nonoverlapping arrays of protein molecules.128 | ||
How can individual molecules be immobilized on S-layers and used for the purpose of single-molecule arrays? The attachment of molecules to the lattices can, similar to the top-down approach, proceed in direct and indirect fashion. In the context of bottom-up, it implies linking the cargo to the S-layer subunits either before or after lattice assembly, respectively.
Given the defined nanoscale and repetitive structure, S-layer lattices can immobilize proteins or vaccine epitopes in high nanoscale density120 thereby mimicking one of their natural functions as immobilization matrices for exoenzymes.129,130 In another application, the single-molecule periodicity of the engineered S-layers was more fully exploited to provide a reference structure to evaluate the resolution of an AFM technique.131,132 Specifically, a lattice displaying peptide tags helped demonstrate an imaging mode where the cognate receptor is attached to the AFM tip. By using read-out via simultaneous topography and recognition imaging, the nanoarray confirmed the validity of the high-resolution approach by yielding maps of the affinity sites with a positional accuracy of 1.5 nm. In a related study, protein affinity arrays were prepared by cloning streptavidin domains.133 While useful, one of the drawbacks of S-layers is their somewhat limited and locked-in lattice periodicity. Consequently, there is interest to obtain biomolecular arrays with tunable spacing. Different types of protein arrays other than the S-layer are available114 but these are similarly restrained in lattice symmetries.
An attractive alternative to protein-based lattices are DNA nanoarrays.134 These nanostructures are more flexible with regard to spacing and geometry. Structurally defined DNA nanoarrays can be generated by sequence-specific self-assembly of component DNA strands. Assembly can be pursued following two main routes that differ with regard to the type of building blocks. In the first case, the smaller units are DNA tiles of about 10 nm size which are assembled from synthetic DNA oligonucleotides. One widely used type of DNA tile contains two parallel aligned duplexes connected by a double-crossover (DX) motif116 but other tiles with alternating geometries exist.116,117 To facilitate assembly into larger structures, DNA tiles can be equipped with DNA overhangs of complementary sequence.135 The resulting pattern can be around hundreds of micrometers large118,135 but are usually highly repetitive.136,137 Indeed, the tiles can be assembled into highly ordered 3D lattices suitable for analysis by X-ray crystallography.138
A second type of DNA assembly can overcome this repetitiveness. Unlike tiles, the so-called origami nanostructures are formed by hybridizing at least one long DNA scaffold strand and several smaller oligonucleotide staple strands.139–141 Benefits of the origami design include a larger unit size of up to 100 nm and the engineering of unique non-repetitive sites which stem from the use of unique DNA sequences. Due to their large size, the origami structures already constitute arrays. However, they can also be used as building blocks to assemble larger lattices, similar to DNA tiles. This can be achieved by connecting origami either via sticky DNA ends,142 complementary concave and convex edges similar to pieces of a jigsaw puzzle,143 or by using a preformed DNA-based scaffold frame onto which smaller origami units can latch.144 The DNA scaffold can also be designed to achieve curved shapes.145
In one variant route, DNA origami-type structures can be assembled without the use of a long DNA scaffold thereby increasing flexibility in design. Rather, the architectures are formed solely from single-stranded DNA molecules. These small bricks have no ordered structure before they are incorporated into the superstructure.146 Nevertheless, hundreds of bricks were incorporated into a 2D lattice comparable in size to that of classical 2D DNA origami.147 The brick-based strategy is also suitable to form 3D architectures148 including membrane-spanning nanopores which require chemically modified DNA strands.149,150
How can individual molecules be attached to DNA arrays? Cargo molecules can be placed at predefined sites via reactive chemical functional or bioaffinity groups. These functional moieties can be incorporated into the lattice by means of chemically modified DNA oligonucleotides that carry thiol, amino, and azide groups, or bioaffinity tags such as biotin or DNA strands. Cargo can be incorporated into the array either prior to or after assembly, corresponding to the direct or indirect route, respectively. The former strategy is ideal for small molecules such as fluorescent dyes, while the latter is preferred for large cargoes which would otherwise interfere with lattice assembly. Indeed, DNA lattices have been used as templates to form arrays of gold nanoparticles,151,152 quantum dots,153 or other nanoparticles,154,155 as well as proteins156–159 and DNA.118,135
What can DNA arrays be used for? Placing individual molecules in regular 2D crystalline lattices has been exploited in structural biology to aid the investigation of a protein's molecular architecture.128 Usually, cryomicroscopy examines non-crystalline protein samples by image reconstruction from noisy snap-shots of individual proteins. The random distribution of the sample is, however, associated with problems in image processing. Positioning thousands of proteins in a defined orientation via a DNA tile template (Fig. 3B) overcomes these hurdles by greatly facilitating data collection, as illustrated with a G-protein-coupled membrane receptor, a soluble G-protein, and their binary complex.128
In other applications, DNA arrays were used as molecular pegboards for placing fewer molecules at pre-defined yet tunable sites and distances. Particularly versatile in this respect are planar DNA origami squares or rectangles of around 100 nm in length, although other shapes are possible.140,160 These platforms have opened up many exciting applications in biophysics, chemistry, fluorescence microscopy, and nanobiotechnology. In biophysics, a flat DNA rectangle was equipped with arrays of two molecular recognition motifs separated by variable distance. This helped probe the spacing-dependent binding of bivalent analytes and shed light on the biophysics of this process.161 In chemistry, diffusion of a reactive oxygen species was studied as a function of the nanoscale distance between a light-activated photosensitizer and corresponding photocleavable moieties suitable for AFM read-out.162 For super-resolution fluorescence microscopy, DNA arrays were equipped with single fluorophores at defined nanoscale distances to serve as reference standards.29 Fluorophore pairs were