Alignment of nematic and bundled semiflexible polymers in cell-sized confinement†
José
Alvarado‡
,
Bela M.
Mulder
and
Gijsje H.
Koenderink
*
FOM Institute AMOLF, Science Park 104, 1098 XG Amsterdam, Netherlands. E-mail: g.koenderink@amolf.nl
First published on 20th December 2013
Abstract
The finite size of cells poses severe spatial constraints on the network of semiflexible filaments called the cytoskeleton, a main determinant of cell shape. At the same time, the high packing density of cytoskeletal filaments poses mutual packing constraints. Here we investigate the competition between excluded volume interactions in the bulk and surface packing constraints on the orientational ordering of confined actin filaments as a function of filament density and the presence of crosslinks. We grow fluorescently labeled actin filaments in shallow (thickness dz 3 μm), rectangular microchambers with a systematically varied length (dy between 5 and 100 μm) and in-plane aspect ratio (dx/dy between 1 and 10). We determine the nematic director field by image analysis of fluorescence confocal images. We find that high-density (nematic) solutions respond sensitively to changes in the size and aspect ratio of the chambers. In small chambers (dy ≤ 20 μm), filaments align parallel to the long walls as soon as the aspect ratio is ≥1.5, indicating that surface-induced ordering dominates. In larger chambers, the filaments instead align along the chamber diagonal, indicating that bulk packing constraints dominate. The nematic order parameter is maximal in small and highly anisometric chambers. In contrast to the nematic solutions, low-density (isotropic) solutions are rather insensitive to confinement. Bundled actin solutions behave similarly to nematic solutions, but are less well-ordered. Our observations imply that the orientational order of actin filaments in flat confining geometries is primarily determined by a balance between bulk and surface packing constraints with a minimal effect of the enthalpic cost of filament bending. Our assay provides an interesting platform for the future reconstitution of more complex, active cytoskeletal systems with actively treadmilling filaments or molecular motors.
Introduction
The mechanical properties of eukaryotic cells are largely determined by the cytoskeleton, a meshwork of filamentous proteins with unique mechanical properties.1 Three cytoskeletal components, microtubules, actin and intermediate filaments, are known to significantly contribute to cell stiffness, cell shape and internal organization.2,3 As cytoskeletal filaments can reach lengths in excess of 10 μm, the finite size of cells (in the range of ∼10 μm for yeast cells to ∼50 μm for plant and animal cells) poses spatial constraints on their organization. In plants, the cytoplasm, and hence the cytoskeleton, is confined to a thin layer pressed between the rigid cellulose cell wall and the vacuole, which occupies 90% of the cell volume.4 In animal cells, the cytoplasm is generally more three-dimensional, but anchoring to the plasma membrane often results in a thin actin cytoskeletal cortex.5,6 The plasma membrane of animal cells is in principle soft and deformable, but still the cell shape in many tissues is strongly constrained, either due to the anchoring of a rigid pericellular coat on the membrane exterior,7,8 or due to close packing of cells within epithelial layers.9,10Recent biophysical research has begun to address the effect of spatial confinement on intracellular organization by culturing individual cells or confluent cell monolayers on micropatterned substrates.11,12 External confinement has been shown to strongly affect the spatial organization of the cytokinetic contractile ring,13 mitotic spindle,14 and nucleus,15 and thereby strongly affect cell fate.16 In addition to global cell shape confinement, a variety of thin cellular extensions also locally constrain the cytoskeleton. On flat substrates and in some tissues, cells migrate using a protrusive flat sheet-like array of actively treadmilling actin filaments called the lamellipodium.17–19 Cells also extend actin filaments and microtubules into linear protrusions, such as filopodia,20–23 stereocilia,24–26 flagella,27,28 and neurite processes.29 A mechanistic understanding of the effect of geometrical confinement on the spatial organization of cytoskeletal filaments remains elusive.
In vitro experiments with reconstituted cytoskeletal polymer solutions provide a convenient way to study the role of confinement in cytoskeletal organization in the absence of complicating factors such as active processes and biochemical regulation. In dilute suspensions of microtubules, confinement to small geometries with rigid boundaries forces filaments to align along the walls to minimize both entropy loss due to excluded volume and bending energy.30 Narrow channels have been shown to stretch filaments of actin,31–33 intermediate filaments,34 and DNA35 by constraining their thermal bending undulations. Specific geometries of actin or microtubule filament nucleation imposed by micropatterned substrates can result in spontaneous bundle formation in the presence of depletion agents.36,37
In addition to external confining geometries, mutual packing interactions between semiflexible filaments at high packing densities also strongly affect the organization of polymer solutions. For rigid filaments with only steric interactions, the lowest free energy state corresponds to a situation where the entropy is maximized. At low densities, the entropy is maximized by an isotropic distribution of rod orientations that maximizes the orientational entropy. However, above a critical packing density, filaments spontaneously align to reduce their mutual excluded volume and form a nematic liquid-crystalline phase. Seminal theoretical work by Onsager showed that the critical density depends solely on the aspect ratio of filaments if the filaments are rigid and interact exclusively by steric repulsion,38 although both filament flexibility39–41 and length polydispersity41,42 are predicted to increase the critical density. Onsager's theory and modifications thereof have been highly successful in predicting the phase behavior of rod-shaped filamentous viruses like tobacco mosaic virus43 and fd-virus44 as well as actin filaments45,46 and microtubules.47 For instance, reducing actin filament length with the capping protein gelsolin has been shown to increase the critical density of nematic liquid crystalline ordering in close agreement with the Onsager theory.48–52
Until now, the combination of packing constraints arising both from mutual filament interactions and external confinement has been little explored. Entangled filamentous (F-)actin solutions confined in spherical emulsion droplets or vesicles spontaneously form peripheral shells when the confinement diameter is less than ∼10–15 μm.53,54 This behavior indicates that the enthalpy cost associated with filament bending becomes more important than the orientational entropy term once the confinement size approaches the persistence length of the actin filaments. Confinement of entangled F-actin solutions in microchambers was shown to cause spontaneous nematic alignment even at densities below the bulk isotropic–nematic (Onsager) transition.55