also used for FRET studies.163 An exciting development is the use of DNA arrays to enhance fluorescence by metallic nanoparticles which set up a plasmonic hotspot of zeptoliter volume. These nanoantennas were prepared by attaching two gold nanoparticles onto a pillar-like DNA origami structure, which also incorporated a docking site for a single fluorescent dye in the gap between the two particles.164 In nanobiotechnology, the placement of individual protein140,165 and DNA molecules onto defined sites of DNA origami has led to artistically ambitious or dynamic nanopatterns. In graphically inspired experiments, individual proteins were used as pixels to draw a nanoscale version of a smiling human face.140,165 By comparison, in a dynamic patterning approach166 single stranded DNA extensions functioned as tracks to guide the movement of individual molecular spiders. The legs of the spider were single stranded DNA which hybridized to the DNA guides. The resulting duplexes could be enzymatically cleaved to release the full-length spider legs to achieve directional movement.167 DNA arrays displaying individual DNA molecules have been exploited for other forms of robotic movement.168,169
Conclusions and prospects
This Highlight has described the exciting interface between single-molecule sensing and biomolecular arrays. This combination of two research areas has led to many new scientific insights and methodological developments. But how widespread is the ultrasensitive analysis of conventional microarrays in research settings? Which role do single-molecule arrays play in the competitive commercial markets of sequencing and diagnostics as well as in academic laboratories?As detailed above, the single-molecule readout of conventional microarrays was pioneered several years ago and yielded tangible analytical benefits in terms of detection limit and dynamic range. But scanning of slides down to the single-molecule level has not been taken up by many other research groups. Possible reasons include the technological lock-in onto conventional array formats which can use glass substrates of several mm thickness. By contrast, the ultrasensitive scanning via wide-field fluorescence microscopy requires glass thicknesses of around 150 μm. Their more wide-spread use requires the development of easy-to-use single-molecule microarray analysis platforms. For hypothesis-free screening on unknown or mutated targets, microarrays are being gradually replaced by modern sequencing techniques.
By comparison, single-molecule arrays have made inroads into sequencing applications. The high through-put for single-molecule analysis has been exploited in DNA-nanoball arrays, zero-mode waveguides, and highly parallel nanopore recordings. Other advantages for sequencing in array format include lowered reagent consumption, as well as reduced and amplification-free processing of samples. The more wide-spread use of these single-molecule techniques is also facilitated by the fact that sequencing technology is usually located in a few sequencing centers and operated by dedicated staff. While the methods are either close to or already commercially available, they face strong competition from other non-single-molecule sequencing techniques.106
Commercial interest into single-molecule arrays also exists in the field of diagnostics. Here, one trend is the use of bead-based arrays in combination with an integrated read-out mode to span the extremes between single molecules and saturating analyte concentrations. For example, a dynamic sensing range of over 6 orders of magnitude was achieved for the diagnostically important prostate-specific antigen.170,171 The impressive results were achieved by capturing the analytes on the beads with a conventional immune-sandwich assay, singulating individual beads within an array of 50 femtoliter wells, and detecting bead-associated enzymatic activity by fluorescence scanning. At lower analyte concentrations, individual molecules were counted while at higher concentrations the fluorescence intensity per bead was averaged.170,171 The spread of single-molecule diagnostics into point-of-care settings will likely be facilitated by several other developments including single-molecule detectors with a small footprint,172 fluorescence-enhancing nanostructured metal surfaces to ease detection with less costly equipment,173 and software to simplify the processing of large single-molecule data sets for improved format standardization, easier dissemination, and transparent analysis.174
Within academic laboratories, single-molecule arrays have been used by many groups for biophysical and, to a lesser extent, for cell biological research. While it is difficult to predict specific topics in future research, existing studies suggest that arrays of single molecules with microscale periodicities can boost cell biological investigations. Once successfully prepared, the proposed arrays could also fill gaps in the range of currently available formats. Possible technological routes towards the micro single-molecule arrays will likely rely on the synergistic combination of top-down patterns of 50 nm feature size with bottom-up atomically precise DNA nanostructures of 50 nm size. For example, the top-down-generated nanopatterned surfaces could be decorated with structurally fine-tuned DNA nanoarchitectures displaying individual molecules. Existing reports have demonstrated the principle of this route175,176 but further research is required to attain the proposed single-molecule arrays.
In conclusion, the ultrasensitive scanning of arrays and the development of single-molecule arrays has had an impact on scanning, sequencing and diagnostics and opened up the opportunity to address many previously unexplored biophysical and biological phenomena.
Acknowledgements
This work has been supported by the Leverhulme Trust (RPG-170), the EPSRC (Institutional Sponsorship Award), UCL Chemistry, the National Physical Laboratory, the Austrian Science Foundation (project P25730-B21), the government of Upper Austria, the European Funds for regional development (EFRE), and the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 278403.Notes and references
- M. Schena, D. Shalon, R. W. Davis and P. O. Brown, Science, 1995, 270, 467–470 CAS.
- S. P. Fodor, R. P. Rava, X. C. Huang, A. C. Pease, C. P. Holmes and C. L. Adams, Nature, 1993, 364, 555–556 CrossRef CAS PubMed.
- G. MacBeath and S. L. Schreiber, Science, 2000, 289, 1760–1763 CAS.
- J. P. Pellois, X. Zhou, O. Srivannavit, T. Zhou, E. Gulari and X. Gao, Nat. Biotechnol., 2002, 20, 922–926 CrossRef CAS PubMed.
- S. Fukui, T. Feizi, C. Galustian, A. M. Lawson and W. Chai, Nat. Biotechnol., 2002, 20, 1011–1017 CrossRef CAS PubMed.
- J. Zlatanova and K. van Holde, Mol. Cell, 2006, 24, 317–329 CrossRef CAS PubMed.