In contrast to in vitro systems, living cells do not appear to exhibit solutions of cytoskeletal filaments in a nematic phase despite the rather high concentrations of actin in the cytoplasm.56 Rather, cells actively regulate filament organization by orchestrating a wide array of accessory proteins that specifically interact with actin filaments to produce (often transient) networks, bundles, and specialized structures such as the mitotic spindle or actin contractile ring.57,58 Bundling likely acts together with spatial confinement to form functional cell structures such as filopodia.20,59In vitro, the combination of packing constraints from cross-linker-mediated interactions and external confinement has again been little explored. In reconstituted actin networks confined to microchambers or emulsion droplets, interesting organized patterns of bundles were observed when the solvent was allowed to slowly evaporate.60–62 In narrow microchannels, actin bundles are efficiently aligned along the channel.33
Here we seek to quantify the effect of spatial confinement on the spatial organization of semiflexible polymer solutions that form either isotropic, nematic, or bundled phases in bulk solution. To that end we create external confining geometries by fabricating shallow, square microchambers with a systematically varied size and in-plane aspect ratio. Inside these chambers, we grow actin filaments at two different concentrations that in bulk solution would lead to an isotropic or a nematic phase. Moreover, we grow solutions of actin filaments bundled by various agents in the microchambers. We visualize these solutions by fluorescence confocal microscopy and quantify the spatial patterns of filament orientations using a pixel-based method related to previous methods that quantified filament orientation in the lamellipodium of migrating cells.63,64 We find that high-density (nematic) solutions respond sensitively to changes in the size and aspect ratio of the chambers, whereas low-density (isotropic) solutions are rather insensitive to the confinement. Bundled actin solutions behave similarly to nematic solutions, but are less well-ordered. We interpret our findings in terms of a competition of the energy costs associated with bulk nematic deformation and misalignment with the confining walls. Overall, our results show that external boundaries with even slight anisometries can strongly affect the orientation of semiflexible filament solutions. This finding is likely relevant in many different cellular contexts as well as outside biology, for soft matter systems such as wormlike micelles65,66 and carbon nanotubes.67
Results
In order to quantify the effect of confinement on the spatial organization of semiflexible filament solutions, we prepare microchambers with a well-controlled rectangular geometry (Fig. 1a; see Methods). Chambers were designed with lengths dy in the range 5–100 μm and widths dx such that the aspect ratio dx/dy varied in the range 1–10 (Fig. 1b). The chambers are shallow with a constant thickness dz of only 3 μm. Inside these chambers, we grow solutions of fluorescently labeled actin filaments under various conditions. When polymerized in bulk solution, the filaments have an approximately exponential length distribution with a mean length of 13 ± 5 μm (ref. 68) of the same order as the in-plane size of the chambers and much longer than the chamber depth, creating a quasi-2D confinement. We visualize the confined solutions by fluorescence confocal microscopy. For solutions grown at high-density (4 mg mL−1), we observe fine aligned textures indicating nematic alignment (Fig. 1c). Due to the small mesh size (∼150 nm) and small diameter of the filaments (∼7 nm), we are unable to resolve and track individual actin filaments. Instead, we quantify the mean orientation of filaments in each image pixel, which measures the local orientation θ of the nematic director. We investigate the effect of chamber dimensions on the director field based on the anisotropy of the fluorescence intensity of each image pixel (see detailed description in the ESI†). From these orientation measurements, we determine the preferred orientation, 〈θ〉, and the order-parameter, S, of the nematic director from the second-order tensor order-parameter S2 (see Methods). | ||
| Fig. 1 Experimental model system of actin filament solutions confined to microchambers. (a) Snapshot of the chip patterned with microchambers, acquired by scanning electron microscopy. Scale bar 500 μm. (b) Close up of the area in the black box in panel a (scale bar 50 μm). White arrows denote the chamber length dy and width, dx, from which the chamber aspect ratio follows as dx/dy. The chamber height is always 3 μm. (c) Confocal fluorescence image of the square microchamber (dy = 50 μm, dx/dy = 1) filled with a suspension of fluorescently labeled actin filaments prepared at [actin] = 4 mg mL−1, a concentration at which the solution forms a nematic phase in the bulk. Note that there is diagonal filament alignment in the center of the chamber. Scale bar 10 μm. | ||
Confined high-density (nematic) solutions of actin filaments
To investigate the competing effects of bulk liquid crystalline ordering and wall-induced ordering on the orientational order of actin filaments, we grow actin filaments at a concentration of 4 mg mL−1 in cell-sized microchambers (Fig. 2a). At this high concentration, bulk solutions of actin filaments self-organize to form a nematic liquid crystal.50,45 In square geometries (aspect ratio dx/dy = 1) with length dy = 30 μm, we observe clear alignment of the filaments with a preferred orientation along the chamber diagonal (±45°). This is evident in Fig. 2b (bottom-left), where green pixels in the interior of the chamber report orientations around −45°. (Due to chamber symmetry, +45° and −45° are equivalent.) As the chamber aspect ratio increases to 10 while the length dy remains fixed at 30 μm, the orientation angle of the pixels gradually decreases. This is evident in a shift from green pixels to cyan pixels, which shows orientations approaching 0° (where 0° corresponds to the long axis of the chambers oriented along the x-axis). For the highest aspect ratio chambers, the average angle is close to the angle of 5° corresponding to the chamber diagonal. | ||
| Fig. 2 High-density solutions of actin filaments confined to microchambers, prepared at [actin] = 4 mg mL−1, a concentration at which the solution forms a nematic phase in the bulk. Chamber length dy = 15 μm. (a) Typical images of actin filaments in microchambers with varying aspect ratios dx/dy (outlined text) ranging from 1 (square) to 10 (long rectangle). Scale bar 20 μm. (b) Maps of the orientations of fluorescence intensity in each pixel. Color corresponds to orientation θ of fluorescence intensity distribution (see calibration wheel, right). Note the gradual change from green (diagonal) to cyan (near-horizontal) as the aspect ratio increases. Black lines separate the chamber interior from the periphery. Pixels at the periphery are dominated by edge effects related to the sudden decrease in fluorescence intensity at the chamber boundaries and are hence excluded from the analysis. (c) Probability distribution function of pixel orientations θ for all chambers investigated with length dy = 30 μm. Color corresponds to different aspect ratios (cf. panel a). Note that the example chambers shown in panels a and b agree well with the orientation probability distributions for chambers with dy = 30 μm rather than dy = 15 μm (see Fig. S1†). | ||