- R. Drmanac, A. B. Sparks, M. J. Callow, A. L. Halpern, N. L. Burns, B. G. Kermani, P. Carnevali, I. Nazarenko, G. B. Nilsen, G. Yeung, F. Dahl, A. Fernandez, B. Staker, K. P. Pant, J. Baccash, A. P. Borcherding, A. Brownley, R. Cedeno, L. Chen, D. Chernikoff, A. Cheung, R. Chirita, B. Curson, J. C. Ebert, C. R. Hacker, R. Hartlage, B. Hauser, S. Huang, Y. Jiang, V. Karpinchyk, M. Koenig, C. Kong, T. Landers, C. Le, J. Liu, C. E. McBride, M. Morenzoni, R. E. Morey, K. Mutch, H. Perazich, K. Perry, B. A. Peters, J. Peterson, C. L. Pethiyagoda, K. Pothuraju, C. Richter, A. M. Rosenbaum, S. Roy, J. Shafto, U. Sharanhovich, K. W. Shannon, C. G. Sheppy, M. Sun, J. V. Thakuria, A. Tran, D. Vu, A. W. Zaranek, X. Wu, S. Drmanac, A. R. Oliphant, W. C. Banyai, B. Martin, D. G. Ballinger, G. M. Church and C. A. Reid, Science, 2010, 327, 78–81 CrossRef CAS PubMed.
- A. Ishijima and T. Yanagida, Trends Biochem. Sci., 2001, 26, 438–444 CrossRef CAS.
- E. A. Josephs and T. Ye, Nano Lett., 2012, 12, 5255–5261 CrossRef CAS PubMed.
- E. A. Josephs and T. Ye, J. Am. Chem. Soc., 2010, 132, 10236–10238 CrossRef CAS PubMed.
- A. Graslund, R. Rigler and J. Widengren, Single Molecule Spectroscopy in Chemistry, Physics, and Biology, Springer, Berlin, 2010 Search PubMed.
- A. W. Peterson, L. K. Wolf and R. M. Georgiadis, J. Am. Chem. Soc., 2002, 124, 14601–14607 CrossRef CAS PubMed.
- J. D. Ding, J. H. Huang, S. V. Grater, F. Corbellinl, S. Rinck, E. Bock, R. Kemkemer, H. Kessler and J. P. Spatz, Nano Lett., 2009, 9, 1111–1116 CrossRef PubMed.
- H. Agheli, J. Malmstrom, E. M. Larrson, M. Textor and D. S. Sutherland, Nano Lett., 2006, 6, 1165–1171 CrossRef CAS PubMed.
- K. L. Christman, V. Vazquez-Dorbatt, E. Schopf, C. M. Kolodziej, R. C. Li, R. M. Broyer, Y. Chen and H. D. Maynard, J. Am. Chem. Soc., 2008, 130, 16585–16591 CrossRef CAS PubMed.
- V. Vogel and M. Sheetz, Nat. Rev. Mol. Cell Biol., 2006, 7, 265–275 CrossRef CAS PubMed.
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433–441 CrossRef CAS PubMed.
- D. Aydin, V. C. Hirschfeld-Warneken, I. Louban and J. P. Spatz, in Intelligent Surfaces in Biotechnology: Scientific and Engineering Concepts, Enabling Technologies, and Translation to Bio-Oriented Applications, ed. H. M. Grandin and M. Textor, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2012, pp. 291–319 Search PubMed.
- R. Schlapak, P. Pammer, D. Armitage, R. Zhu, P. Hinterdorfer, M. Vaupel, T. Fruhwirth and S. Howorka, Langmuir, 2006, 22, 277–285 CrossRef CAS PubMed.
- J. Hesse, M. Sonnleitner, A. Sonnleitner, G. Freudenthaler, J. Jacak, O. Höglinger, H. Schindler and G. J. Schutz, Anal. Chem., 2004, 76, 5960–5964 CrossRef CAS PubMed.
- J. Hesse, J. Jacak, M. Kasper, G. Regl, T. Eichberger, M. Winklmayr, F. Aberger, M. Sonnleitner, R. Schlapak, S. Howorka, L. Muresan, A. M. Frischauf and G. J. Schutz, Genome Res., 2006, 16, 1041–1045 CrossRef CAS PubMed.
- M. Wiesbauer, R. Wollhofen, B. Vasic, K. Schilcher, J. Jacak and T. A. Klar, Nano Lett., 2013, 13, 5672–5678 CrossRef CAS PubMed.
- E. B. Shera, N. K. Seitzinger, L. M. Davis, R. A. Keller and S. A. Soper, Chem. Phys. Lett., 1990, 174, 553–557 CrossRef CAS.
- J. Jacak, H. Schnidar, L. Muresan, G. Regl, A. M. Frischauf, F. Aberger, G. J. Schütz and J. Hesse, J. Biotechnol., 2013, 164, 525–530 CrossRef CAS PubMed.
- R. Lebofsky, R. Heilig, M. Sonnleitner, J. Weissenbach and A. Bensimon, Mol. Biol. Cell, 2006, 17, 5337–5345 CrossRef CAS PubMed.
- R. Schlapak, H. Kinns, C. Wechselberger, J. Hesse and S. Howorka, ChemPhysChem, 2007, 8, 1618–1621 CrossRef CAS PubMed.
- M. J. Levene, J. Korlach, S. W. Turner, M. Foquet, H. G. Craighead and W. W. Webb, Science, 2003, 299, 682–686 CrossRef CAS PubMed.