In order to quantify the effect of chamber geometry on the degree of nematic order and on the preferred orientation of the nematic director, we plot histograms of orientation measurements for different chamber aspect ratios (Fig. 2c). In all cases, we observe a peaked distribution, indicative of nematic order. The orientation distribution of actin solutions in square chambers (aspect ratio 1, light blue curve) is peaked around −45°. As the chamber aspect ratio increases, the peaks shift towards values close to 5°. This shift is also evident in Fig. 3a (lower panel), where the preferred orientation 〈θ〉 of the nematic director is plotted against chamber aspect ratio for all chambers investigated (black dots). The mean value of 〈θ〉 averaged over all chambers for each condition (black horizontal lines) clearly decreases with increasing chamber aspect ratio. Furthermore, these mean values agree well with the angle formed between the diagonals of the chamber with the long (x-)axis (green squares). We also note that in chambers with small aspect ratios (1 and 1.5), there is a rather wide spread of 〈θ〉-values across the chambers, whereas in more elongated chambers the distribution of 〈θ〉-values sharpens. We find similar behavior for chambers with larger sizes of 50 or 70 μm (see ESI Fig. S1 and S2†). However, in the larger chambers, the spread of 〈θ〉-values across the chambers is substantially larger than in the 30 μm chambers, even at large chamber aspect ratio.
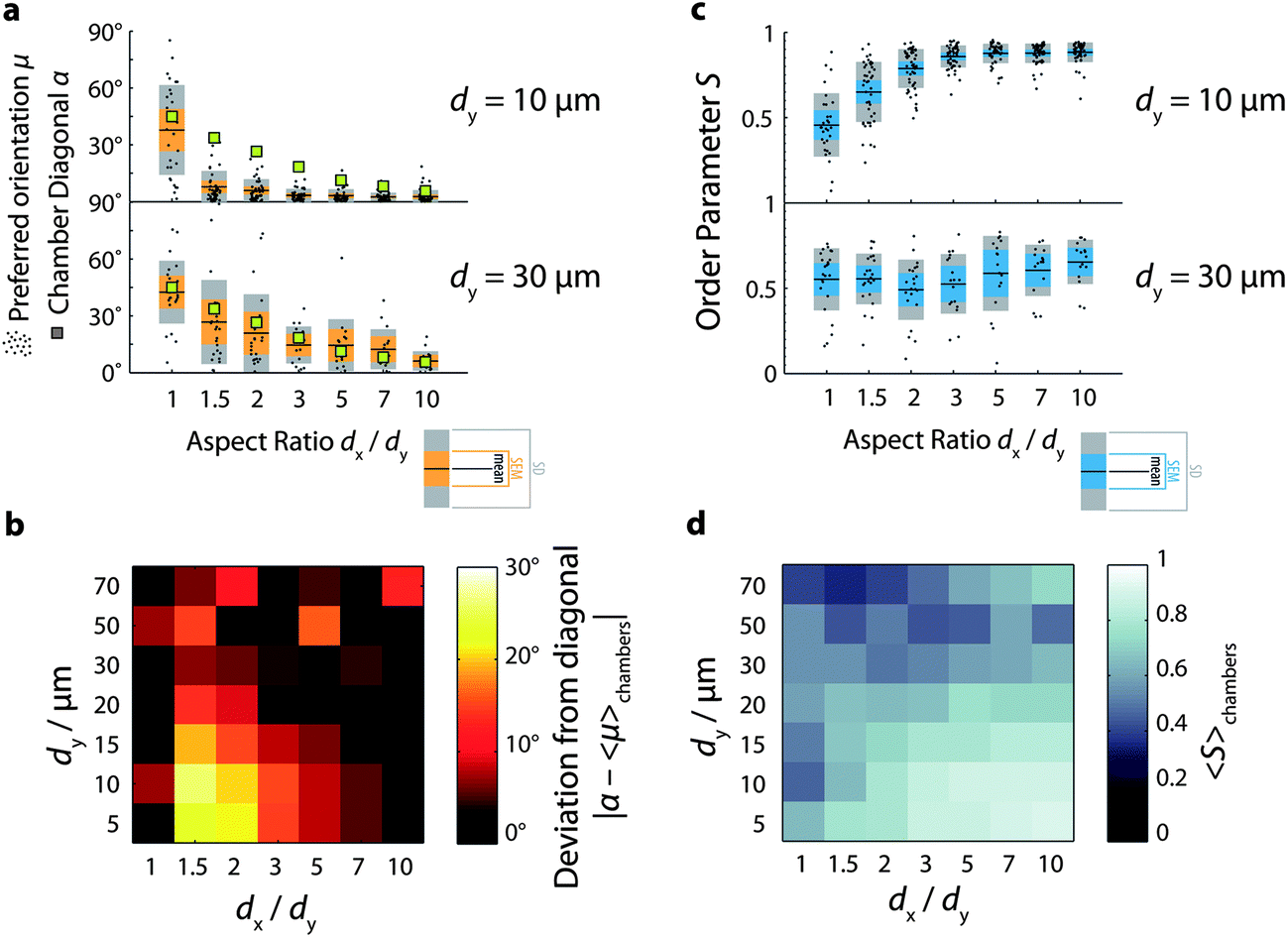 | ||
| Fig. 3 Orientational order of confined nematic actin solutions. (a) Preferred orientation μ = 〈θ〉 as a function of chamber aspect ratio for two given chamber lengths (10 and 30 μm). Black dots denote preferred orientation μ of a single chamber. Black lines denote mean preferred orientation averaged across all chambers. Orange bars denote standard error of the mean (SEM). Gray bars denote standard deviation (SD). Green squares denote the angle of the two diagonals of the chamber, given by arctan(dy/dx). (b) Color map of the deviation of the mean preferred orientation from the chamber diagonal for all aspect ratios and chamber lengths. Brighter shades denote greater deviation (see the calibration bar, right). (c) Order parameter S as a function of chamber aspect ratio. Black dots denote order parameter S of a single chamber. Black lines denote the mean order parameter averaged across all chambers. Cyan bars denote SEM. Gray bars denote SD. (d) Color map of the mean order parameter for all aspect ratios and chamber lengths. Brighter shades denote the greater order parameter (see calibration bar, right). | ||
We conclude that in chambers with sizes between 30 and 70 μm, the filaments preferentially align along the longest available dimension of the chamber for all chamber aspect ratios in the range of 1 to 10. There are two ways to interpret this finding. The first interpretation is that minimization of the free energy is dominated by a competition between the energy cost associated with nematic deformation in the bulk and that associated with rod misalignment with the walls. Liquid crystal continuum theory for rods confined in square geometries predicts that rods will align along the diagonal when the cost of bulk nematic deformation dominates, whereas they will align along the walls when the cost of misalignment with the wall dominates.69 For vertically vibrated granular rods confined to quasi-2D containers, both extreme patterns could be observed by systematically varying the rod packing density and aspect ratio.69 In the case of nematic actin solutions, the relevant elastic constants (here, in the quasi 2D setting primarily the ones for splay and bend) are not well characterized,70 hampering an a priori prediction of the correlation length that governs the influence of wall induced order on the bulk. An alternative interpretation is that the preferential diagonal ordering may minimize the energy cost associated with filament bending. Indeed, one expects an important enthalpic contribution to the free energy based on the measured length of the filaments when polymerized in bulk solution, where the mean length is 13 μm. However, based on previous experiments on actin solutions confined in spherical emulsion droplets and vesicles53,54 we expect that enthalpic costs would promote peripheral accumulation rather than diagonal alignment. Strikingly, the actin solutions in the microchambers are homogeneous for all chamber aspect ratios, with no sign of peripheral accumulation. Even in the smallest chambers we investigated, with a length of only 5 μm, the F-actin solutions are homogeneous. Given that the filaments are polymerized in situ within the chambers, it is possible that confinement effects cause the filaments to be shorter compared to filaments grown at the same density in bulk solution.71 Alternatively, the energy costs associated with deformation of the bulk nematic may dominate the total free energy.
If rod orientational order is governed by a competition of bulk nematic ordering and wall-induced ordering, we expect that stronger confinement should shift the balance to wall-induced ordering. To test this hypothesis, we observe actin solutions in smaller chambers. In line with our hypothesis, we observe that decreasing the chamber length to dy = 20 μm or less changes the orientational ordering of the filaments (Fig. S1 and S2†). Fig. 3a (upper panel) illustrates this for chambers with dy = 10 μm: in square chambers, the mean preferred orientation (black horizontal lines) is still close to the diagonal, but as soon as the aspect ratio is increased to 1.5, the mean preferred orientation drops down to values close to zero, corresponding to alignment along the longest walls of the chambers. For all aspect ratios between 1.5 and 10, the mean preferred orientation remains close to zero, and is significantly different from the expected diagonal orientation (green squares). Similar to the larger chambers, however, the distribution of mean angles across chambers is broad in square chambers, but increasingly narrow as the chamber aspect ratio increases.
Fig. 3b summarizes the deviation between the measured orientation (averaged over all chambers for each condition) and the diagonal orientation expected if the energy cost of bulk nematic deformation dominates, as shown in the form of a color map for all chamber sizes and aspect ratios. The deviation is maximal in the lower-left hand corner, where the aspect ratio dx/dy ≥ 1.5 and the chamber length dy ≤ 20 μm. For small chamber lengths (dy ≤ 20 μm), the deviation decreases systematically as the aspect ratio increases. In contrast, for large chamber lengths (dy ≥ 30 μm), the deviation does not systematically vary with aspect ratio, indicating a minimal effect of confinement on the orientation of the network. We conclude that in small rectangular chambers (≤20 μm) surface effects favoring filament alignment parallel to the longest walls dominate as soon as the chamber is slightly anisometric. In intermediate-sized chambers (30 and 50 μm), bulk packing effects favoring diagonal orientation appear to dominate, and in the largest chambers (≥70 μm) we find nematic alignment, but with a highly variable preferred orientation across different chambers.
In small chambers, the nematic order parameter depends strongly on chamber aspect ratio: it increases from an average value of around 0.5 in square chambers to limiting values close to 0.9 in anisometric chambers (Fig. 3c, upper panel). Interestingly, a modest increase of the aspect ratio from 1 to 1.5 already results in a measurable increase of the order parameter, indicating that filament alignment is very sensitive to the imposed confinement. In larger chambers (dy ≥ 30 μm), the order parameter is smaller (fluctuating between 0.5 and 0.6) and does not show a systematic dependence on chamber aspect ratio, as illustrated in Fig. 3c, lower panel for dy = 30 μm. We furthermore observe that the spread in S-values across chambers is substantially smaller in small (dy ≤ 20 μm) chambers than in larger chambers (Fig. S3†). The color map in Fig. 3d summarizing the average order parameter measured for all chamber sizes and aspect ratios clearly shows that orientational order is maximal in small (dy ≤ 10 μm) and anisometric chambers.