- J. Eid, A. Fehr, J. Gray, K. Luong, J. Lyle, G. Otto, P. Peluso, D. Rank, P. Baybayan, B. Bettman, A. Bibillo, K. Bjornson, B. Chaudhuri, F. Christians, R. Cicero, S. Clark, R. Dalal, A. Dewinter, J. Dixon, M. Foquet, A. Gaertner, P. Hardenbol, C. Heiner, K. Hester, D. Holden, G. Kearns, X. Kong, R. Kuse, Y. Lacroix, S. Lin, P. Lundquist, C. Ma, P. Marks, M. Maxham, D. Murphy, I. Park, T. Pham, M. Phillips, J. Roy, R. Sebra, G. Shen, J. Sorenson, A. Tomaney, K. Travers, M. Trulson, J. Vieceli, J. Wegener, D. Wu, A. Yang, D. Zaccarin, P. Zhao, F. Zhong, J. Korlach and S. Turner, Science, 2009, 323, 133–138 CrossRef CAS PubMed.
- J. Vogelsang, C. Steinhauer, C. Forthmann, I. H. Stein, B. Person-Skegro, T. Cordes and P. Tinnefeld, ChemPhysChem, 2010, 11, 2475–2490 CrossRef CAS PubMed.
- S. W. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780–782 CrossRef CAS.
- M. Dyba and S. W. Hell, Phys. Rev. Lett., 2002, 88, 163901 CrossRef.
- T. A. Klar, S. Jakobs, M. Dyba, A. Egner and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8206–8210 CrossRef CAS.
- C. Eggeling, C. Ringemann, R. Medda, G. Schwarzmann, K. Sandhoff, S. Polyakova, V. N. Belov, B. Hein, C. von Middendorff, A. Schönle and S. W. Hell, Nature, 2009, 457, 1159–1162 CrossRef CAS PubMed.
- S. T. Hess, T. P. K. Girirajan and M. D. Mason, Biophys. J., 2006, 91, 4258–4272 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and F. H. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- H. Shroff, C. G. Galbraith, J. A. Galbraith and E. Betzig, Nat. Methods, 2008, 5, 417–423 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–795 CrossRef CAS PubMed.
- E. Lubeck and L. Cai, Nat. Methods, 2012, 9, 743–748 CrossRef CAS PubMed.
- B. Huang, W. Wang, M. Bates and X. Zhuang, Science, 2008, 319, 810–813 CrossRef CAS PubMed.
- S. A. Jones, S.-H. Shim, J. He and X. Zhuang, Nat. Methods, 2011, 8, 499–508 CrossRef CAS PubMed.
- M. Heilemann, S. van de Linde, M. Schüttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2008, 47, 6172–6176 CrossRef CAS PubMed.
- R. Wombacher, M. Heidbreder, S. van de Linde, M. P. Sheetz, M. Heilemann, V. W. Cornish and M. Sauer, Nat. Methods, 2010, 7, 717–719 CrossRef CAS PubMed.
- E. Betzig and J. K. Trautman, Science, 1992, 257, 189–195 CAS.
- E. Betzig, A. Lewis, A. Harootunian, M. Isaacson and E. Kratschmer, Biophys. J., 1986, 49, 269–279 CrossRef CAS.
- E. Betzig and R. J. Chichester, Science, 1993, 262, 1422–1425 CAS.
- J. M. Kim, T. Ohtani, J. Y. Park, S. M. Chang and H. Muramatsu, Ultramicroscopy, 2002, 91, 139–149 CrossRef CAS.
- C. Manzo, T. S. van Zanten and M. F. Garcia-Parajo, Biophys. J., 2011, 100, L8–L10 CrossRef CAS PubMed.
- D. R. Walt, Anal. Chem., 2013, 85, 1258–1263 CrossRef CAS PubMed.
- P. Holzmeister, G. P. Acuna, D. Grohmann and P. Tinnefeld, Chem. Soc. Rev., 2014 10.1039/C3CS60207A.
- G. Binnig, C. F. Quate and C. Gerber, Phys. Rev. Lett., 1986, 56, 930–933 CrossRef.
- V. Dupres, F. D. Menozzi, C. Locht, B. H. Clare, N. L. Abbott, S. Cuenot, C. Bompard, D. Raze and Y. F. Dufrene, Nat. Methods, 2005, 2, 515–520 CrossRef CAS PubMed.
- C. Stroh, H. Wang, R. Bash, B. Ashcroft, J. Nelson, H. Gruber, D. Lohr, S. M. Lindsay and P. Hinterdorfer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12503–12507 CrossRef CAS PubMed.
- V. T. Moy, E. L. Florin and H. E. Gaub, Science, 1994, 266, 257–259 CAS.
- P. Hinterdorfer, W. Baumgartner, H. J. Gruber, K. Schilcher and H. Schindler, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 3477–3481 CrossRef CAS.
- A. F. Oberhauser, P. E. Marszalek, H. P. Erickson and J. M. Fernandez, Nature, 1998, 393, 181–185 CrossRef CAS PubMed.
- M. Rief, H. Clausen-Schaumann and H. E. Gaub, Nat. Struct. Biol., 1999, 6, 346–349 CrossRef CAS PubMed.
- R. Merkel, P. Nassoy, A. Leung, K. Ritchie and E. Evans, Nature, 1999, 397, 50–53 CrossRef CAS PubMed.
- F. Oesterhelt, D. Oesterhelt, M. Pfeiffer, A. Engel, H. E. Gaub and D. J. Muller, Science, 2000, 288, 143–146 CrossRef CAS.
- T. Ando, Nanotechnology, 2012, 23, 062001 CrossRef PubMed.
- I. Casuso, F. Rico and S. Scheuring, Curr. Opin. Chem. Biol., 2011, 15, 704–709 CrossRef CAS PubMed.
- K. L. Christman, V. D. Enriquez-Rios and H. D. Maynard, Soft Matter, 2006, 2, 928–939 RSC.
- C. M. Niemeyer, Science, 2002, 297, 62–63 CrossRef CAS PubMed.
- C. M. Niemeyer and C. A. Mirkin, Nanobiotechnology: Concepts, applications and perspectives, John Wiley & Sons, 2004 Search PubMed.
- C. A. Mirkin and C. M. Niemeyer, Nanobiotechnology II: More concepts and applications, John Wiley & Sons, 2007 Search PubMed.
- K. Salaita, Y. Wang, J. Fragala, R. A. Vega, C. Liu and C. A. Mirkin, Angew. Chem., Int. Ed., 2006, 45, 7220–7223 CrossRef CAS PubMed.
- A. Bruckbauer, L. Ying, A. M. Rothery, A. I. Shevchuk, C. Abell, Y. E. Korchev and D. Klenerman, J. Am. Chem. Soc., 2002, 124, 8810–8811 CrossRef CAS PubMed.
- S. K. Kufer, E. M. Puchner, H. Gumpp, T. Liedl and H. E. Gaub, Science, 2008, 319, 594–596 CrossRef CAS PubMed.
- W. T. S. Huck, H. W. Li, B. V. O. Muir and G. Fichet, Langmuir, 2003, 19, 1963–1965 CrossRef.
- H. Schmid and B. Michel, Macromolecules, 2000, 33, 3042–3049 CrossRef CAS.
- P. Pammer, R. Schlapak, M. Sonnleitner, A. Ebner, R. Zhu, P. Hinterdorfer, O. Höglinger, H. G. Schindler and S. Howorka, ChemPhysChem, 2005, 6, 900–903 CrossRef CAS PubMed.
- A. Tinazli, J. Piehler, M. Beuttler, R. Guckenberger and R. Tampe, Nat. Nanotechnol., 2007, 2, 220–225 CrossRef CAS PubMed.
- H. Artelsmair, F. Kienberger, A. Tinazli, R. Schlapak, R. Zhu, J. Preiner, J. Wruss, M. Kastner, N. Saucedo-Zeni, M. Hoelzl, C. Rankl, W. Baumgartner, S. Howorka, D. Blaas, H. Gruber, R. Tampe and P. Hinterdorfer, Small, 2008, 4, 847–854 CrossRef CAS PubMed.
- C. A. Mirkin, D. S. Ginger and H. Zhang, Angew. Chem., Int. Ed., 2004, 43, 30–45 CrossRef PubMed.
- R. Valiokas, T. Rakickas, M. Gavutis, A. Reichel, J. Piehler and B. Liedberg, Nano Lett., 2008, 8, 3369–3375 CrossRef PubMed.
- S. Lenhert, S. Sekula, J. Fuchs, S. Weg-Remers, P. Nagel, S. Schuppler, J. Fragala, N. Theilacker, M. Franueb, C. Wingren, P. Ellmark, C. A. K. Borrebaeck, C. A. Mirkin and H. Fuchs, Small, 2008, 4, 1785–1793 CrossRef PubMed.
- J. Rundqvist, J. H. Hoh and D. B. Haviland, Langmuir, 2006, 22, 5100–5107 CrossRef CAS PubMed.
- R. Michel, I. Reviakine, D. Sutherland, C. Fokas, G. Csucs, G. Danuser, N. D. Spencer and M. Textor, Langmuir, 2002, 18, 8580–8586 CrossRef CAS.
- N. Reynolds, J. D. Tucker, P. A. Davison, J. A. Timney, C. N. Hunter and G. J. Leggett, J. Am. Chem. Soc., 2009, 131, 896–897 CrossRef CAS PubMed.
- S. Lata, A. Reichel, R. Brock, R. Tampe and J. Piehler, J. Am. Chem. Soc., 2005, 127, 10205–10215 CrossRef CAS PubMed.
- R. Schlapak, J. Danzberger, T. Haselgrubler, P. Hinterdorfer, F. Schaffler and S. Howorka, Nano Lett., 2012, 12, 1983–1989 CrossRef CAS PubMed.
- M. Bhagawati, S. Lata, R. Tampé and J. Piehler, J. Am. Chem. Soc., 2010, 132, 5932–5933 CrossRef CAS PubMed.
- C. M. Kolodziej and H. D. Maynard, Chem. Mater., 2012, 24, 774–780 CrossRef CAS.
- V. N. Truskett and M. P. C. Watts, Trends Biotechnol., 2006, 24, 312–317 CrossRef CAS PubMed.
- W. Shim, A. B. Braunschweig, X. Liao, J. Chai, J. K. Lim, G. Zheng and C. A. Mirkin, Nature, 2011, 469, 516–520 CrossRef CAS PubMed.
- S. K. Kufer, M. Strackharn, S. W. Stahl, H. Gumpp, E. M. Puchner and H. E. Gaub, Nat. Nanotechnol., 2009, 4, 45–49 CrossRef CAS PubMed.
- J. Chai, L. S. Wong, L. Giam and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19521–19525 CrossRef CAS PubMed.
- T. Plenat, C. Tardin, P. Rousseau and L. Salome, Nucleic Acids Res., 2012, 40, e89 CrossRef CAS PubMed.
- M. J. Barrett, P. M. Oliver, P. Cheng, D. Cetin and D. Vezenov, Anal. Chem., 2012, 84, 4907–4914 CrossRef CAS PubMed.
- R. Schlapak, J. Danzberger, D. Armitage, D. Morgan, A. Ebner, P. Hinterdorfer, P. Pollheimer, H. J. Gruber, F. Schaffler and S. Howorka, Small, 2012, 8, 89–97 CrossRef CAS PubMed.
- D. Stamou, C. Duschl, E. Delamarche and H. Vogel, Angew. Chem., Int. Ed., 2003, 42, 5580–5583 CrossRef CAS PubMed.
- S. Sakakihara, S. Araki, R. Iino and H. Noji, Lab Chip, 2010, 10, 3355–3362 RSC.
- H. Zhang, S. Nie, C. M. Etson, R. M. Wang and D. R. Walt, Lab Chip, 2012, 12, 2229–2239 RSC.