Confined low-density (isotropic) solutions of actin filaments
To determine whether solutions of semiflexible polymers lacking nematic order also respond to cell-sized confinement, we grow low-density (0.5 mg mL−1) suspensions of actin filaments in the microchambers (Fig. 4a). At these low concentrations, actin filaments form isotropic solutions in the bulk. We find that these dilute actin solutions do not seem to exhibit a strongly preferred orientation, as evident from Fig. 4b, where a wide variety of colors represents a wide distribution of orientations. The orientation distributions are indeed broad, relative to nematic chambers. This is particularly the case for low aspect ratio chambers, where the distributions do not exhibit a clear peak (Fig. 4c and S4†). However, for increasing aspect ratios ≥2, the distributions do develop a more distinct peak around 0°, indicating that confinement does promote increased orientational order, even in low-density solutions. Similar to the high-density solutions, the low-density actin solutions are homogeneous, with no signs of peripheral filament accumulation, suggesting that enthalpic energy costs associated with filament bending do not play a major role.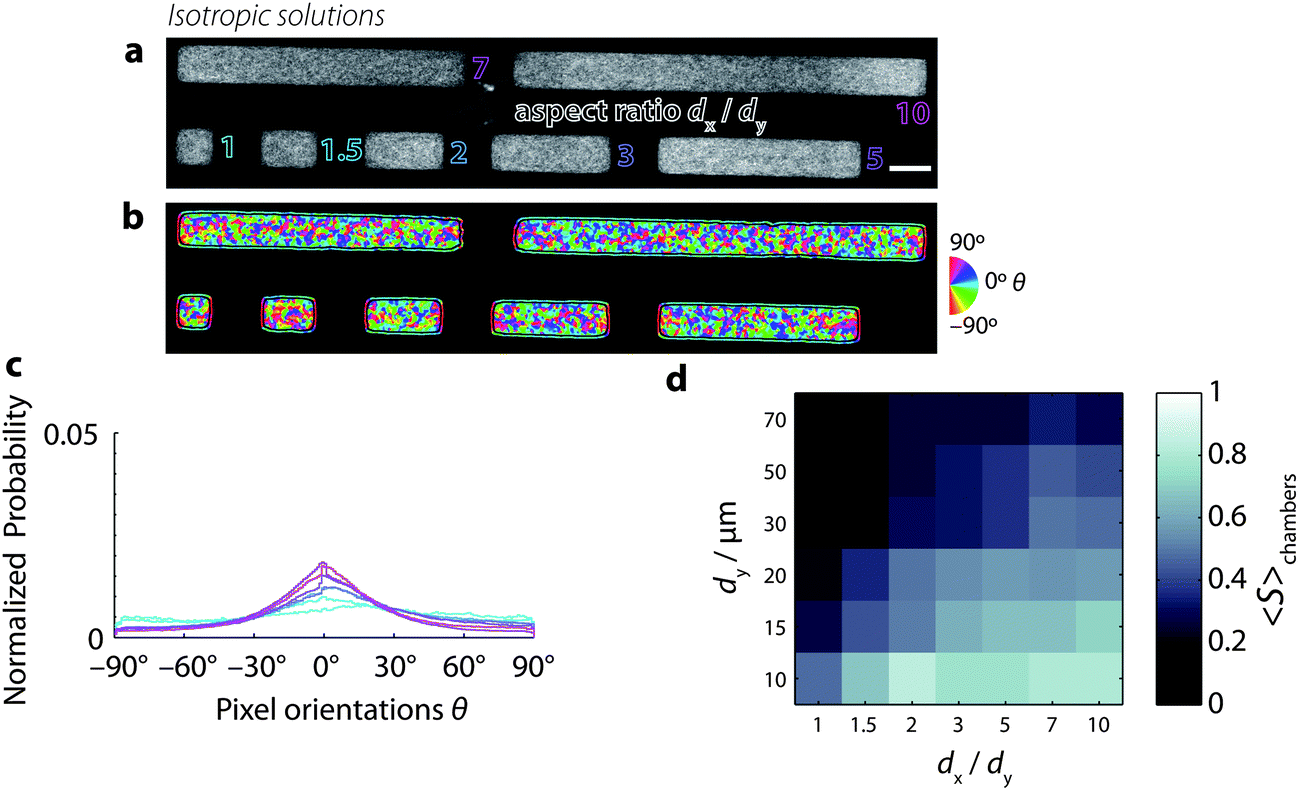 | ||
| Fig. 4 Low-density solutions of actin filaments confined to microchambers, prepared at [actin] = 0.5 mg mL−1, a concentration where the solutions are isotropic in the bulk. Chamber length dy = 20 μm. (a) Snapshots of actin filaments in microchambers with varying aspect ratios dx/dy (outlined text) ranging from 1 (square) to 10 (long rectangle). Scale bar 20 μm. (b) Orientation of fluorescence intensity. Color denotes orientations of fluorescence intensity in each pixel (see the calibration wheel, right). Black lines separate the chamber interior from the periphery. Pixels at the periphery are dominated by edge effects related to the sudden decrease in fluorescence intensity at the chamber boundaries and are hence excluded from analysis. (c) Probability distribution function of pixel orientations θ for the seven different regions of interest from panel b. Color corresponds to different aspect ratios (cf. panel a). (d) Color map of the mean order parameter for all aspect ratios and chamber lengths, as in Fig. 3d. | ||
Fig. 4d shows a color map summarizing the order parameter S (in each case averaged across many chambers) as a function of chamber length and aspect ratio. We find that the order parameter depends on chamber aspect ratio similarly to nematic chambers. S is maximal for small, long chambers. However, the values that S attains for low-density solutions are lower than in the nematic case. These lower order parameter values reflect the wide distribution of orientations found in low-density solutions, both within chambers (cf.Fig. 4b) as well as across chambers (Fig. S5 and S6†).
Confined solutions of bundled actin filaments
In order to investigate the combined effect of crosslink-induced organization and confinement-induced organization, we grow bundles of actin filaments in the microchambers. We prepare bundles of actin using two different techniques.First, we prepare bundles of actin by polymerizing the actin filaments at a concentration of 0.5 mg mL−1 in the presence of 20 mM magnesium chloride (MgCl2). The positive, divalent magnesium ions accumulate around the negatively charged actin filaments to form a cloud of counterions; above a critical concentration of counterions, filaments share counterion clouds, which establishes attractive interactions between filaments and drives bundling72 and raft formation.73Fig. 5a shows example images of actin bundles confined to microchambers, and Fig. 5b shows the corresponding bundle orientations. We see that in square chambers, some bundles align mostly to the chamber diagonal at approximately +45° (dark blue-violet pixels). However, other bundles are oriented in different directions, as indicated by the wide variety of colors found inside chambers. In particular, bundles that are perpendicular to the longest chamber walls can be found in most chambers (90°, red pixels). However, as the chamber aspect ratio increases, bundles that align closer to 0° qualitatively appear longer and occur more frequently than bundles aligned along 90°.
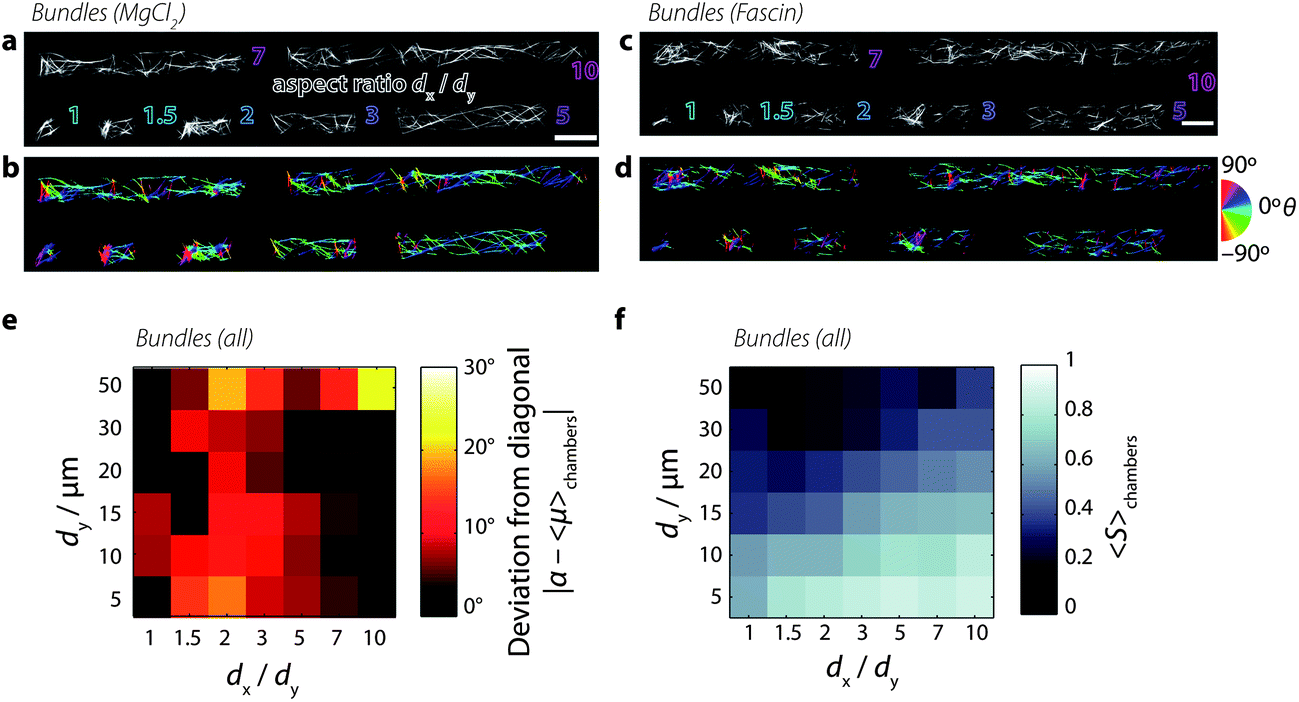 | ||
| Fig. 5 Solutions of bundled actin filaments confined to microchambers. [actin] = 0.5 mg mL−1. (a) Snapshots of actin filaments in microchambers, bundled by 20 mM magnesium chloride. Outlined text denotes chamber aspect ratios dx/dy from 1 (square) to 10 (long rectangle). Scale bar 20 μm. (b) Maps of the orientations of fluorescence intensity in each pixel. Color corresponds to orientation θ of fluorescence intensity distribution (see the calibration wheel, right). Orientation image was masked by an Otsu threshold of panel a. (c) Snapshots of actin filaments in microchambers, bundled by 1.2 μM fascin. (d) Orientation image of panel c. (e) Color map of the deviation of the mean preferred orientation from the chamber diagonal, as in Fig. 3b. (f) Color map of the mean order parameter, as in Fig. 3d. | ||
We obtain similar results when bundling actin filaments with the physiological crosslink protein fascin. Fascin is enriched in cellular structures called filopodia:21 linear extensions that protrude the plasma membrane in migrating cells. Fascin proteins simultaneously bind two actin filaments via two actin-binding sites,74 forming bundles with a well-controlled maximum number of 19 filaments.75 We find that fascin bundles formed by polymerizing actin filaments at 0.5 mg mL−1 in the presence of 1.2 μM fascin crosslinks organize similarly to the magnesium-chloride bundles (Fig. 5c and d): as the chamber aspect ratio increases, long bundles aligned along 0° dominate.
The preferred bundle orientations show a similar dependence on chamber dimension as for the orientation of nematic liquid crystalline phases of the non-bundled actin solutions (Fig. 5e). The deviation of preferred orientation in the lower-left corner, however, reaches only ∼10° (red values in the color map, Fig. 5e) as opposed to 30° (yellow values, cf.Fig. 3b) for nematic solutions. As shown in Fig. 5f, the order parameter qualitatively shows a similar dependence on chamber dimensions as seen for nematic solutions, being maximal in small chambers with high aspect ratios. This observation is consistent with prior observations showing that actin filaments bundled by α-actinin confined to narrow microchannels align along the channel.33 The maximal values of S for the bundled solutions are ∼0.8, which is slightly lower than those attained by nematic solutions, which gave ∼0.9. The lowest values of S attained by bundled solutions in large chambers (dy ≥ 50 μm) are around ∼0.2, which is lower than nematic solutions which only decrease to ∼0.4. Additionally, there is a larger variability in the preferred orientation (Fig. S8†) and in the nematic order parameter (Fig. S9†) in bundled solutions compared to nematic solutions. These observations indicate a lower degree of order and responsiveness to confinement for bundled solutions.
Discussion
We have shown that an external confining geometry can strongly influence the spatial organization of semiflexible polymer solutions when the confinement dimensions are comparable to the persistence and contour lengths of the filaments. High-density (nematic) solutions align parallel to the long walls of anisometric microchambers when chamber lengths are small (dy ≤ 20 μm) and anisometric (aspect ratio dx/dy ≥ 1.5), whereas they align along the chamber diagonal when the chambers are intermediate in length (dy ≥ 30 μm). In large chambers, nematic solutions exhibit a wide distribution of orientations. In square chambers, we find a large variability in the preferred orientation for all chamber sizes and aspect ratios. Furthermore, the nematic order parameter in small chambers increases strongly when the aspect ratio is increased from 1 to 10, whereas the nematic order parameter in large chambers does not depend on chamber aspect ratio. Low-density (isotropic) solutions appear to respond similarly to high-density (nematic) solutions to the length scale of confinement. But the nematic order parameter attains lower values of S. These lower values of dilute solutions compared to nematic solutions can be a concentration effect, but it is also possible that a difference in filament length distribution contributes as well. The fact that high order parameters were measured for low-density solutions suggests the possibility that confinement can induce some alignment, even in isotropic solutions. However, two observations suggest that this alignment may be weak: first, S does not depend systematically on chamber size for low-density solutions (while high-density solutions exhibit a relatively smoothly increasing S as aspect ratio increases and chamber length decreases); and second, the spread in S-values across identical chambers is large for low-density solutions (compared to high-density solutions).Taken together, our observations suggest that the orientational order of actin filaments in rectangular microchambers is determined by a balance between the free energy cost associated with deformation of the bulk nematic and that associated with filament misalignment with the walls. In small anisometric chambers, surface-induced ordering dominates, causing the filaments to align along the longest walls, whereas in larger chambers bulk liquid crystalline ordering dominates, causing the filaments to align along the chamber diagonal. In the largest chambers (dy ≥ 70 μm), we find a large variability in preferred orientations across chambers, indicating that confinement has a minimal effect. For the actin solutions employed here (with a concentration of 4 mg mL−1 and an average length of 13 μm when polymerized in bulk solution) the crossover confinement size L* between bulk-dominated and surface-dominated orientational ordering is around 20 μm, which is of the same order as the persistence length of F-actin. Given that the elastic constants associated with liquid crystalline phases of actin filaments are currently unknown, it is difficult to predict a priori the expected crossover size. The experimental data presented here in combination with liquid crystal theory may potentially give insight into these currently unknown elastic constants.69
Intriguingly, both the low-density and high-density actin solutions were spatially homogeneous in terms of their density for all chamber sizes (down to 5 μm) and all aspect ratios, with no evidence of edge accumulation. This observation is consistent with our earlier observations of actin solutions in shallow microchambers of varying shapes.55 These observations suggest that enthalpic energy costs associated with filament bending play a minor role for actin filaments confined in shallow microchambers, since this energy term is expected to cause peripheral accumulation of actin filaments. Prior studies of low-density (isotropic) actin solutions confined in spherical vesicles54 and emulsion droplets53 revealed a spontaneous formation of a dense cortical layer when the confinement diameter was less than 10–15 μm, which coincides with the persistence length of actin filaments.76 It is possible that the quasi-2D confinement influences actin polymerization proceeding in the shallow microchambers (which have a depth of 3 μm), such that the filaments polymerized in microchambers are shorter than expected from measurements of filament length in bulk solution. Unfortunately, it is extremely challenging to measure filament length distributions in situ. Overcoming this experimental challenge would allow us to better understand the results presented here, as well as in our previous work.55 An alternative avenue could be to perform numerical calculations of actin polymerization in confining geometries, which has been performed for dynamic microtubules.77
Since actin filaments in living cells are often assembled into bundles by a variety of actin-binding proteins, we also investigated the combined effect of crosslinks and confinement on filament organization. We find that actin bundles formed either by divalent cations (20 mM Mg2+) or by the physiological bundling protein fascin (1.2 μM) respond to rectangular confinement similarly to nematic actin solutions. As the chamber aspect ratio increases, long bundles that align along 0° dominate. However, the maximal values of S for the bundled solutions are around 0.8, which is slightly lower than those attained by nematic solutions. Indeed, the bundles exhibit a wider distribution of orientations, with a distinct population of short bundles perpendicular to the long axis of rectangular chambers. The bundle patterns may represent equilibrium structures resulting from a balance of boundary effects and bulk linker-assisted filament aggregation,78,79 but may also be influenced by kinetic trapping, depending on the balance of the kinetics of actin filament polymerization and bundling.61,80–82
Our assay provides an interesting platform for the future reconstitution of more complex, physiologically relevant cytoskeletal systems. One particularly interesting question to address is how dynamic actin filaments that are able to treadmill will respond to confinement. In our experiments, actin filaments were prevented from treadmilling by stabilization with the toxin phalloidin. Recent simulations of dynamic microtubules confined to the cortex of dividing plant cells show that the microtubules spontaneously reorient due to an interplay between growth dynamics and imposed boundary conditions.83 Our model system provides a way to test such predictions experimentally using a well-controlled system. Furthermore, it would be interesting to add molecular motors to the confined actin-filament solutions. Recent studies of nematic solutions of microtubules mixed with kinesin motors showed intriguing patterning and internal flows upon confinement in emulsion droplets.84
To quantify the degree of order in the actin chambers, we introduced a method based on conventional fluorescence microscopy imaging combined with automated image analysis to determine the local anisotropy for each image pixel. We measured typical values of the order parameter S of 0.5–0.6, which are comparable to the range of values (0.4–0.75) measured previously by optical birefringence and X-ray scattering46 and single filament dynamics85 for bulk actin nematics. Similar values were also measured for bulk nematic suspensions of tobacco-mosaic virus,86 bacteriophage fd,87 and microtubules.88 Interestingly, we also measured significantly higher values of close to 0.9 for actin solutions confined in small (dy ≤ 10 μm) chambers with large aspect ratios, suggesting that confinement strengthens nematic ordering. However, we should note that the order parameter we measured here is not necessarily equivalent to the order parameter measured by birefringence, scattering, or polarized fluorescence techniques, which are sensitive to the differences in filament orientation on microscopic length scales.89,49 In contrast, the measurements of the angle θ reported here were determined by computing the average orientation of a window of 3 pixels radius around each pixel, corresponding to a window of approximately 1 μm in diameter. Therefore, our order parameter S does not directly report the microscopic arrangements of filaments. Rather, it measures the variance of measurements of θ across all pixels investigated.