- Z. Li, R. B. Hayman and D. R. Walt, J. Am. Chem. Soc., 2008, 130, 12622–12623 CrossRef CAS PubMed.
- W. L. Hwang, M. Chen, B. Cronin, M. A. Holden and H. Bayley, J. Am. Chem. Soc., 2008, 130, 5878–5879 CrossRef CAS PubMed.
- M. A. Holden, D. Needham and H. Bayley, J. Am. Chem. Soc., 2007, 129, 8650–8655 CrossRef CAS PubMed.
- G. Maglia, A. J. Heron, W. L. Hwang, M. A. Holden, E. Mikhailova, Q. H. Li, S. Cheley and H. Bayley, Nat. Nanotechnol., 2009, 4, 437–440 CrossRef CAS PubMed.
- J. T. Groves, Curr. Opin. Drug Discovery Dev., 2002, 5, 606–612 CAS.
- T. Lohmuller, L. Iversen, M. Schmidt, C. Rhodes, H. L. Tu, W. C. Lin and J. T. Groves, Nano Lett., 2012, 12, 1717–1721 CrossRef CAS PubMed.
- S. G. Boxer, Curr. Opin. Chem. Biol., 2000, 4, 704–709 CrossRef CAS.
- J. S. Hovis and S. G. Boxer, Langmuir, 2001, 17, 3400–3405 CrossRef CAS.
- F. Roder, O. Birkholz, O. Beutel, D. Paterok and J. Piehler, J. Am. Chem. Soc., 2013, 135, 1189–1192 CrossRef CAS PubMed.
- T. Osaki, H. Suzuki, B. Le Pioufle and S. Takeuchi, Anal. Chem., 2009, 81, 9866–9870 CrossRef CAS PubMed.
- A. Kleefen, D. Pedone, C. Grunwald, R. Wei, M. Firnkes, G. Abstreiter, U. Rant and R. Tampe, Nano Lett., 2010, 10, 5080–5087 CrossRef CAS PubMed.
- G. Baaken, N. Ankri, A. K. Schuler, J. Ruhe and J. C. Behrends, ACS Nano, 2011, 5, 8080–8088 CrossRef CAS PubMed.
- D. Branton, D. W. Deamer, A. Marziali, H. Bayley, S. A. Benner, T. Butler, M. Di Ventra, S. Garaj, A. Hibbs, X. H. Huang, S. B. Jovanovich, P. S. Krstic, S. Lindsay, X. S. S. Ling, C. H. Mastrangelo, A. Meller, J. S. Oliver, Y. V. Pershin, J. M. Ramsey, R. Riehn, G. V. Soni, V. Tabard-Cossa, M. Wanunu, M. Wiggin and J. A. Schloss, Nat. Biotechnol., 2008, 26, 1146–1153 CrossRef CAS PubMed.
- D. H. Stoloff and M. Wanunu, Curr. Opin. Biotechnol., 2013, 24, 699–704 CrossRef CAS PubMed.
- Z. Chen, Y. Jiang, D. R. Dunphy, D. P. Adams, C. Hodges, N. Liu, N. Zhang, G. Xomeritakis, X. Jin, N. R. Aluru, S. J. Gaik, H. W. Hillhouse and C. J. Brinker, Nat. Mater., 2010, 9, 667–675 CrossRef CAS PubMed.
- G. A. Chansin, R. Mulero, J. Hong, M. J. Kim, A. J. DeMello and J. B. Edel, Nano Lett., 2007, 7, 2901–2906 CrossRef CAS PubMed.
- R. dela Torre, J. Larkin, A. Singer and A. Meller, Nanotechnology, 2012, 23, 385308 CrossRef PubMed.
- W. Xu, J. G. Wang, M. F. Jacobsen, M. Mura, M. Yu, R. E. A. Kelly, Q. Q. Meng, E. Laegsgaard, I. Stensgaard, T. R. Linderoth, J. Kjems, L. N. Kantorovich, K. V. Gothelf and F. Besenbacher, Angew. Chem., Int. Ed., 2010, 49, 9373–9377 CrossRef CAS PubMed.
- J. P. Hill, Y. Wakayama, M. Akada and K. Ariga, J. Phys. Chem. C, 2007, 111, 16174–16180 CAS.
- D. W. Schmidtke, Z. R. Taylor, K. Patel, T. G. Spain, J. C. Keay, J. D. Jernigen, E. S. Sanchez, B. P. Grady and M. B. Johnson, Langmuir, 2009, 25, 10932–10938 CrossRef PubMed.
- T. Pavkov-Keller, S. Howorka and W. Keller, in Progress in Molecular Biology and Translational Science: Molecular Assembly in Natural and Engineered Systems, ed. S. Howorka, Academic Press, London, 2011, vol. 103, pp. 73–130 Search PubMed.
- S. Howorka, Curr. Opin. Biotechnol., 2011, 22, 485–491 CrossRef CAS PubMed.
- D. Papapostolou and S. Howorka, Mol. BioSyst., 2009, 5, 723–732 RSC.
- N. C. Seeman, Annu. Rev. Biochem., 2010, 79, 65–87 CrossRef CAS PubMed.
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS PubMed.
- A. M. Hung, H. Noh and J. N. Cha, Nanoscale, 2010, 2, 2530–2537 RSC.
- S. Howorka, Langmuir, 2013, 29, 7344–7353 CrossRef CAS PubMed.
- U. B. Sleytr, M. Sára, D. Pum, B. Schuster, P. Messner and C. Schäffer, in Biopolymers, ed. A. Steinbüchel and S. R. Fahnestock, Wiley-VCH, Weinheim, 2002, vol. 7, pp. 285–338 Search PubMed.
- S. Scheuring, H. Stahlberg, M. Chami, C. Houssin, J. L. Rigaud and A. Engel, Mol. Microbiol., 2002, 44, 675–684 CrossRef CAS.
- A. Ebner, F. Kienberger, C. Huber, A. S. M. Kamruzzahan, V. P. Pastushenko, J. Tang, G. Kada, H. J. Gruber, U. B. Sleytr, M. Sára and P. Hinterdorfer, ChemBioChem, 2006, 7, 588–591 CrossRef CAS PubMed.
- U. B. Sleytr, C. Huber, N. Ilk, D. Pum, B. Schuster and E. M. Egelseer, FEMS Microbiol. Lett., 2007, 267, 131–144 CrossRef CAS.
- U. B. Sleytr, E. Egelseer, D. Pum and B. Schuster, in Nanobiotechnology, ed. C. M. Niemeyer and C. A. Mirkin, Wiley-VCH, Weinheim, 2004, pp. 77–92 Search PubMed.
- E. Baranova, R. Fronzes, A. Garcia-Pino, N. Van Gerven, D. Papapostolou, G. Péhau-Arnaudet, E. Pardon, J. Steyaert, S. Howorka and H. Remaut, Nature, 2012, 487, 119–122 CAS.
- T. Pavkov, E. M. Egelseer, M. Tesarz, D. I. Svergun, U. B. Sleytr and W. Keller, Structure, 2008, 16, 1226–1237 CrossRef CAS PubMed.
- R. P. Fagan, O. Qazi, D. I. Svergun, K. A. Brown and N. Fairweather, Mol. Microbiol., 2009, 71, 1308–1322 CrossRef CAS PubMed.
- D. N. Selmi, R. J. Adamson, H. Attrill, A. D. Goddard, R. J. Gilbert, A. Watts and A. J. Turberfield, Nano Lett., 2011, 11, 657–660 CrossRef CAS PubMed.
- E. Egelseer, I. Schocher, M. Sára and U. B. Sleytr, J. Bacteriol., 1995, 177, 1444–1451 CAS.
- M. J. Ford, J. F. Nomellini and J. Smit, J. Bacteriol., 2007, 189, 2226–2237 CrossRef CAS PubMed.
- J. L. Tang, A. Ebner, H. Badelt-Lichtblau, C. Vollenkle, C. Rankl, B. Kraxberger, M. Leitner, L. Wildling, H. J. Gruber, U. B. Sleytr, N. Ilk and P. Hinterdorfer, Nano Lett., 2008, 8, 4312–4319 CrossRef CAS PubMed.
- J. Tang, A. Ebner, N. Ilk, H. Lichtblau, C. Huber, R. Zhu, D. Pum, M. Leitner, V. Pastushenko, H. J. Gruber, U. B. Sleytr and P. Hinterdorfer, Langmuir, 2008, 24, 1324–1329 CrossRef CAS PubMed.
- D. Moll, C. Huber, B. Schlegel, D. Pum, U. B. Sleytr and M. Sára, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14646–14651 CrossRef CAS PubMed.
- N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS.
- K. Fujibayashi, R. Hariadi, S. H. Park, E. Winfree and S. Murata, Nano Lett., 2008, 8, 1791–1797 CrossRef CAS PubMed.
- S. H. Park, C. Pistol, S. J. Ahn, J. H. Reif, A. R. Lebeck, C. Dwyer and T. H. LaBean, Angew. Chem., Int. Ed., 2006, 43, 735–739 CrossRef PubMed.
- W. Liu, H. Zhong, R. Wang and N. C. Seeman, Angew. Chem., Int. Ed., 2011, 50, 246–247 Search PubMed.
- J. Zheng, J. J. Birktoft, Y. Chen, T. Wang, R. Sha, P. E. Constantinou, S. L. Ginell, C. Mao and N. C. Seeman, Nature, 2009, 461, 74–77 CrossRef CAS PubMed.
- P. W. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- B. Sacca and C. M. Niemeyer, Angew. Chem., Int. Ed., 2012, 51, 58–66 CrossRef CAS PubMed.
- T. Tørring, N. V. Voigt, J. Nangreave, H. Yan and K. V. Gothelf, Chem. Soc. Rev., 2011, 40, 5636–5646 RSC.
- N. Stephanopoulos, M. H. Liu, G. J. Tong, Z. Li, Y. Liu, H. Yan and M. B. Francis, Nano Lett., 2010, 10, 2714–2720 CrossRef CAS PubMed.
- M. Endo, T. Sugita, Y. Katsuda, K. Hidaka and H. Sugiyama, Chem.–Eur. J., 2010, 16, 5362–5368 CrossRef CAS PubMed.
- Z. Zhao, Y. Liu and H. Yan, Nano Lett., 2011, 11, 2997–3002 CrossRef CAS PubMed.
- H. Dietz, S. M. Douglas and W. M. Shih, Science, 2009, 325, 725–730 CrossRef CAS PubMed.
- P. Yin, R. F. Hariadi, S. Sahu, H. M. Choi, S. H. Park, T. H. Labean and J. H. Reif, Science, 2008, 321, 824–826 CrossRef CAS PubMed.
- B. Wei, M. Dai and P. Yin, Nature, 2012, 485, 623–626 CrossRef CAS PubMed.
- Y. Ke, L. L. Ong, W. M. Shih and P. Yin, Science, 2012, 338, 1177–1183 CrossRef CAS PubMed.
- J. R. Burns, K. Göpfrich, J. W. Wood, V. V. Thacker, E. Stulz, U. F. Keyser and S. Howorka, Angew. Chem., Int. Ed., 2013, 52, 12069–12072 CrossRef CAS PubMed.
- J. Burns, E. Stulz and S. Howorka, Nano Lett., 2013, 13, 2351–2356 CrossRef CAS PubMed.
- J. D. Le, Y. Pinto, N. C. Seeman, K. Musier-Forsyth, T. A. Taton and R. A. Kiehl, Nano Lett., 2004, 4, 2343–2347 CrossRef CAS.