An important advantage of our analysis over conventional techniques used to measure nematic order in liquid crystalline systems is that it can be generally applied to confocal microscopy images of any filamentous solution, including in vitro cytoskeletal solutions and also solutions of synthetic fibers like carbon nanotubes.66,90 Thus, alignment and nematic ordering can be determined without a need for sophisticated techniques such as the PolScope52,89,91 or polarized fluorescence,49 which may be especially difficult to combine with confinement in microchambers, emulsion droplets, or giant unilamellar vesicles. Importantly, our analysis is applicable to a broad range of different images, being able to extract orientation information even when the length scales of the filaments or the filament mesh size are below the diffraction limit. Our analysis could be applied also to images of the cytoskeleton in cells. This would for instance be interesting in the context of the orientation distribution of actin filaments in the lamellipodium of migrating cells, which protrude by directed polymerization of a branched actin solution,19,63,92,93 where our analysis could provide information complementary to more labor-intensive filament reconstruction of electron tomography data.64,94–97
Methods
Protein preparation
Lyophilized monomeric G-actin was purchased from Cytoskeleton (via Tebu-Bio). Resuspended G-actin was stored in G-buffer (2 mM Tris, 0.2 mM sodium adenosine triphosphate, 0.2 mM calcium chloride, 0.2 mM dithiothreitol, pH 8.0), stored at 0 °C, and used within one week. Actin monomers were labeled with Alexa Fluor 594 (Invitrogen) and mixed with unlabeled monomers to yield a 10% molar ratio of dye to protein. Recombinant mouse fascin was prepared from T7 pGEX E. coli, as described elsewhere.98 The fascin plasmid was a kind gift from Scott Hansen and R. Dyche Mullins (UC, San Francisco). Protein concentrations were determined by measuring the solution absorbance at 280 nm with a NanoDrop 2000 (ThermoScientific, Wilmington, DE, USA), using extinction coefficients, in M−1 cm−1, of 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 (actin99), and 66280 (fascin, computed from the amino acid sequence100).
600 (actin99), and 66280 (fascin, computed from the amino acid sequence100).
Microchamber preparation
Microchambers were assembled using a standard photolithographic technique.101 In short, glass cover slips were spin-coated with a layer of photoresist and exposed to UV light patterned by a customized mask. Before spin-coating, cover slips (thickness #1, Menzel Gläser) were cleaned with base piranha (water, 30% ammonium hydroxide, 30% hydrogen peroxide at a ratio of 5:1:1; 75 °C for 15 min), followed by rinsing with water and baking (200 °C, 5 min). Cover slips were coated (Delta 80 GYRSET, SUSS MicroTec; spin speed 3000 rpm) with a negative photoresist (SU-8 2005, MicroChem). Layer thickness was adjusted by diluting photoresist with cyclopentanone by approximately 10–20% and measured using a profilometer (Alpha-Step 500, KLA-Tencor). Coated cover slips were then baked (95 °C, 5 min) before exposure to ultraviolet light (only wavelengths above 365 nm, BG-12 bandpass filter, Schott) in a mask aligner (MJB, Karl Süss; typical dosage: 50–100 mJ cm−2). Patterning was achieved through a custom-designed mask of chromium features printed on soda-lime glass (DeltaMask). Mask design included square and rectangular geometries with various dimensions. Chamber lengths dy were 3, 5, 10, 15, 20, 30, 50, 70, and 100 μm. For each length dy, various aspect ratios dx/dy were designed, ranging from 1, 1.5, 2, 3, 5, 7, and 10. This results in 56 possible geometries. However, an upper bound of resolution in the range of ∼1–3 μm prevents proper formation of the smallest and thinnest patterns. Exposed cover slips were baked (95 °C, 5 min), developed (2-methoxy-1-methylethyl acetate, MicroChem; 1–2 min), rinsed with isopropanol, and ultimately hard-baked (150 °C, 2 h). This process results in a glass substrate with photoresist microchambers. Next, lids were created by coating microscope slides (Menzel Gläser) with a layer (∼1 mm thick) of polydimethylsiloxane rubber (Sylgard 184, Dow Corning; 10:1 base–curing-agent w/w ratio; 120 °C, 5 min). Rubber-coated glass was rendered hydrophilic by corona discharge (BD-20V high-frequency generator, Electro-Technic Products) and soaked overnight in G-buffer + 0.1% amphiphilic block copolymer Pluronic F-127 (Sigma-Aldrich) to block nonspecific adsorption of actin filaments to the surface of the chambers. Saturation of the PDMS with buffer prevented drying of the sample.
Confinement assay
To polymerize actin filaments, we added a solution containing salts and buffer to a tube containing monomeric G-actin (10% label-to-monomer molar ratio). Samples were mixed to yield a final buffer composition of 20 mM imidazole pH 7.4, 50 mM potassium chloride, 1 mM dithiothreitol, and 0.1 mM adenosine triphosphate. In addition, 1 mM trolox, 2 mM protocatechuic acid, and 0.1 μM protocatechuate 3,4-dioxygenase were added to minimize photobleaching. Furthermore, actin filaments were stabilized with phalloidin in all cases. Freshly mixed samples were immediately mixed with phalloidin (in an amount equimolar to actin monomers) dried on a patterned glass slide, and pressed against the rubber-coated glass to form closed microchambers. After hermetically sealing glass edges with VALAP (equal parts vaseline, lanolin, and paraffin wax), samples were left to equilibrate for 30 min and subsequently visualized by fluorescence microscopy. Only chambers that were well-sealed were considered, amounting to approximately 40% of all chambers. Chambers that were not well-sealed were evident by the presence of fluorescently labeled actin between chambers at the interface between the patterned glass and rubber lid.Fluorescence microscopy
Microchambers were visualized with a confocal point scanner (Nikon) on an inverted microscope (Ti, Nikon) with a photomultiplier tube detector (A1, Nikon). Labeled actin filaments were excited with 561 nm laser light (Coherent). Images were acquired over several fields of view which were automatically acquired and stitched (NIS Elements, Nikon). The orientations θ of all image pixels were determined by image analysis with the freely available plugin OrientationJ (http://bigwww.epfl.ch/demo/orientation/), using a Gaussian window with radius σ = 3 px (see ESI† and ref. 55). The orientation distributions serve as input for calculating the order parameter and preferred orientation.Scalar order parameter
Given a collection of orientation measurements θ in the range (−90°, 90°), we first compute the second-order tensor order-parameter S2:102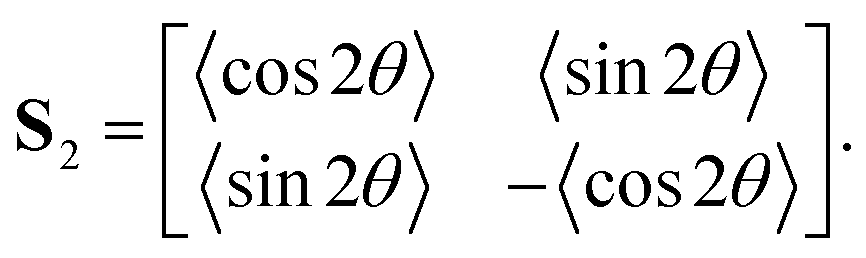
Angle brackets 〈 〉 denote averages over all measurements. The tensor S2 is symmetric and traceless. Solving the eigenvalue problem for S2 yields two eigenvalues:
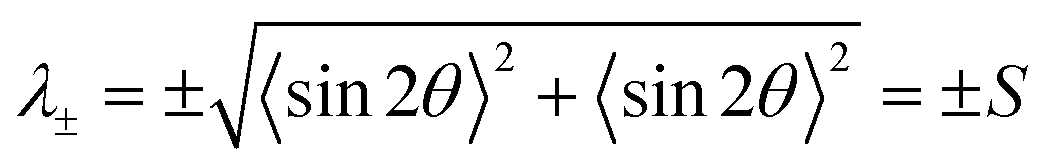
where e+,x and e+,y denote the x- and y-components of the eigenvector e+.
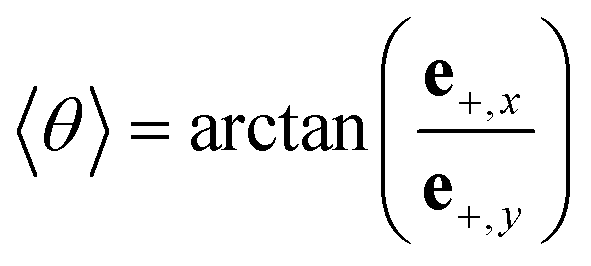
Acknowledgements
We thank Ioana Garlea (AMOLF, Amsterdam) for insightful discussions, Daniel Sage (EPFL, Lausanne) for OrientationJ software, Florian Huber (AMOLF, Amsterdam) for critical reading of the manuscript, and Marjolein Kuit-Vinkenoog (AMOLF, Amsterdam) for assistance with actin purification.References
- C. Storm, J. J. Pastore, F. C. MacKintosh, T. C. Lubensky and P. A. Janmey, Nature, 2005, 435, 191–194 CrossRef CAS PubMed.
- E. Paluch and C.-P. Heisenberg, Curr. Biol., 2009, 19, R790–R799 CrossRef CAS PubMed.
- R. D. Mullins, Cold Spring Harbor Perspect. Biol., 2010, 2, a003392 Search PubMed.
- B. E. S. Gunning and M. W. Steer, Plant Cell Biology, Jones & Bartlett Learning, 1996 Search PubMed.
- G. Salbreux, G. Charras and E. Paluch, Trends Cell Biol., 2012, 22, 536–545 CrossRef CAS PubMed.
- A. G. Clark, K. Dierkes and E. K. Paluch, Biophys. J., 2013, 105, 570–580 CrossRef CAS PubMed.
- M. Tanaka, F. Rehfeldt, M. F. Schneider, G. Mathe, A. Albersdörfer, K. R. Neumaier, O. Purrucker and E. Sackmann, J. Phys.: Condens. Matter, 2005, 17, S649–S663 CrossRef CAS.
- N. Nijenhuis, D. Mizuno, J. A. E. Spaan and C. F. Schmidt, Phys. Biol., 2009, 6, 025014 CrossRef PubMed.
- R. Fernandez-Gonzalez, S. de M. Simoes, J.-C. Röper, S. Eaton and J. A. Zallen, Dev. Cell, 2009, 17, 736–743 CrossRef CAS PubMed.
- C. Guillot and T. Lecuit, Science, 2013, 340, 1185–1189 CrossRef CAS PubMed.
- T. Vignaud, L. Blanchoin and M. Théry, Trends Cell Biol., 2012, 22, 671–682 CrossRef CAS PubMed.
- S. R. K. Vedula, M. C. Leong, T. L. Lai, P. Hersen, A. J. Kabla, C. T. Lim and B. Ladoux, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12974–12979 CrossRef CAS PubMed.
- M. Théry, V. Racine, A. Pépin, M. Piel, Y. Chen, J.-B. Sibarita and M. Bornens, Nat. Cell Biol., 2005, 7, 947–953 CrossRef PubMed.
- J. Fink, N. Carpi, T. Betz, A. Bétard, M. Chebah, A. Azioune, M. Bornens, C. Sykes, L. Fetler, D. Cuvelier and M. Piel, Nat. Cell Biol., 2011, 13, 771–778 CrossRef CAS PubMed.
- M. Versaevel, T. Grevesse and S. Gabriele, Nat. Commun., 2012, 3, 671 CrossRef PubMed.
- C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Science, 1997, 276, 1425–1428 CrossRef CAS.
- J. V. Small, M. Herzog and K. Anderson, J. Cell Biol., 1995, 129, 1275–1286 CrossRef CAS.
- A. B. Verkhovsky, O. Y. Chaga, S. Schaub, T. M. Svitkina, J.-J. Meister and G. G. Borisy, Mol. Biol. Cell, 2003, 14, 4667–4675 CrossRef CAS PubMed.
- A. Mogilner and G. Oster, Biophys. J., 2003, 84, 1591–1605 CrossRef CAS.
- A. Mogilner and B. Rubinstein, Biophys. J., 2005, 89, 782–795 CrossRef CAS PubMed.
- T. M. Svitkina, E. A. Bulanova, O. Y. Chaga, D. M. Vignjevic, S.-I. Kojima, J. M. Vasiliev and G. G. Borisy, J. Cell Biol., 2003, 160, 409–421 CrossRef CAS PubMed.
- P. J. Bryant, Curr. Biol., 1999, 9, R655–R657 CrossRef CAS.
- R. W. Davenport, P. Dou, V. Rehder and S. B. Kater, Nature, 1993, 361, 721–724 CrossRef CAS PubMed.
- M. S. Tilney, L. G. Tilney, R. E. Stephens, C. Merte, D. Drenckhahn, D. A. Cotanche and A. Bretscher, J. Cell Biol., 1989, 109, 1711–1723 CrossRef CAS.
- L. Zheng, G. Sekerková, K. Vranich, L. G. Tilney, E. Mugnaini and J. R. Bartles, Cell, 2000, 102, 377–385 CrossRef CAS.
- U. Manor and B. Kachar, Semin. Cell Dev. Biol., 2008, 19, 502–510 CrossRef PubMed.
- D. J. Luck, J. Cell Biol., 1984, 98, 789–794 CrossRef CAS.
- D. Nicastro, C. Schwartz, J. Pierson, R. Gaudette, M. E. Porter and J. R. McIntosh, Science, 2006, 313, 944–948 CrossRef CAS PubMed.
- L. Dehmelt and S. Halpain, Cytoskeleton, 2003, 58, 18–33 Search PubMed.
- M. Cosentino Lagomarsino, C. Tanase, J. W. Vos, A. M. C. Emons, B. M. Mulder and M. Dogterom, Biophys. J., 2007, 92, 1046–1057 CrossRef PubMed.
- S. Köster, D. Steinhauser and T. Pfohl, J. Phys.: Condens. Matter, 2005, 17, S4091–S4104 CrossRef.
- M. Choi, C. Santangelo, O. Pelletier, J. Kim, S. Kwon, Z. Wen, Y. Li, P. Pincus, C. Safinya and M. Kim, Macromolecules, 2005, 38, 9882–9884 CrossRef CAS.
- L. S. Hirst, E. R. Parker, Z. Abu-Samah, Y. Li, R. Pynn, N. C. MacDonald and C. R. Safinya, Langmuir, 2005, 21, 3910–3914 CrossRef CAS PubMed.
- B. Noding and S. Köster, Phys. Rev. Lett., 2012, 108, 088101 CrossRef.
- J. O. Tegenfeldt, C. Prinz, H. Cao, S. Chou, W. W. Reisner, R. Riehn, Y. M. Wang, E. C. Cox, J. C. Sturm, P. Silberzan and R. H. Austin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10979–10983 CrossRef CAS PubMed.
- A.-C. Reymann, J.-L. Martiel, T. Cambier, L. Blanchoin, R. Boujemaa-Paterski and M. Théry, Nat. Mater., 2010, 9, 827–832 CrossRef CAS PubMed.
- D. Portran, J. Gaillard, M. Vantard and M. Théry, Cytoskeleton, 2013, 70, 12–23 CrossRef CAS PubMed.
- L. Onsager, Ann. N. Y. Acad. Sci., 1949, 51, 627–659 CrossRef CAS.
- A. R. Khokhlov and A. N. Semenov, Physica A, 1982, 112, 605–614 CrossRef.
- Z. Y. Chen, Macromolecules, 1993, 26, 3419–3423 CrossRef CAS.
- T. Odijk, Macromolecules, 1986, 19, 2313–2329 CrossRef CAS.
- P. J. Flory and A. Abe, Macromolecules, 1978, 11, 1119–1122 CrossRef CAS.
- F. C. Bawden, N. W. Pirie, J. D. Bernal and I. Fankuchen, Nature, 1936, 138, 1051–1052 CrossRef.
- Z. Dogic and S. Fraden, Curr. Opin. Colloid Interface Sci., 2006, 11, 47–55 CrossRef CAS PubMed.
- J. Käs, H. Strey, J. X. Tang, D. Finger, R. Ezzell, E. Sackmann and P. A. Janmey, Biophys. J., 1996, 70, 609–625 CrossRef.
- E. Helfer, P. Panine, M.-F. Carlier and P. Davidson, Biophys. J., 2005, 89, 543–553 CrossRef CAS PubMed.
- A. L. Hitt, A. R. Cross and R. C. Williams, J. Biol. Chem., 1990, 265, 1639–1647 CAS.
- A. Suzuki, T. Maeda and T. Ito, Biophys. J., 1991, 59, 25–30 CrossRef CAS.
- C. M. Coppin and P. C. Leavis, Biophys. J., 1992, 63, 794–807 CrossRef CAS.
- R. Furukawa, R. Kundra and M. Fechheimer, Biochemistry, 1993, 32, 12346–12352 CrossRef CAS.
- T. Oda, K. Makino, I. Yamashita, K. Namba and Y. Maéda, Biophys. J., 1998, 75, 2672–2681 CrossRef CAS.
- J. Viamontes and J. Tang, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 040701 CrossRef.
- M. M. A. E. Claessens, R. Tharmann, K. Kroy and A. R. Bausch, Nat. Phys., 2006, 2, 186–189 CrossRef CAS.
- L. Limozin, M. Bärmann and E. Sackmann, Eur. Phys. J. E, 2003, 10, 319–330 CrossRef CAS PubMed.
- M. Soares e Silva, J. Alvarado, J. Nguyen, N. Georgoulia, B. M. Mulder and G. H. Koenderink, Soft Matter, 2011, 7, 10631–10641 RSC.
- S. A. Koestler, K. Rottner, F. Lai, J. Block, M. Vinzenz and J. V. Small, PLOS One, 2009, 4, e4810 Search PubMed.
- E. S. Chhabra and H. N. Higgs, Nat. Cell Biol., 2007, 9, 1110–1121 CrossRef CAS PubMed.
- D. Vignjevic, D. Yarar, M. D. Welch, J. Peloquin, T. Svitkina and G. G. Borisy, J. Cell Biol., 2003, 160, 951–962 CrossRef CAS PubMed.
- E. Atilgan, D. Wirtz and S. X. Sun, Biophys. J., 2006, 90, 65–76 CrossRef CAS PubMed.
- L. Vonna, L. Limozin, A. Roth and E. Sackmann, Langmuir, 2005, 21, 9635–9643 CrossRef CAS PubMed.
- F. Huber, D. Strehle and J. Käs, Soft Matter, 2012, 8, 931–936 RSC.
- S. Deshpande and T. Pfohl, Biomicrofluidics, 2012, 6, 034120 CrossRef PubMed.
- J. Weichsel and U. S. Schwarz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6304–6309 CrossRef CAS PubMed.
- J. Weichsel, E. Urban, J. V. Small and U. S. Schwarz, Cytometry, Part A, 2012, 81, 496–507 CrossRef PubMed.
- P. V. D. Schoot and M. E. Cates, Europhys. Lett., 1994, 25, 515–520 CrossRef.
- M. F. Islam, A. M. Alsayed, Z. Dogic, J. Zhang, T. C. Lubensky and A. G. Yodh, Phys. Rev. Lett., 2004, 92, 088303 CrossRef CAS.
- E. Hobbie and D. Fry, Phys. Rev. Lett., 2006, 97, 036101 CrossRef CAS.
- K. Carvalho, F. C. Tsai, E. Lees, R. Voituriez, G. H. Koenderink and C. Sykes, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(41), 16456–16461 CrossRef CAS PubMed.
- J. Galanis, D. Harries, D. Sackett, W. Losert and R. Nossal, Phys. Rev. Lett., 2006, 96, 028002 CrossRef.
- G. H. Lai, J. C. Butler, O. V. Zribi, I. I. Smalyukh, T. E. Angelini, K. R. Purdy, R. Golestanian and G. C. L. Wong, Phys. Rev. Lett., 2008, 101, 218303 CrossRef.
- B. Zelinski, N. Müller and J. Kierfeld, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 86, 041918 CrossRef.
- J. Tang, S. Wong, P. Tran and P. Janmey, Bunsen-Ges. Phys. Chem., Ber., 1996, 100, 796–806 CrossRef CAS.
- G. C. L. Wong, A. Lin, J. X. Tang, Y. Li, P. A. Janmey and C. R. Safinya, Phys. Rev. Lett., 2003, 91, 018103 CrossRef.
- S. Jansen, A. Collins, C. Yang, G. Rebowski, T. Svitkina and R. Dominguez, J. Biol. Chem., 2011, 286, 30087–30096 CrossRef CAS PubMed.
- M. M. A. E. Claessens, C. Semmrich, L. Ramos and A. R. Bausch, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8819–8822 CrossRef CAS PubMed.
- F. Gittes, B. Mickey, J. Nettleton and J. Howard, J. Cell Biol., 1993, 120, 923–934 CrossRef CAS.
- I. V. Gregoretti, G. Margolin, M. S. Alber and H. V. Goodson, J. Cell Sci., 2006, 119, 4781–4788 CrossRef CAS PubMed.
- L. Borukhov, R. Bruinsma, W. Gelbart and A. Liu, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3673–3678 CrossRef PubMed.
- C. J. Cyron, K. W. Müller, K. M. Schmoller, A. R. Bausch, W. A. Wall and R. F. Bruinsma, Europhys. Lett., 2013, 102, 38003 CrossRef.
- M. Kiemes, P. Benetatos and A. Zippelius, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 021905 CrossRef.
- K. Schmoller, O. Lieleg and A. Bausch, Soft Matter, 2008, 4, 2365–2367 RSC.
- T. T. Falzone, M. Lenz, D. R. Kovar and M. L. Gardel, Nat. Commun., 2012, 3, 861 CrossRef PubMed.
- E. E. Deinum, Simple models for complex questions on plant development, PhD thesis, Wageningen University, Ch. 9, 2013, www.amolf.nl/publications/theses.
- T. Sanchez, D. T. N. Chen, S. J. DeCamp, M. Heymann and Z. Dogic, Nature, 2012, 491, 431–434 CrossRef CAS PubMed.
- J. Viamontes, S. Narayanan, A. R. Sandy and J. X. Tang, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 73, 061901 CrossRef.
- S. Fraden, G. Maret and D. Caspar, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 1993, 48, 2816–2837 CrossRef CAS.
- K. Purdy, Z. Dogic, S. Fraden, A. Rühm, L. Lurio and S. Mochrie, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 031708 CrossRef.
- W. Bras, G. P. Diakun, J. F. Díaz, G. Maret, H. Kramer, J. Bordas and F. J. Medrano, Biophys. J., 1998, 74, 1509–1521 CrossRef CAS.
- B. Gentry, D. Smith and J. Käs, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 79, 031916–32011 CrossRef.
- N. Puech, E. Grelet, P. Poulin, C. Blanc and P. van der Schoot, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 82, 020702 CrossRef.
- J. Viamontes, P. W. Oakes and J. X. Tang, Phys. Rev. Lett., 2006, 97, 118103 CrossRef.
- T. E. Schaus, E. W. Taylor and G. G. Borisy, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7086–7091 CrossRef CAS PubMed.
- D. A. Quint and J. M. Schwarz, J Math Biol, 2011, 63, 735–755 CrossRef CAS PubMed.
- E. Urban, S. Jacob, M. Nemethova, G. P. Resch and J. V. Small, Nat. Cell Biol., 2010, 12, 429–435 CrossRef CAS PubMed.
- C. Yang and T. Svitkina, Nat. Cell Biol., 2011, 13, 1012–1013 CrossRef CAS PubMed.
- I. V. Maly and G. G. Borisy, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11324–11329 CrossRef CAS PubMed.
- A. B. Verkhovsky, O. Y. Chaga, S. Schaub, T. M. Svitkina, J.-J. Meister and G. G. Borisy, Mol. Biol. Cell, 2003, 14, 4667–4675 CrossRef CAS PubMed.
- B. S. Gentry, S. Meulen, P. Noguera, B. Alonso-Latorre, J. Plastino and G. H. Koenderink, Eur. Biophys. J., 2012, 41, 979–990 CrossRef CAS PubMed.
- J. D. Pardee and J. A. Spudich, Methods Enzymol., 1982, 85 Pt B, 164–181 CAS.
- P. Artimo, M. Jonnalagedda, K. Arnold, D. Baratin, G. Csardi, E. de Castro, S. Duvaud, V. Flegel, A. Fortier, E. Gasteiger, A. Grosdidier, C. Hernandez, V. Ioannidis, D. Kuznetsov, R. Liechti, S. Moretti, K. Mostaguir, N. Redaschi, G. Rossier, I. Xenarios and H. Stockinger, Nucleic Acids Res., 2012, 40, W597–W603 CrossRef CAS PubMed.
- Micromachining Technology for Micro-Optics and Nano-Optics IV, ed. E. G. Johnson, G. P. Nordin and T. J. Suleski, Proceedings of SPIE, 2006 Search PubMed.
- S. Hess and W. Köhler, Formeln zur Tensor-Rechnung, Palm & Enke, Erlangen, 1980 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sm52421c |
| ‡ Current address: Department of Mechanical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, MA 02139, USA. |
| This journal is © The Royal Society of Chemistry 2014 |