- J. P. Zhang, Y. Liu, Y. G. Ke and H. Yan, Nano Lett., 2006, 6, 248–251 CrossRef CAS PubMed.
- J. Sharma, Y. G. Ke, C. X. Lin, R. Chhabra, Q. B. Wang, J. Nangreave, Y. Liu and H. Yan, Angew. Chem., Int. Ed., 2008, 47, 5157–5159 CrossRef CAS PubMed.
- Y. Y. Pinto, J. D. Le, N. C. Seeman, K. Musier-Forsyth, T. A. Taton and R. A. Kiehl, Nano Lett., 2005, 5, 2399–2402 CrossRef CAS PubMed.
- J. W. Zheng, P. E. Constantinou, C. Micheel, A. P. Alivisatos, R. A. Kiehl and N. C. Seeman, Nano Lett., 2006, 6, 1502–1504 CrossRef CAS PubMed.
- S. H. Park, P. Yin, Y. Liu, J. H. Reif, T. H. LaBean and H. Yan, Nano Lett., 2005, 5, 729–733 CrossRef CAS PubMed.
- Y. He, Y. Tian, A. E. Ribbe and C. D. Mao, J. Am. Chem. Soc., 2006, 128, 12664–12665 CrossRef CAS PubMed.
- R. Chhabra, J. Sharma, Y. G. Ke, Y. Liu, S. Rinker, S. Lindsay and H. Yan, J. Am. Chem. Soc., 2007, 129, 10304–10305 CrossRef CAS PubMed.
- B. A. R. Williams, K. Lund, Y. Liu, H. Yan and J. C. Chaput, Angew. Chem., Int. Ed., 2007, 46, 3051–3054 CrossRef CAS PubMed.
- B. Sacca, R. Meyer, M. Erkelenz, K. Kiko, A. Arndt, H. Schroeder, K. S. Rabe and C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 9378–9383 CrossRef CAS PubMed.
- S. Rinker, Y. G. Ke, Y. Liu, R. Chhabra and H. Yan, Nat. Nanotechnol., 2008, 3, 418–422 CrossRef CAS PubMed.
- S. Helmig, A. Rotaru, D. Arian, L. Kovbasyuk, J. Arnbjerg, P. R. Ogilby, J. Kjems, A. Mokhir, F. Besenbacher and K. V. Gothelf, ACS Nano, 2010, 4, 7475–7480 CrossRef CAS PubMed.
- I. H. Stein, C. Steinhauer and P. Tinnefeld, J. Am. Chem. Soc., 2011, 133, 4193–4195 CrossRef CAS PubMed.
- G. P. Acuna, F. M. Moller, P. Holzmeister, S. Beater, B. Lalkens and P. Tinnefeld, Science, 2012, 338, 506–510 CrossRef CAS PubMed.
- W. Q. Shen, H. Zhong, D. Neff and M. L. Norton, J. Am. Chem. Soc., 2009, 131, 6660–6661 CrossRef CAS PubMed.
- H. Z. Gu, J. Chao, S. J. Xiao and N. C. Seeman, Nat. Nanotechnol., 2009, 4, 245–248 CrossRef CAS PubMed.
- K. Lund, A. J. Manzo, N. Dabby, N. Michelotti, A. Johnson-Buck, J. Nangreave, S. Taylor, R. J. Pei, M. N. Stojanovic, N. G. Walter, E. Winfree and H. Yan, Nature, 2010, 465, 206–210 CrossRef CAS PubMed.
- S. F. Wickham, J. Bath, Y. Katsuda, M. Endo, K. Hidaka, H. Sugiyama and A. J. Turberfield, Nat. Nanotechnol., 2012, 7, 169–173 CrossRef CAS PubMed.
- S. F. Wickham, M. Endo, Y. Katsuda, K. Hidaka, J. Bath, H. Sugiyama and A. J. Turberfield, Nat. Nanotechnol., 2011, 6, 166–169 CrossRef CAS PubMed.
- D. H. Wilson, D. W. Hanlon, G. K. Provuncher, L. Chang, L. Song, P. P. Patel, E. P. Ferrell, H. Lepor, A. W. Partin, D. W. Chan, L. J. Sokoll, C. D. Cheli, R. P. Thiel, D. R. Fournier and D. C. Duffy, Clin. Chem., 2011, 57, 1712–1721 CAS.
- D. M. Rissin, D. R. Fournier, T. Piech, C. W. Kan, T. G. Campbell, L. Song, L. Chang, A. J. Rivnak, P. P. Patel, G. K. Provuncher, E. P. Ferrell, S. C. Howes, B. A. Pink, K. A. Minnehan, D. H. Wilson and D. C. Duffy, Anal. Chem., 2011, 83, 2279–2285 CrossRef CAS PubMed.
- J. M. Emory, Z. Peng, B. Young, M. L. Hupert, A. Rousselet, D. Patterson, B. Ellison and S. A. Soper, Analyst, 2012, 137, 87–97 RSC.
- W. J. Cho, Y. Kim and J. K. Kim, ACS Nano, 2012, 6, 249–255 CrossRef CAS PubMed.
- M. Greenfeld, D. S. Pavlichin, H. Mabuchi and D. Herschlag, PLoS One, 2012, 7, e30024 CAS.
- R. J. Kershner, L. D. Bozano, C. M. Micheel, A. M. Hung, A. R. Fornof, J. N. Cha, C. T. Rettner, M. Bersani, J. Frommer, P. W. K. Rothemund and G. M. Wallraff, Nat. Nanotechnol., 2009, 4, 557–561 CrossRef CAS PubMed.
- A. M. Hung, C. M. Micheel, L. D. Bozano, L. W. Osterbur, G. M. Wallraff and J. N. Cha, Nat. Nanotechnol., 2010, 5, 121–126 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |