 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyelectrolyte adsorption, interparticle forces, and colloidal aggregation
Istvan
Szilagyi
,
Gregor
Trefalt
,
Alberto
Tiraferri
,
Plinio
Maroni
and
Michal
Borkovec
*
Department of Inorganic and Analytical Chemistry, University of Geneva, Sciences II, Quai Ernest-Ansermet 30, 1205 Geneva, Switzerland. E-mail: michal.borkovec@unige.ch
First published on 15th January 2014
Abstract
This review summarizes the current understanding of adsorption of polyelectrolytes to oppositely charged solid substrates, the resulting interaction forces between such substrates, and consequences for colloidal particle aggregation. The following conclusions can be reached based on experimental findings. Polyelectrolytes adsorb to oppositely charged solid substrates irreversibly up to saturation, whereby loose and thin monolayers are formed. The adsorbed polyelectrolytes normally carry a substantial amount of charge, which leads to a charge reversal. Frequently, the adsorbed films are laterally heterogeneous. With increasing salt levels, the adsorbed mass increases leading to thicker and more homogeneous films. Interaction forces between surfaces coated with saturated polyelectrolyte layers are governed at low salt levels by repulsive electric double layer interactions, and particle suspensions are stable under these conditions. At appropriately high salt levels, the forces become attractive, principally due to van der Waals interactions, but eventually also through other forces, and suspensions become unstable. This situation can be rationalized with the classical theory of Derjaguin, Landau, Verwey, and Overbeek (DLVO). Due to the irreversible nature of the adsorption process, stable unsaturated layers form in colloidal particle suspensions at lower polyelectrolyte doses. An unsaturated polyelectrolyte layer can neutralize the overall particle surface charge. Away from the charge reversal point, electric double layer forces are dominant and particle suspensions are stable. As the charge reversal point is approached, attractive van der Waals forces become important, and particle suspensions become unstable. This behaviour is again in line with the DLVO theory, which may even apply quantitatively, provided the polyelectrolyte films are sufficiently laterally homogeneous. For heterogeneous films, additional attractive patch–charge interactions may become important. Depletion interactions may also lead to attractive forces and suspension destabilization, but such interactions become important only at high polyelectrolyte concentrations.
 Istvan Szilagyi | Istvan Szilagyi is a senior lecturer at the University of Geneva since 2009. He got his PhD in inorganic chemistry at the University of Szeged, Hungary, in 2006. He was then a postdoctoral researcher at the Murdoch University, Perth, Australia. His research focuses on inorganic and colloid chemistry and the development of novel functional materials. |
 Gregor Trefalt | Gregor Trefalt is a postdoctoral researcher at the University of Geneva. He studied chemistry at the University of Ljubljana, Slovenia. He stayed in Ljubljana to obtain his PhD in 2012 at Jožef Stefan Institute to carry out research in the area of colloidal processing of ceramic materials. He now focuses on colloidal interactions, particle aggregation, and light scattering. |
 Alberto Tiraferri | Alberto Tiraferri is a Marie Curie postdoctoral fellow at the University of Geneva. He studied environmental engineering at the Politecnico di Torino, Italy, and received his PhD in 2012 at Yale University, New Haven, USA, in the area of membrane-based separation processes for water treatment. His current project explores the adsorption of macromolecules to surfaces and implication of these processes to membrane fouling and environmental remediation. |
 Plinio Maroni | Plinio Maroni works as a senior scientist at the University of Geneva. He studied physics at the University of Pisa, Italy and he got his PhD in 2005 at the Swiss Federal Institute of Technology, Lausanne, in the area of molecular reactivity of molecules adsorbed on surfaces. His current research interests center around surface spectroscopy, surface sensitive optical and acoustic techniques, and local probe microscopy. |
 Michal Borkovec | Michal Borkovec is a full professor of chemistry at the University of Geneva and a member of the Swiss National Research Council. He received his PhD in 1986 at Columbia University, New York, USA, and then worked as a lecturer at the Swiss Federal Institute of Technology, Zürich. Later, he became an associate professor at Clarkson University, Potsdam, USA, and in 2001 he accepted his current position at Geneva. His research interests include physical chemistry of colloids, interfaces, and polyelectrolytes. |
1 Introduction
Charged polymers or polyelectrolytes (PEs) are widely employed to modify properties of surfaces or of colloidal particle suspensions. They are industrially used in water purification, papermaking, mineral separation, or to control flow properties of particle slurries.1–6 Emerging industrial applications of PEs include, for example, stabilization of metallic iron particles for environmental remediation, use as additives in the chemical–mechanical polishing process, or in biomedical applications.7–11 PEs are further employed to create protective or functional surface coatings, whereby important approaches include adsorption of PE monolayers,7,12–14 fabrication of multilayers of oppositely charged PEs,13,15–18 or the preparation of PE brushes by grafting or by adsorption of block copolymers.19–21 Such coatings may be used to control surface properties, including wetting, lubrication, adhesion, or biological resistance.22,23 Certain PEs offer the possibility to tune these properties through external stimuli, such as temperature or solution composition.24,25PEs interact strongly with solid substrates, and in turn, they may substantially alter the respective surface characteristics. Thereby, the interaction forces between such surfaces can be modified, and as a consequence, properties of particle suspensions can be controlled. Understanding of the relationship between PE adsorption, particle interactions, and the stability of the resulting suspensions is critical for further development of functional PE additives. The present review attempts to draw a systematic picture of these processes for the relevant situation when PEs adsorb onto oppositely charged solid substrates.
PE adsorption to solid substrates and the resulting charging behaviour were investigated by numerous experimental techniques. Planar substrates were probed with optical reflectivity, ellipsometry, quartz crystal microbalance, and streaming potential techniques,26–28 while particle suspensions were characterized by means of classical batch depletion techniques, light, X-ray and neutron scattering, and electrophoresis.29–32 This review will focus on linear or branched homopolymers, including dendrimers, in monovalent electrolyte solutions. We will further investigate properties of PE films in the same electrolyte solution as that used for the adsorption process and discuss the influence of salt concentration and effects of charge densities of the PEs and of the substrate. PE adsorption will be mainly interpreted in terms of the random sequential adsorption (RSA) model and its variants,33,34 while the self-consistent field approach and computer simulation studies will be addressed only briefly.35–41
Adsorbed PEs modify interaction forces acting between substrates, and for this reason, they are frequently used as additives to control the stability of colloidal suspensions or to tune their rheological properties.1–4,7 The resulting interaction forces between surfaces or particles in the presence of PEs were investigated with the surface forces apparatus (SFA),42,43 total internal reflection microscope,44–46 or the colloidal probe technique based on the atomic force microscope (AFM).47,48 Particle aggregation phenomena were investigated with turbidity measurements, time-resolved light scattering, or rheology.4,49–51 We only focus on interactions in symmetric systems involving the same type of interfaces or particles, and correspondingly on homoaggregation processes.
An interpretation of the underlying mechanisms of the adsorption process, interaction forces, and particle aggregation will be put forward. We explore to what extent interaction forces can be rationalized in terms of the classical theory of Derjaguin, Landau, Verwey, and Overbeek (DLVO).52–54 The role of specific forces induced by PEs, in particular, steric, bridging, and depletion interactions will be equally discussed.54 The relevance of these forces in controlling the suspension stability in the presence of PEs was proposed early on.55 We will further discuss the patch–charge interactions resulting from the lateral heterogeneity of the adsorbed PE layers.56 Simple models will be used to clarify the mechanisms of the interactions involved.
There are various important topics involving PE adsorption that will not be addressed in this review. We shall skip the interesting aspects related to adsorption of comb polymers, such as bottlebrush and dendronized polymers57–59 or copolymers, such as block copolymers and proteins.60–66 The response of adsorbed PE films when exposed to solutions of variable compositions or containing multivalent ions will not be addressed either.67–75 We will not discuss mixed adsorbed PE films, which are especially important for multilayers prepared by the layer-by-layer deposition process,13,15–18,24,76,77 as well as the adsorption of neutral polymers or PEs with the same sign of charge as the substrate.35,73,75,78 Neither the adsorption of PEs to interfaces under applied external electric potential, nor to instable or fluid interfaces (e.g., air–water and oil–water) will be addressed.79–82 We also skip the discussion of interaction forces in asymmetric systems involving different types of surfaces, heteroaggregation, particle deposition, or growth of particle–PE multilayers.83–87 Finally, we make no attempt to provide a detailed review of the numerous theoretical developments and computer simulations addressing PE adsorption and the resulting interaction forces. For these topics, we refer the interested reader to the appropriate literature.36–38,88
The present review is organized as follows. Section 2 summarizes the current understanding of the PE adsorption phenomena and the underlying charging process, whereby planar substrates as well as colloidal particles are discussed. Interaction forces between the same type of interfaces, either involving planar substrates, colloidal particles, or both, are addressed in Section 3. Particle aggregation phenomena involving the same type of particles are addressed in Section 4, whereby the main focus is on early stages of the homoaggregation process. An outlook highlighting open questions and possible research directions concludes the review.
2 Polyelectrolyte adsorption
This section focuses on adsorption of polyelectrolytes (PEs) to oppositely charged water–solid interfaces. These interfaces might be realized by means of a planar substrate or through the internal surface in a colloidal particle suspension. PEs normally have a linear architecture and some examples are summarized in Fig. 1. PEs can also be branched and corresponding examples include dendritic poly(amido amine) (PAMAM) or branched poly(ethylene imine) (BPEI). One refers to anionic and cationic PEs when they are negatively and positively charged, respectively. PEs with a permanent charge are called strong PEs, and they include sodium poly(styrene sulfonate) (PSS) and poly(diallyldimethyl ammonium) (PDDA). The charge of weak PEs varies with solution pH and ionic strength, and examples include poly(vinyl amine) (PVA), poly(L-lysine) (PLL), poly(acrylic acid) (PAA), linear poly(ethylene imine) (LPEI) or PAMAM. Their degree of ionization can be measured by potentiometric titrations and modelled with mean field or site binding models.89 In monovalent electrolyte solutions, the gyration radius Rg of linear PEs obeys the classical scaling law90| Rg ∝ Mα | (1) |
 | ||
| Fig. 1 Structural formulae of various fully ionized PEs discussed in this review. The names and acronyms refer to the ionized forms of the strong PEs, while to the neutral forms of the weak PEs. | ||
2.1 Irreversible nature of the adsorption process
Highly charged PEs adsorb strongly to oppositely charged interfaces. This affinity is primarily caused by attractive electrostatic forces acting between the oppositely charged PE and substrate. Since the PE backbone is hydrophobic, however, attractive van der Waals and hydration forces are important as well. This subsection will compare adsorption to planar substrates and in colloidal suspensions and rationalize this process with simple models.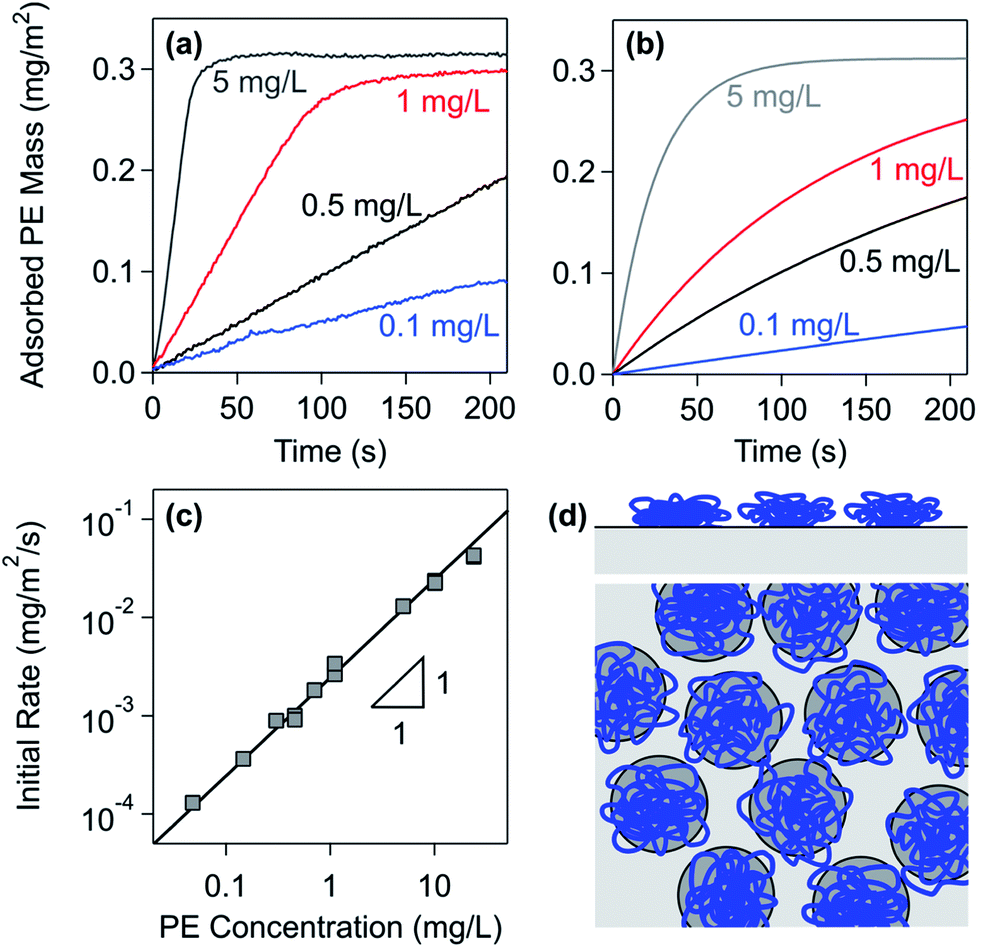 | ||
| Fig. 2 Adsorption kinetics of PDDA to the silica surface in a solution of 50 mM monovalent salt at pH 4. (a) Measurements with optical reflectivity at different PE concentrations.91 (b) Results of the simple irreversible adsorption model with the Langmuir blocking function. (c) Initial adsorption rate versus different PE concentrations. (d) Graphical illustration of the RSA model. | ||
Partial desorption could be only demonstrated for PEs in special circumstances, for example, for PEs of very low molecular mass, typically below 10 kg mol−1.27,95 Desorption of PEs of higher molecular mass has been reported to be induced by changes in the solution composition or by exchange with other PEs.27,58,99 However, the adsorption process is irreversible provided the composition of the electrolyte solution is not changed during the experiment.
With increasing concentration of the PE, the initial adsorption rate increases.91 The rate law can be identified by plotting the initial rate versus the PE concentration as shown in Fig. 2c. Since this rate is proportional to the PE concentration, the adsorption process follows a first-order rate law in the PE concentration. Similar dependencies were observed with other PEs.34,97,100
Converting the data shown in Fig. 2c into the adsorbed number density, one finds an adsorption rate coefficient of ka = 2.5 × 10−6 m s−1. This rate coefficient can also be calculated from the cell geometry by assuming laminar flow and perfect sink conditions.34,101 Based on the known flow rate and the hydrodynamic radius of PDDA of about 26 nm as estimated from light scattering experiments,90,102 the perfect sink model predicts a rate coefficient of ka = 4.9 × 10−6 m s−1. This value is only about a factor two larger than the one observed experimentally. The remaining discrepancy could be related to forces acting between the PE and the substrate and to hydrodynamic interactions. A similar agreement between experimental and calculated adsorption rate coefficients was reported in other PE systems.34,97,100
The plateau value is independent of the PE concentration to good approximation. However, this value may increase with increasing PE concentration somewhat. This increase can be rationalized by the finite relaxation time of the polymer chains.28,76,91 With increasing PE concentration, the lateral relaxation of the individual adsorbing PEs is increasingly hindered by the rapidly arriving neighbouring PEs. A similar relaxation mechanism was also suggested to be present for protein adsorption.103
| θjam ≃ 0.55 | (2) |
| θ = πa2Γ | (3) |
 | (4) |
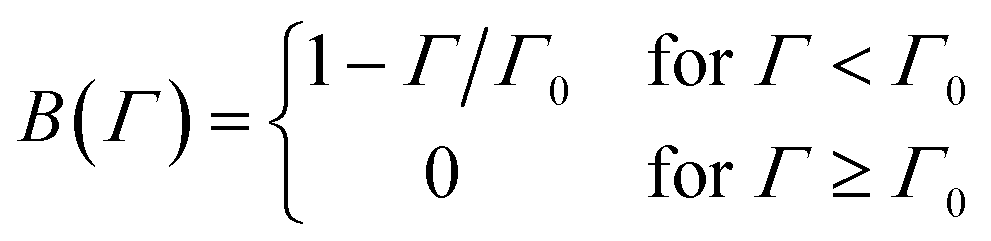 | (5) |
However, the model predicts a too gradual transition from the initial stages to saturation. An analysis of the RSA model leads to a blocking function, which suggests an even slower approach to saturation. Alternative blocking functions have been proposed to remedy this problem.66,105,106 However, they were mainly used to describe irreversible adsorption of particles and proteins and have not been applied to model PE adsorption so far.
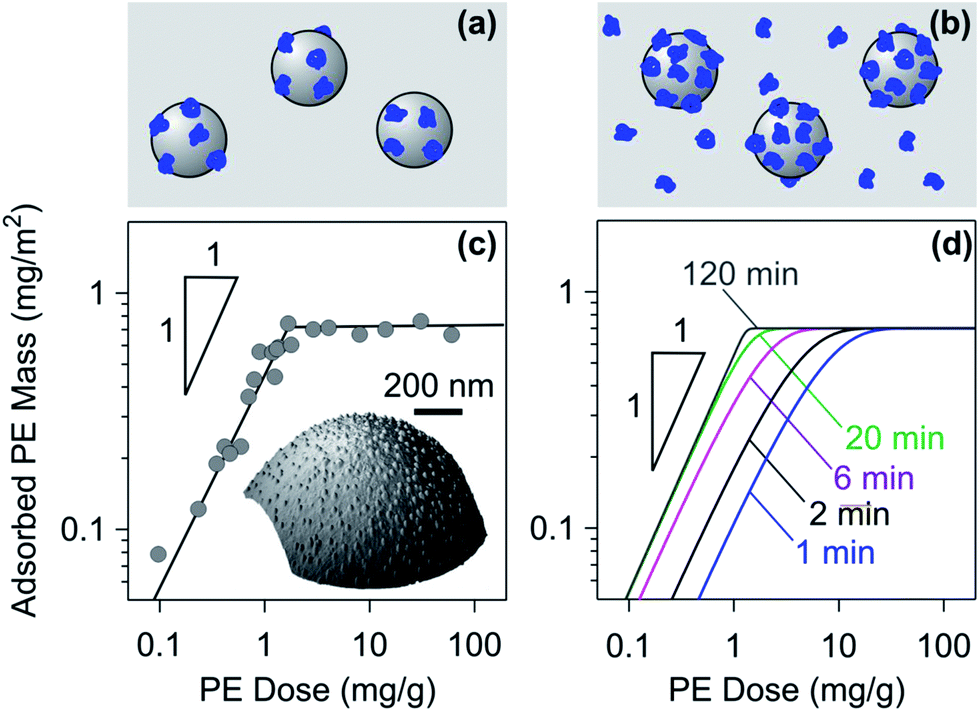 | ||
| Fig. 3 Adsorption of PEs in a colloidal suspension of oppositely charged particles. Schematic representation of (a) the unsaturated layer that depletes the solution of the free PE and (b) the saturated layer with excess PE in solution. (c) Overnight adsorption of dendritic PAMAM of 935 kg mol−1 to sulfate latex particles of diameter 3.1 μm in a colloidal suspension of pH 4.0 without salt added. The adsorbed amount was obtained by counting from AFM images shown in the inset.108 The solid line represents the expected adsorbed amount given the saturation value. (d) Representative results with the kinetic RSA model where the effect of finite adsorption time is indicated. | ||
The irreversible nature of the adsorption can be confirmed experimentally with batch adsorption experiments as well.31,107Fig. 3c shows results of adsorption of dendritic PAMAM to sulfate latex particles, where the adsorbed amount was obtained by counting the adsorbed single molecules with the AFM.108 The adsorbed mass is plotted versus the PE dose, which reflects the mass of PE relative to the mass of the particles (mg g−1).
At high PE doses, the adsorbed mass is constant due to saturation. The fact that the total adsorbed mass is independent of the solution concentrations was also experimentally confirmed with PVA and PDDA adsorbing to latex particles.30,31,107 At low doses, the entire quantity of PE added is adsorbed, but is insufficient to achieve saturation, meaning that the plateau is not reached. The PE dose can be also expressed as the mass of the PE per unit particle surface area (mg m−2). These units are useful in the unsaturated regime, where the dose simply reflects the adsorbed mass. The fact that adsorption in the unsaturated regime is quantitative can be also demonstrated by electrophoresis, and this technique will be discussed in Section 2.3.
Deviations from this idealized picture occur due to the kinetics of the adsorption process. This process can be particularly slow when the PE concentration is close to the one needed to reach saturation. The simple model summarized in eqn (4) and (5) can be also used to model adsorption in suspensions, and the corresponding results are illustrated in Fig. 3d. When the adsorption time is too short such that adsorption cannot be completed, the plot of the adsorbed mass versus the PE concentration will be rounded. Such dependencies might be wrongly interpreted by an equilibrium adsorption isotherm.
When a substrate is continuously flushed with PE solution in a flow-through cell, one always obtains a saturated layer due to a sufficient supply of PEs. Unsaturated layers can be formed in a flow-through cell too. In that case, however, the PE feed solution must be changed to a pure electrolyte solution before the saturation plateau is reached.
2.2 Properties of adsorbed polyelectrolyte layers
Let us now discuss the main characteristics of saturated PE layers, particularly, the adsorbed mass, their thickness, water content, and lateral heterogeneity. Not much is known concerning the unsaturated layers, but we will also comment on those. The RSA model will be further generalized to introduce effects of electrostatic interactions.The mass of adsorbed PE depends on several factors related to the characteristics of the PE, those of the substrates, and the solution composition. Here, we discuss effects of the molecular mass, salt concentration, as well as the influence of the charge density of the PE and the substrate. These findings will be then explained in terms of a modified RSA model that includes electrostatic interactions between adsorbed molecules.
The adsorbed mass at saturation depends only weakly on the molecular mass for linear PEs.28,32 This observation can be also rationalized with the RSA model. Based on eqn (1) and (3), this model suggests that the adsorbed number density scales as
| Γ ∝ M−2α | (6) |
The adsorbed mass normally increases with increasing concentration of the added monovalent electrolyte. Fig. 4 illustrates this trend for various PEs and substrates. This increase has been observed for a wide range of cationic and anionic PEs adsorbed on oppositely charged substrates, and typically results in an increase by a factor 2–4 when the salt concentration is increased by 4 orders of magnitude.28,32,34,91,97,98,110,112,114–116 This salt dependence only reverses for very weakly charged PEs and high salt concentrations, whereby the adsorbed mass goes through a maximum, and finally decreases.97,98,117–119
 | ||
| Fig. 4 Adsorbed mass of PEs on oppositely charged substrates versus salt concentration at saturation. (a) Adsorbed mass versus the salt concentration for dendritic PAMAM and linear PDDA on silica measured by reflectivity and AFM and comparison with the RSA model (solid lines).34,91 (b) Dimensionless representation of the coverage θ versus screening parameter κa of the same data as shown in (a). The grey region corresponds to radii between 5 and 50 nm. (c) Further experimental results obtained with different substrates, namely mica, silica, amino-functionalized silica (AS), and amidine latex (AL). The solid lines serve as a guide to the eye. (d) Schematic representation of the electrostatic RSA model. The diffuse layer is indicated in purple. | ||
Trends concerning the variation of the charge densities are less clearly established. The charge density of PEs can be varied by studying copolymers involving charged and neutral monomers, but other properties of the PEs may change at the same time (e.g., hydrophobicity).31,111 The charge density of the substrates has been sometimes varied by investigating different oxides, but other characteristics of these substrates are altered in this fashion as well.26,27 A good way to tune the charge density of PEs and of the substrates is through weak ionizable groups and the respective pH adjustments. Nevertheless, the charges of the isolated components may not correspond to the ones in the adsorbed state, since ionization may occur upon adsorption.117,120,121 The adsorbed mass normally increases with increasing charge density of the substrate and with decreasing charge density of the PE.29,34,91,111 With decreasing PE charge density, the adsorbed mass may go through a maximum at very low charge densities.29,111,119,122
This situation can be captured by a simple modification of the RSA model.106,123,124 Since the adsorbing polymers repel each other due to overlapping electric double layers, one can model this phenomenon as an increase in the radius of the adsorbing disks to an effective radius aeff, which now also includes the range of the repulsion of the surrounding diffuse layer. The situation is schematically depicted in Fig. 4d. The resulting surface coverage is now given by
 | (7) |
| u(2aeff) ≃ kBT | (8) |
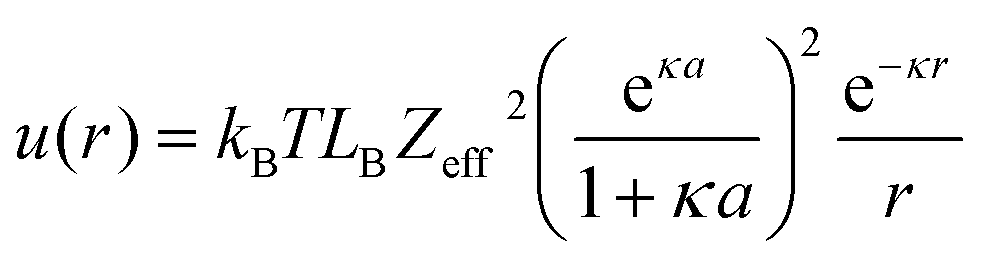 | (9) |
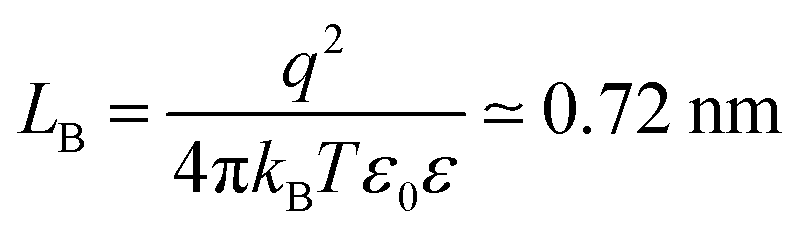 | (10) |
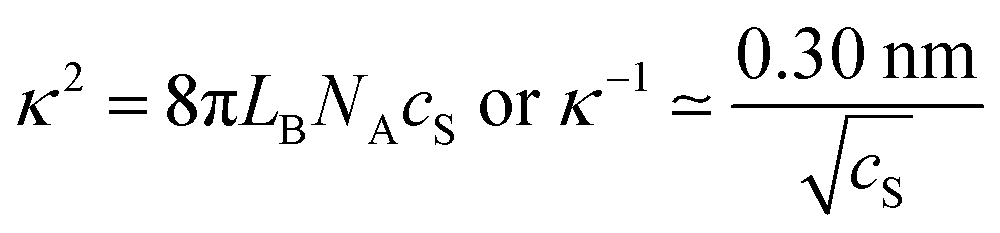 | (11) |
 | (12) |
Results obtained from this modified RSA model are shown in Fig. 4a. One observes that this model predicts very similar dependencies to the ones observed for the adsorption of PDDA and dendritic PAMAM.34,91Fig. 4b shows a dimensionless representation of the surface coverage θ versus the screening parameter κa where the curves almost collapse on a master curve.124,125 This model can qualitatively explain the characteristic increase of the adsorbed mass with decreasing charge density of the PE. In this case, decreasing the effective charge Zeff will lead to smaller effective radii aeff and therefore to larger adsorbed mass.
The RSA model can be further extended to rationalize the increase of the adsorbed mass with increasing charge density of the substrate.34 At charged water–solid interfaces, electrical double layers form, and the diffuse layer contains a higher concentration of counterions than the one in the bulk. When two PE chains interact close to the interface, the higher concentration of counterions close to the interface will enhance the screening of the electrostatic interaction. Therefore, the electrostatic repulsion between the adsorbing chains will be weaker and result in a larger adsorbed mass. This effect can be included into the RSA model, and the modified model can explain the increase of the adsorbed amount of dendritic PAMAM with the solution pH quite well.34 The more substantial adsorbed amounts of PAMAM compared to other PEs at very low salt concentrations shown in Fig. 4c can be probably rationalized through the same mechanism. The spirit of the electrostatic RSA model is similar to the treatment of the dilute 2-d Wigner regime, which makes the assumption that PEs adsorb individually and that their mutual interactions are dominated by diffuse layer repulsion.126
The RSA model is unable to predict the adsorption maximum that is observed for weakly charged PEs and high salt concentrations.97,117,118 This maximum is related to the fact that PEs will not adsorb to oppositely charged substrates beyond a critical salt concentration threshold, if solely electrostatic forces are present.39,119,127 At high salt levels, a weakly charged PE will be strongly screened. Therefore, attractive electrostatic forces acting between the PE and the substrate will be not sufficient to overcome the thermal motion, and the PE will no longer adsorb. Since the adsorbed amount increases with increasing salt levels, but vanishes above the salt threshold, a sharp maximum in the adsorbed amount results. In reality, however, additional attractive forces act between the PE chain and the substrate (e.g., van der Waals and hydration). These non-electrostatic forces may be quite important, as evidenced by adsorption of neutral polymers. For PEs, however, the forces responsible for the adsorption weaken substantially beyond the salt threshold. Therefore, one observes a wider maximum with respect to the one expected from electrostatic forces alone. Under these conditions, the adsorption process may no longer be irreversible, and the simple RSA model is expected to fail. Alternative models capable of describing this situation are discussed below.
Fig. 5 summarizes layer thickness measurements of adsorbed PE films with two different techniques.28,32 The first technique is based on dynamic light scattering (DLS) in a colloidal particle suspension, where the layer thickness is inferred from the difference between the hydrodynamic radii of the particles in the presence and in the absence of the PE; see Fig. 5a.32,128 The second technique relies on a combination of optical reflectivity and quartz crystal microbalance measurements on planar substrates; see Fig. 5b.28,129 Both techniques yield comparable results. PE layers adsorbed on oppositely charged substrates are extremely thin, merely a few nm. Considering the fact that the diameter of PE chains in solution typically is 20–100 nm, the PEs are strongly flattened in the adsorbed state. Based on these thickness measurements, one further concludes that these films are rather compact and contain only 20–60% of water.28,32 At higher salt levels, however, these films become more swollen and porous. Few additional reports confirm that PE films adsorbed on oppositely charged substrates are very thin indeed.18,111,130 One also finds that the layer thickness increases with increasing salt levels and with increasing molar mass, especially at high salt concentrations.28 An increase in thickness was also reported with the decreasing charge of the PE, and this quantity also passes through a maximum at very low charge densities.111
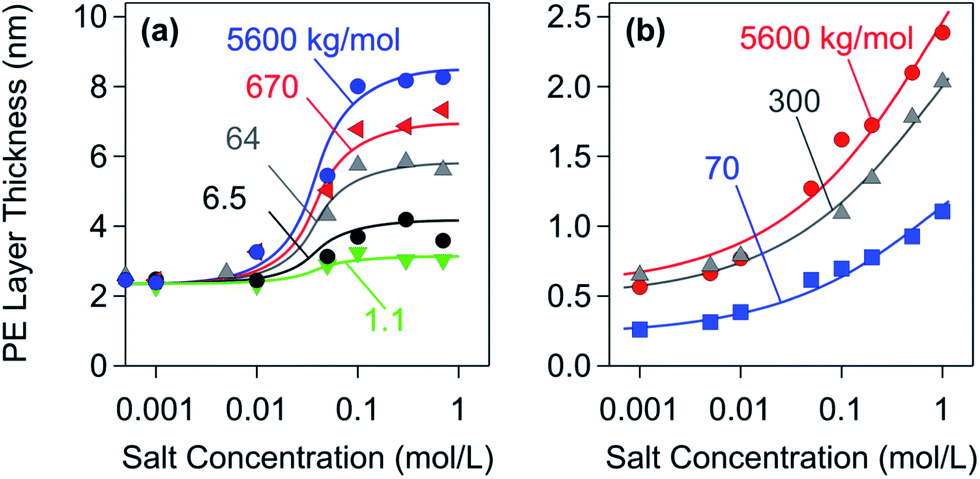 | ||
| Fig. 5 Layer thickness of saturated PSS layers versus salt concentration adsorbed on oppositely charged substrates for different molecular masses of the sodium salt. (a) Amidine latex particles probed with DLS.32 (b) Planar amino-functionalized silica probed with reflectivity and quartz crystal microbalance.28 | ||
While data shown in Fig. 5 clearly support the picture of flat adsorbed PE films, one observes that DLS measurements yield a larger thickness than the ones measured by the surface sensitive techniques. Moreover, the latter data suggest a more gradual swelling of the film. While the differences in the substrates used may lead to these differences, they might also be related to the two sub-layer structure of an adsorbed PE film.94 The thickness of these sub-layers may vary upon solution conditions and lead to the different salt dependencies observed with the two different techniques. One should also realize that thickness measurements for such extremely thin films are difficult and prone to systematic errors. Some of the available results in the literature should be thus considered with caution.
The lateral heterogeneity of adsorbed PE films can be best confirmed by AFM imaging. Such non-uniformities are particularly pronounced for highly charged PEs and low salt levels. Fig. 6 shows such images of adsorbed PEs recorded in the dry state. Fig. 6a shows adsorbed dendritic PAMAM, and the individual molecules can be clearly distinguished. Note that this layer is saturated, and no further adsorption will occur, in spite of the unoccupied surface in between individual molecules. This low coverage results from the strong electrostatic repulsion between the adsorbing dendrimers. The other images show linear PEs adsorbed to different substrates. Fig. 6b refers to a saturated layer of PSS on amidine latex particles.131Fig. 6c and d show unsaturated layers of poly(vinyl pyridine) (PVP) on mica.132
 | ||
| Fig. 6 AFM images of various PEs adsorbed on oppositely charged substrates recorded in air. Saturated layers formed with (a) dendritic PAMAM on mica in pH 4.0 solution without added salt and (b) PSS on amidine latex particles in 1 mM electrolyte solution.47 Reprinted with permission from J. Phys. Chem. B, 113, 8458. Copyright (2009) American Chemical Society. Unsaturated layers of PVP on mica with (c) small (d) and larger adsorbed amounts.132 Reprinted with permission from J. Phys. Chem. B, 111, 8597. Copyright (2007) American Chemical Society. | ||
While such imaging techniques provide strong evidence that adsorbed PE layers are laterally heterogeneous, quantitative characterization of these heterogeneities is mostly lacking. Exceptions are layers formed with dendritic PAMAM, whereby the individual molecules can be resolved with AFM. They can be described as dilute monolayers and they feature a liquid-like order as indicated by a structural peak in the radial distribution function.34,133 The statistical properties of individual adsorbed linear polyelectrolytes and of nucleic acids were successfully determined with the AFM.74,132,134–136 However, little is known about the detailed structure of saturated layers of adsorbed linear polyelectrolytes and the characteristic distances involved. Direct force measurements have confirmed that adsorbed layers of dendritic PAMAM and linear PSS are heterogeneous.107,131 These techniques have also demonstrated that similar layers formed with LPEI are homogeneous down to about 10 nm, but probably heterogeneous on smaller length scales.48 Weakly charged hydrophobic PEs have been reported to form more homogeneous layers, resembling disordered lamellar phases.74 Similar structures were also predicted on theoretical grounds and were also referred to as the semi-dilute 2-d Wigner regime.88,126
An alternative interpretation of the small layer thicknesses shown in Fig. 5 could be related to the lateral heterogeneity of the films. Typically, surface sensitive and scattering techniques yield a laterally averaged thickness, and dilute, heterogeneous layers would lead to a smaller thickness than the gyration radius of an individual PE even if the adsorption process did not lead to a deformation in the normal direction. However, height measurements of adsorbed dendritic PAMAM with the AFM indicate that even these molecules flatten substantially.67,133 These highly branched molecules have rather compact structures, and therefore linear PEs will flatten much more.
An important class of analytical models is based on density functional theories. These theories normally assume that the adsorbed film is laterally homogeneous, and they attempt to estimate self-consistently the profiles of the concentrations and of the electric potential in the normal direction. Such a self-consistent field (SCF) approach was implemented within a numerical scheme by Fleer and co-workers.29,35,137 Based on a similar formulation, simple scaling laws could be derived.138 These approaches are capable of reproducing the frequently observed increase of the adsorbed amount with increasing salt concentration, increasing charge density of the substrate, and decreasing charge of the PEs. The SCF approach was also able to provide information concerning the distribution of loops, tails, and trains, to rationalize experimentally observed adsorbed amounts, and to reproduce the maxima in the adsorbed amount for weakly charged PEs.137,139 Since the adsorbed PE layers are laterally heterogeneous, results obtained from SCF models that assume laterally homogeneous layers must be interpreted with caution. These models are probably most useful to describe the adsorption of weakly charged PEs, which probably form more homogeneous layers.
Computer simulations have also been used to investigate the adsorption of PEs.39–41 The conformation of a single adsorbed PE chain was studied by considering screened Coulombic interactions only.39 These authors have found that the adsorbed chain is strongly flattened at low salt concentrations, while it swells at higher salt levels. The simulated normal extensions of the adsorbed PE chain show very similar trends to the measured layer thickness shown in Fig. 5b. This finding strongly supports the view that the layer thickness is determined by the dimensions of individual adsorbed PE chains that are well separated at the surface, leading to a laterally heterogeneous layer. When interactions are governed by electrostatic forces only, this study also confirms that PEs do not adsorb at oppositely charged surfaces above a critical salt concentration.39,127
Adsorption of PEs to spherical particles in the presence of salt was recently studied with computer simulations and density functional theories.40 This approach explains the experimentally observed large accumulation of opposite charge to the particle surface. However, these simulations also predict a maximum in the adsorbed amount at very low salt concentrations, which is at odds with the experiment. All Coulombic interactions were explicitly taken into account in another recent computer simulation study of PE adsorption, whereby effects of short-range hydrophobic attractions were also investigated.41 This study confirms the view that adsorbed PEs are strongly flattened and that the adsorbed layer is laterally heterogeneous. Unfortunately, the latter study was carried out in the absence of salt, and these conditions are difficult to realize experimentally.
2.3 Charge balance in adsorbed polyelectrolyte layers
Let us now discuss the charge reversal phenomenon and the charge distribution within the adsorbed PE film.The overall charge of adsorbed PE layers can be best addressed by electrokinetic techniques. Electrophoresis is the method of choice for colloidal particles, whereby the electrophoretic mobility can be converted into the surface potential (ζ-potential) with appropriate models.30,48,140–142,148 For planar substrates, streaming potential or streaming current techniques are being used.33,93 In many situations, this surface potential approximates the diffuse layer potential ψD well. When the surface potential is known, the surface charge density σ can be estimated with the Gouy–Chapman relationship54
 | (13) |
The charge reversal upon PE adsorption is illustrated in Fig. 7a and b, where the surface potentials of bare amidine latex particles are compared to those with a saturated adsorbed PSS layer.131 The data are consistent with the Gouy–Chapman eqn (13) with constant surface charge densities of +5.9 mC m−2 for bare particles and −2.6 mC m−2 for the coated ones. Besides the different signs, however, the particle coated with the PE behaves very similarly to a bare particle. This observation is not surprising given the fact that the adsorbed PE layer is very thin. The adsorbed PEs are indeed capable of reversing the positive charge of the bare particle and even accumulating substantial additional negative charge at the surface. A good agreement between surface potentials estimated from electrophoresis and direct force measurements is frequently found.48,107,131,147 In some cases, however, these results disagree, probably due to surface charge heterogeneities.48
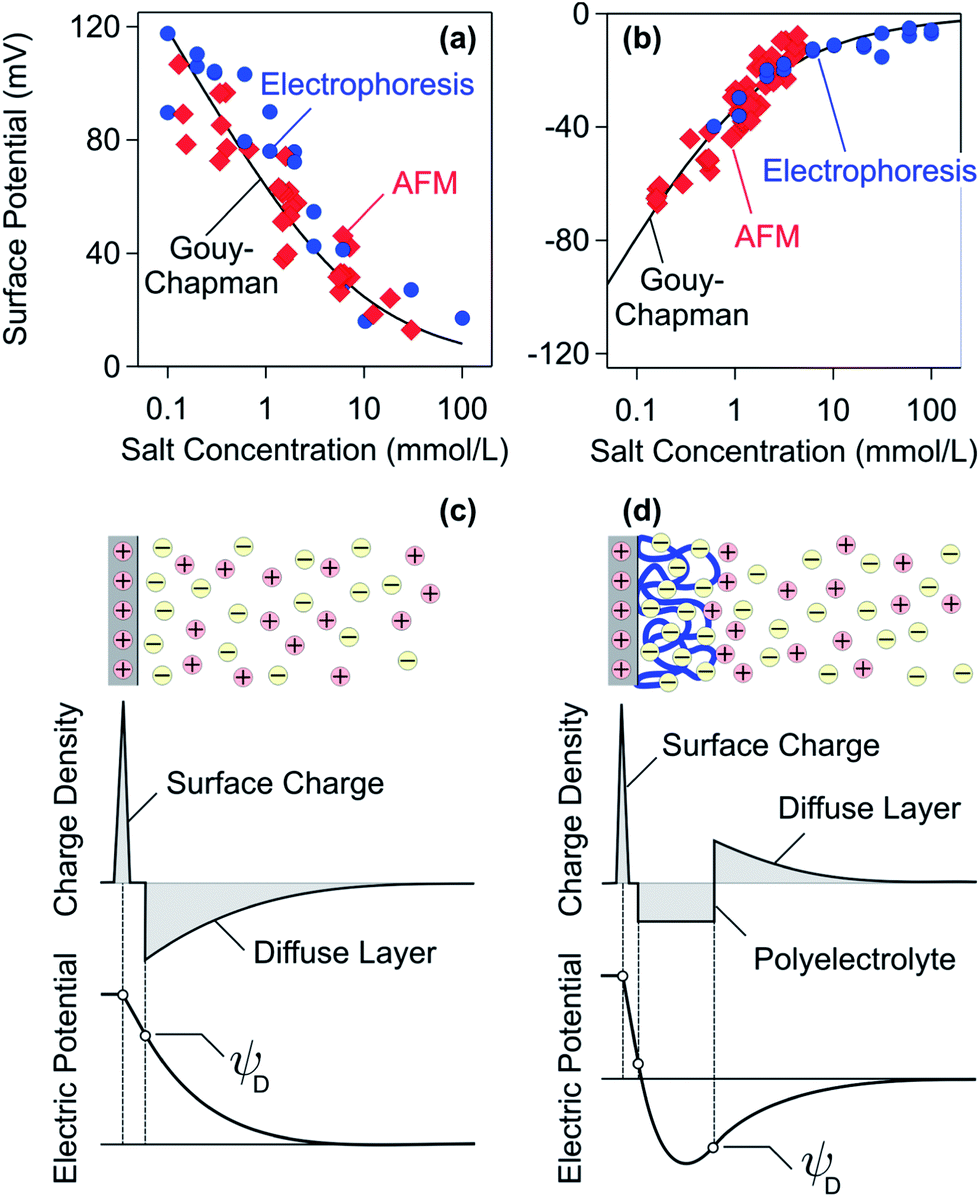 | ||
| Fig. 7 Comparison of surface potentials for amidine latex particles determined by electrophoresis and direct force measurements by AFM. The solid line is the best fit with the Gouy–Chapman equation. (a) Bare particles and (b) coated with a saturated layer of PSS.131 Schematic representation of the charge density and the electric potential profiles where the diffuse layer potential ψD is indicated. (c) Bare charged interface and (d) charged interface with an adsorbed PE of opposite charge. | ||
Fig. 8a illustrates the build-up of this negative charge upon addition of PSS in a suspension of positively charged latex particles.131 At low PE doses, the particles are positively charged. At a particular dose, the surface charge is neutralized by the PE, and no diffuse layer forms. Upon further PE addition, the negative charge continues to accumulate, until one reaches the saturation point. Before that point is reached, the surface is unsaturated and no PE is dissolved in solution. For doses beyond the saturation point, the adsorbed amount remains constant and the excess PE dissolves in solution.
 | ||
| Fig. 8 Charge reversal by adsorbed PEs to oppositely charged substrates in the unsaturated regime as illustrated by surface potential measurements. (a) Amidine latex particles in the presence of PSS of different molecular masses of the sodium salt by electrophoresis and direct force measurements.47 (b) Calculation of the charge reversal with linear superposition relationship. (c) Electrophoresis measurements of carboxylated latex particles in the presence of LPEI at different particle concentrations.149,150 (d) Streaming potential measurements of mica in contact with poly(allyl amine) solutions at different electrolyte concentrations indicated for 20 min.152 Reprinted from J. Colloid Interface Sci., 303, Z. Adamczyk, A. Zembala and A. Michna, PE adsorption layers studied by streaming potential and particle deposition, 353, Copyright (2006), with permission from Elsevier. | ||
For unsaturated layers, no free PE is dissolved in solution. This fact can be confirmed in colloidal particle suspensions with electrophoresis experiments at different particle concentrations. Results of such experiments are illustrated in Fig. 8c with carboxylated latex particles and LPEI.149,150 These particles are negatively charged and adsorbing LPEI leads to a pronounced charge reversal. The collapse of the plots of the electrophoretic mobility versus the PE dose for different particle concentrations confirms that the adsorption is quantitative. If this were not the case, there would be a shift of the corresponding curves due to partitioning between adsorbed and dissolved PEs.151 Adsorbed PSS and PVA on oppositely charged latex particles were shown to behave analogously.31,140
The charge reversal of planar charged surfaces induced by adsorption of PEs can be also followed by streaming potential measurements.33,93,152 These results are illustrated with the adsorption of poly(allyl amine) to mica in Fig. 8d.152 Bare mica is negatively charged. At high PE doses, the surface reverses its sign, and the saturation plateau is reached. To avoid the saturation of the surface, the PE solution was in contact with the surface for only 20 min. At lower polymer doses, the surface is unsaturated and passes through the charge reversal.
The charge reversal phenomenon can be captured with a very simple model. One has to assume that the surface charge density of the substrate originates from two additive contributions107,153
| σ = σ0 + qZeffΓ | (14) |
Adamczyk and coworkers have proposed that this transition is more gradual, which would reflect a decrease of the effective charge with the surface coverage.33 On the other hand, a sharper transition was observed for latex particles with adsorbed PSS and dendritic PAMAM.131,153 In the two latter systems, Zeff appears to be constant at first, then increases in magnitude near the charge neutralization point, and again remains constant after this point. Unfortunately, we currently lack a general picture concerning eventual variations of the effective charge of PEs upon changes in the adsorbed amount.
Let us now discuss to what extent the simplified picture shown in Fig. 7d actually reflects the actual charge distribution between the different adsorbed components.31,109,154 At the charge reversal point, the interface is neutral, and thus the substrate, PE, and the adsorbed salt ions neutralize each other precisely. In some cases, the PE neutralizes the surface exactly, and one refers to stoichiometric adsorption. For other PEs, especially for highly branched ones or for weakly charged surfaces, the counterions of the PEs contribute substantially to the charge balance, and the adsorption is super-stoichiometric. In the case of adsorbed BPEI and PAMAM,31,155 the counterions may be responsible for the neutralization of up to 90% of the charge originating from the adsorbed PE. In saturated layers, the situation is similar, since the overall surface charge that is neutralized by the diffuse layer is normally just a small fraction of the total charge carried by the adsorbed PE. Furthermore, the lateral heterogeneity will lead to lateral undulations of the diffuse layer. These effects might be responsible for the observed variations of the effective charge with the adsorbed amount of the PE.153
A fixed charge stoichiometry of the adsorption process can often be used to rationalize shifts of the charge reversal point. This principle explains why a higher dose of a more weakly charged PE is needed to neutralize the charge of a given surface.31,50 This trend is also reflected in Fig. 8b. Similarly, a lesser amount of a given PE is needed to neutralize a surface of a smaller surface charge.150 Dependencies on the solution pH involving weak PEs can be understood similarly. The charge of a weak cationic PE increases with decreasing pH. For a surface with a fixed charge density, the charge reversal point thus shifts towards a higher pH with an increasing amount of adsorbed PE.143 The same trend is observed for a negatively charged surface with weak acid or amphoteric groups (e.g., silica) in the presence of strong cationic PEs.110,120 Reverse trends are observed for weak anionic PEs adsorbed on a cationic surface of fixed charge density or for strong anionic PEs adsorbed on a positively charged surface with weak bases or amphoteric groups.144,148 More complex phenomena are observed when the charges of the PE and of the surfaces are both pH dependent.4,120,156
The shift of the charge neutralization point for dendritic PAMAM with the molecular mass can be rationalized in a similar way.155 Due to the compact architecture of these PEs, an increasing number of charged groups will be neutralized by their counterions the higher their molecular mass. Therefore, the effective charge increases more slowly than the molecular mass, thus the charge neutralization point shifts toward higher PE doses. However, this effect is absent for linear PEs. They adsorb in a much flatter configuration, and therefore the charge stoichiometry is independent of the molecular mass.
3 Forces induced by polyelectrolytes
In this section, we will discuss measured force profiles between charged surfaces or between particles with adsorbed polyelectrolytes (PEs) of opposite charge. An important observation will be that the classical DLVO theory describes forces in such systems reasonably well. Additional attractive non-DLVO forces have been identified and can be important under certain conditions, especially for PEs of high charge density and high molecular mass. These additional forces are largely of electrostatic origin and result from the laterally heterogeneous patch–charge distribution within the adsorbed PE film. Additional attractive forces may occur due to bridging of PE chains or due to depletion at higher PE concentrations.DVLA forces. The force F acting between two charged objects across an aqueous electrolyte solution is assumed to have two main contributions54,157
| F = FvdW + Fdl | (15) |
 | (16) |
| Fdl = 2πRε0εκψeff2e−κh | (17) |
3.1 Bare surfaces and surfaces with saturated PE layers
As discussed in Section 2.1, saturated layers form when the adsorbing PE is added in sufficiently large quantities. Such layers are normally thin and highly charged. Therefore, forces between these layers mainly originate from double layer interactions. In the following, we will discuss forces between bare surfaces first, and then between surfaces coated with saturated PE layers.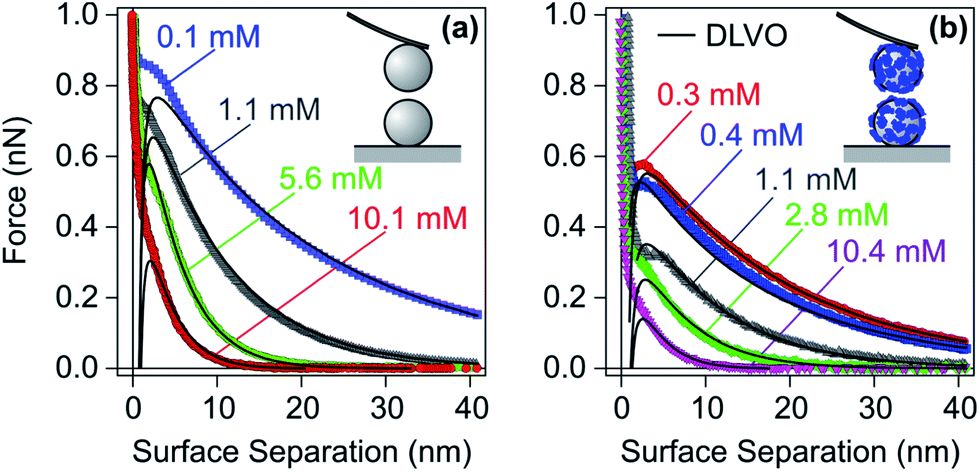 | ||
| Fig. 9 Experimental force profiles for sulfate latex particles in monovalent electrolyte solutions adjusted to pH 4.0 compared with their best fits by DLVO theory. (a) Bare negatively charged particles and (b) the same particles at a LPEI dose of 1.1 mg g−1, which results in a saturated adsorbed PE layer of positive charge.48 | ||
The forces become attractive at shorter distances, but this attraction cannot be well resolved due to the inherent mechanical jump-in instability.162 During this instability, deviations from DLVO theory are mainly caused by hydrodynamic drag. This drag creates additional repulsive forces, which mask the attractive forces at short distances. The force measurements shown were carried out at relatively low salt levels, where double layer forces dominate. Similar force measurements at higher salt levels or for weakly charged particles have been reported more recently, and they reveal the expected attractive van der Waals forces.163 These results confirm that the DLVO theory also correctly describes the transition between attractive and repulsive forces in such systems.
Similar force measurements between charged solid interfaces across aqueous solutions of monovalent electrolytes have been carried out with the SFA and the colloidal probe in the sphere–plate geometry.161,164–166 In the latter case, the symmetry of the system remains difficult to ascertain. Nevertheless, these studies confirm that forces in such systems are consistent with the simple DLVO picture, at least down to distances of few nm.
Numerous other studies confirm that interactions between surfaces coated with saturated PE films are governed by repulsive double layer forces. Such a behaviour was observed for positively charged amidine particles coated with the anionic PSS131 and for negatively charged sulfate latex particles in the presence of cationic LPEI and dendritic PAMAM.48,107 Double layer forces were observed between silica, mica, or functionalized surfaces in the presence of various oppositely charged PEs.72,109,167–175 The strength of double layer forces could also be varied through solution pH.72,147,176 This dependence originates from the resulting variation of the dissociation degree of the PE. In some cases, deviations from DLVO theory have been reported at short distances, and they were either attributed to steric repulsion168,169,177,178 or to patch–charge attraction.48,107,131 However, these contributions are rather weak and one can conclude that double layer forces dominate the interactions between charged substrates with saturated PE layers of opposite charge.
Bridging polymer chains are known to induce additional attractive forces, and this mechanism was suggested to be important for PEs as well.43,179,180 Such bridging processes can be probed directly with the AFM, and this approach is referred to as single molecule force spectroscopy.109,181–183 The principle is illustrated in Fig. 10. The force profiles are normally measured through a repeated approach and retraction cycles of the probe with respect to the surface, and the surfaces remain in contact for short periods of time. When the surfaces are in proximity, some of the PE chains adsorbed to one of the surfaces may adsorb to the other surface and thereby bridge both surfaces. The existence of such bridging PE chains is easily detected during the retraction of the probe, since these chains are being stretched and detached from the surfaces. These processes lead to characteristic spikes or plateaus in the retraction force curves.
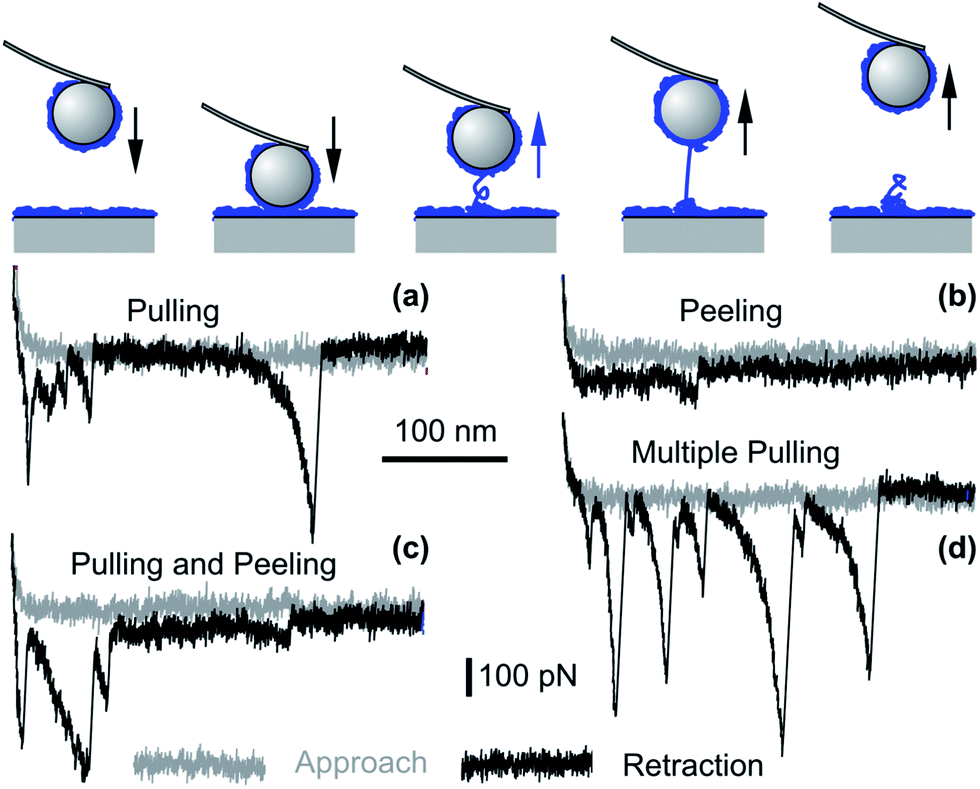 | ||
| Fig. 10 Bridging events as probed by colloidal probe AFM force measurements for saturated PVA layers of molecular mass of 520 kg mol−1 adsorbed to silica in 100 mM electrolyte solution of pH 4.0. Single molecule (a) pulling and (b) peeling events. More complex events involving several molecules may show (c) a combination of pulling and peeling events and (d) multiple pulling events.109 | ||
Bridging events were investigated in detail for saturated layers of adsorbed PVA on silica by force measurements with AFM.109,182 Representative examples are shown in Fig. 10. When the PE chain is anchored strongly to both surfaces, the chain is stretched, which leads to a characteristic spike. This situation is referred to as pulling and is shown in Fig. 10a. When the anchoring to one of the surfaces is weaker, the chain will detach continuously, which will lead to a plateau in the force curve. Here we refer to peeling, which is shown in Fig. 10b. More complex events equally occur and they are illustrated in Fig. 10c and d. Similar experiments were equally used to obtain information about the loop distribution of the adsorbed PEs.184
While such bridging processes are rare at low salt levels, they become rather frequent at higher salt concentrations and for weakly charged PEs.109 Similar observations could be made for saturated BPEI films.169 This trend can be rationalized since the adsorbed layers were thin under low salt conditions, and the PE chains are strongly bound to the substrate by attractive electrostatic forces. As the salt level increases, these attractive forces are screened, thus allowing PE chains to explore the regions further away from the surface. In this case, bridging becomes more frequent. Under these conditions, however, the forces are often completely attractive, and the presence of additional attractive bridging forces may not modify the picture substantially.
3.2 Surfaces coated with unsaturated layers
At lower PE doses, one obtains unsaturated layers, which feature lower adsorbed amounts than the saturated ones. As discussed in Section 2.2, adsorbed unsaturated layers are often laterally heterogeneous, but they can also be more homogeneous in some situations. Nevertheless, the principal contributions to the force can be again understood within DLVO theory. Let us first discuss the forces in the simpler case of laterally homogeneous layers, and later address the more complex situation of heterogeneously adsorbed layers.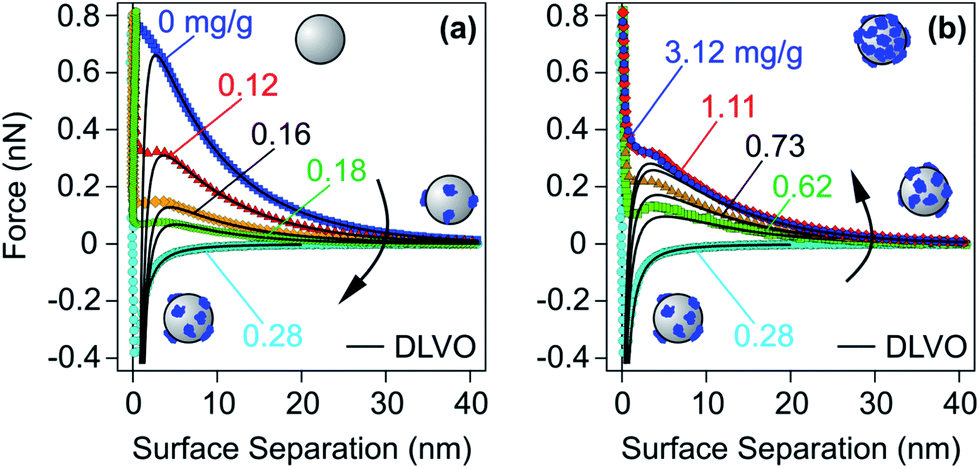 | ||
| Fig. 11 Experimental force profiles for sulfate latex particles for different doses of LPEI at an ionic strength of 1.1 mM adjusted with a monovalent salt at pH 4.0 compared with their best fits by DLVO theory.48 PE doses (a) below and at the charge reversal point and (b) at the charge reversal point and above. | ||
Force measurements for unsaturated PE layers near the charge neutralization point are difficult to perform, and therefore only few such reports are available.48,107,131,176,185 The main obstacle is that charge neutralization can be only achieved in a narrow range of PE doses, and this condition is difficult to realize for the small surface areas available in the currently used force measurement protocols. From this point of view, the SFA or its variants are more advantageous, since the surface area is few cm2. The surface area of a single particle used in the colloidal probe experiment is only few μm2, whereby the necessary PE doses are minute and they cannot be properly controlled. The recently described multi-particle colloidal probe technique circumvents this problem by depositing a larger number of particles to a substrate.48,107,131 In this fashion, one may again reach surface areas of several cm2, for which the necessary dose is simpler to control. Another possibility is to work with low PE concentration and to monitor the force profiles with time.185 In such an experiment, the system initially passes through the charge neutralization point, while the saturated layer forms later.
Another possibility is to prepare a saturated layer with a weak PE and to neutralize the charge by adjusting the solution pH. With this technique, adsorbed PVP layers were shown to interact by pure van der Waals interactions at their charge neutralization point.176 This finding suggests that these PVP films are also laterally homogeneous, similar to the ones formed with LPEI.48 One may hypothesise that partially protonated LPEI and PVP form homogeneous adsorbed layers due to lowering of the PE charge by deprotonation and the presence of additional hydrophobic interactions.
| F = FvdW + Fdl + Fpc | (18) |
Fig. 12 shows interaction forces between amidine latex particles neutralized with PSS at a dose of 1.0 mg g−1 (0.58 mg m−2).131 One observes that the attractive forces are substantially stronger than the van der Waals force expected from DLVO theory, especially for high molecular mass and at low salt levels. Experimental force profiles can be well fitted when this additional non-DLVO force is assumed to be exponential
| Fpc = −Ae−qh | (19) |
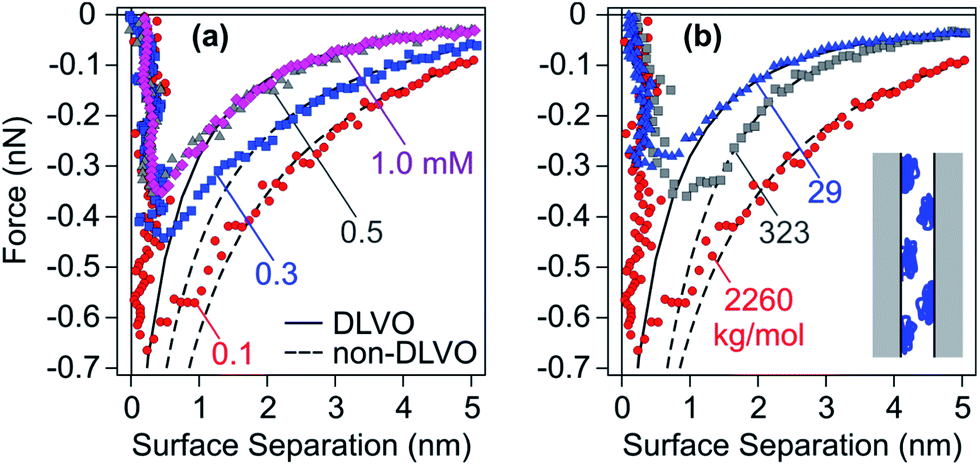 | ||
| Fig. 12 Attractive force profiles for amidine latex particles neutralized with adsorbed PSS in monovalent salt solutions at pH 4.0 together with best fits by DLVO theory (solid lines) and additional patch–charge attraction (dashed lines).47 The force curves might be inaccurate close to contact due to eventual jump-in instability. (a) Molecular mass of 2260 kg mol−1 at different ionic strengths and (b) different molecular masses at an ionic strength of 0.1 mM. The scheme illustrates the patch–charge attraction mechanism. | ||
 | (20) |
Very similar results were obtained by direct force measurements between negatively charged sulfate latex particles in the presence of dendritic PAMAM.107,188 Near the charge neutralization point, forces are attractive, and they are again much stronger than the van der Waals force, especially for high molecular mass and low salt levels. The additional attraction can be again rationalized with the exponential relationship given in eqn (19). The measured corresponding lattice spacing of this particular system is about b = 16 nm, but this value is substantially smaller than the nearest neighbour spacing of the dendrimers at the surface, which is about 50 nm. This discrepancy probably originates from the assumption of a square lattice inherent to the patch–charge model, while the actual surface structure is irregular.
Additional attractive forces near the charge neutralization point were reported in other systems with the SFA or related techniques.185 By exploiting the kinetics of the adsorption process, attractive forces near this point could be observed for PVA films at low salt levels.185 These forces were equally reported to be exponential and substantially more attractive than the van der Waals forces. We suspect that these forces also originate from patch–charge attraction. Similar non-DLVO attractive forces were reported between layers of adsorbed poly((3-methacrylamido)-propyl)trimethylammonium chloride on mica.42
Polymer bridging might also lead to additional attractive forces. As discussed above, bridging events can be detected with the AFM in the retraction part of the force curves, as shown in Fig. 10. The occurrence of such bridging events was analyzed near the charge neutralization point for the LPEI and PSS systems.47,48 While such events could be observed, they occurred at low salt concentrations very rarely. Moreover, forces observed in the PSS and PAMAM systems are similar, in spite of the fact that the PE architectures are very different. If bridging would be important, one expects substantial differences between the forces in these two systems.
At higher salt levels, where bridging forces are expected to be operational, the DLVO theory also predicts attractive forces. Therefore, additional attractive bridging forces may not alter the scenario much. At intermediate salt levels, however, where the strength of double layer forces and van der Waals forces are comparable, additional bridging forces may influence the picture considerably. Similarly, when the charge of the PEs is low, bridging forces might become important in analogy to neutral polymers.189 This suggestion is supported by more frequent occurrences of single molecule bridging events observed with the AFM under these conditions.178,190 However, the precise conditions where bridging forces become important remain unclear to us.
Interactions between surfaces in the presence of PEs were investigated theoretically in detail by density functional theories and computer simulations.191,192 Density functional theories suggest that interactions can be indeed described by DLVO theory at larger distances. Computer simulations were used to model forces between surfaces by approximating the PEs as charged spheres that interact by means of screened Coulombic interactions. These simulations confirmed the importance of DLVO forces, but also suggested additional short-range interactions originating from ordered arrangements of the PEs at the surface.188 A similar study with flexible PE chains, which included all Coulombic interactions explicitly, found similar additional attractive forces, but the range of the forces did strongly depend on the chain flexibility.193 Attractive forces close to charge neutralization and repulsive forces away from this point were also reported.194 These studies demonstrate the importance of additional attractive forces. Other computer simulation studies suggest that bridging forces might also be important.195–198 However, these studies refer to bridging even when PEs are not absorbed at both surfaces and these simulations are carried out under equilibrium conditions. The irreversible nature of the adsorption process and the resulting slow dynamics of the adsorbed chains may modify the nature of the bridging contributions substantially. On the other hand, the adsorbed PE chains may maintain some lateral mobility and equilibrate laterally to some extent.
4 Particle aggregation induced by polyelectrolytes
Aggregation of colloidal particles is governed by the interaction forces acting between the particles. Simplistically, attractive forces lead to rapid particle aggregation, while this process is slowed down by repulsive forces. The elementary step of the aggregation process can be viewed as a chemical reaction| A + A → A2 | (21) |
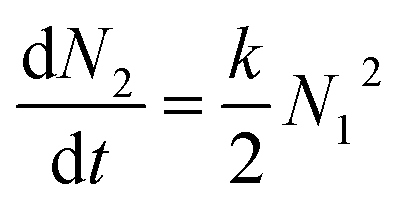 | (22) |
Aggregation rates from DVLO theory. The key contribution of DLVO theory was to derive the aggregation rate coefficient in terms of the interaction potential between colloidal particles. From the steady-state solution of the forced diffusion equation one finds that the rate coefficient is given by52–54,206
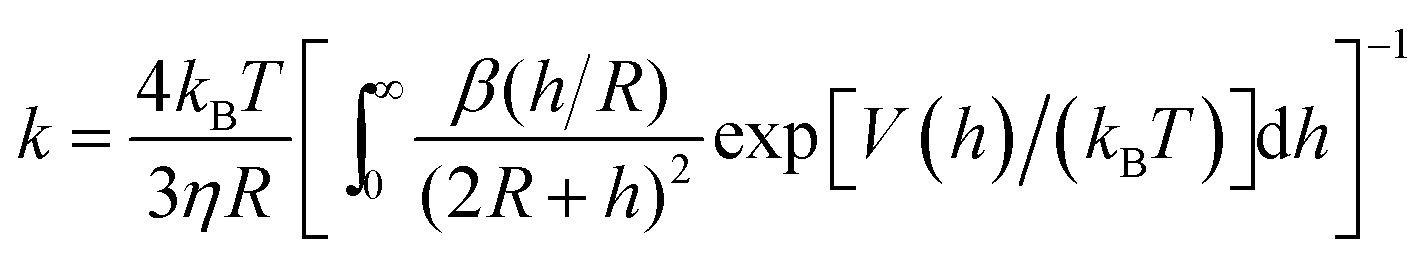 | (23) |
 | (24) |
 | (25) |
DLVO theory predicts two regimes for the aggregation processes.54,206 The first regime, referred to as the fast or diffusion controlled aggregation, typically occurs for high salt concentrations or small surface charge densities. In this situation, the aggregation rate coefficient is approximately given by the Smoluchowski value for hard spheres54,206
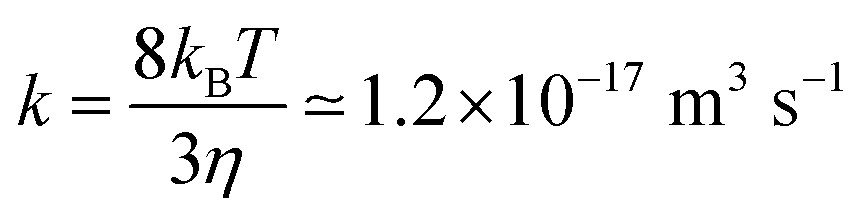 | (26) |
The second regime, referred to as the slow or reaction controlled aggregation, occurs at lower salt concentrations and higher surface charge densities. In this case, the interaction potential develops a barrier, which leads to a small aggregation rate coefficient. The relatively sharp transition between these two regimes is referred to as the critical coagulation concentration (CCC). This transition has been observed in numerous systems, for example, as a function of the salt concentration,215,216 or when the surface charge density was varied by adjusting the solution pH.212–214 The DLVO theory is capable of describing the aggregation rates accurately provided that the surface charge density is not too high.212 At higher surface charge densities, important deviations may occur.
The aggregation rate coefficients are normally reported as the stability ratio defined as54,212
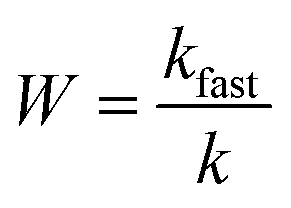 | (27) |
4.1 Bare particles and particles with saturated PE layers
Aggregation rate coefficients of colloidal particles can be accurately measured with time-resolved light scattering, turbidity, or single particle counting.207,217,218 In many situations, the trends observed can be well rationalized by DLVO theory. In the following, we will discuss aggregation rates in suspensions of bare particles first and then in suspensions with particles coated with saturated PE layers.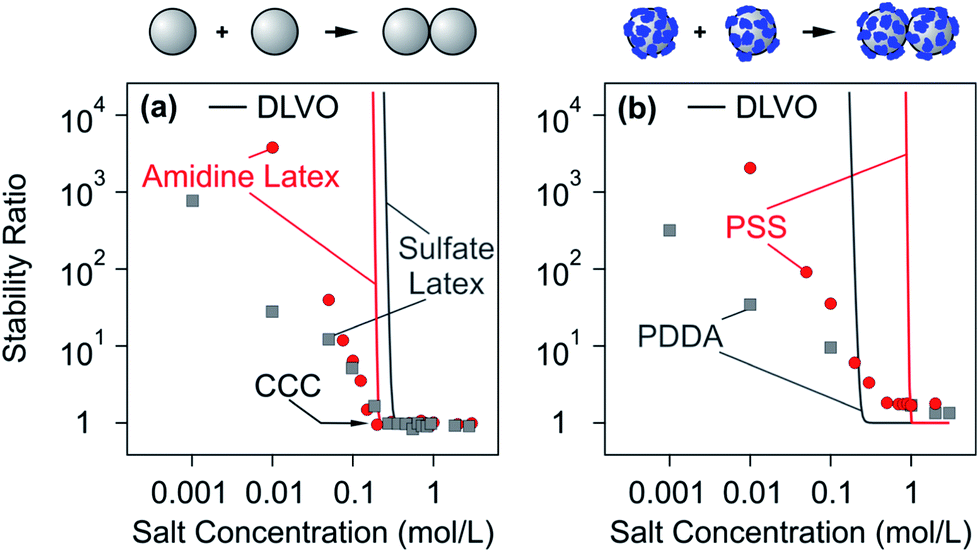 | ||
| Fig. 13 Dependence of experimentally measured stability ratios of latex particles on monovalent salt concentration at pH 4.0 and comparison with DLVO theory. (a) Bare amidine latex and sulfate latex particles and (b) the same particles coated with a saturated layer of PSS and PDDA, respectively.140,142 Schemes on the top illustrate the dimer formation without and in the presence of PEs. | ||
A common difficulty with DLVO theory is that it predicts a substantially stronger dependence of the stability ratio on the salt concentration than that observed experimentally. In this example, this discrepancy is apparent in the slow aggregation regime by the different slopes of experimental data and of the DLVO calculations. A similar behaviour and analogous discrepancies with theoretical predictions have been reported in other systems containing charged particles in the presence of monovalent salts.216,219–223 The origin of this discrepancy is probably related to lateral patch–charge heterogeneities.214,220,223–225 These patch–charge heterogeneities that are also likely present on the bare particles have a different origin than the ones discussed above, which originate from the lateral heterogeneity of the adsorbed PE films. The patch–charge heterogeneities of the bare particles may originate from the discreteness of the charged groups or an uneven distribution of these groups at the surface that result from the synthesis process. Their presence was also evidenced by differential electrophoresis techniques.226 These heterogeneities will equally induce additional attractive forces. Such attractions are expected to be stronger between particles suspended in solution than those measured with the colloidal probe AFM. Suspended particles can rotate freely and they will eventually find a configuration of the patches on the two particles involved that lead to an approach pathway of the lowest free energy.
In the fast regime, DLVO theory predicts an absolute rate constant of 7.1 × 10−18 m3 s−1. The fact that this value is smaller than Smoluchowski's value given in eqn (26) originates from the interplay between van der Waals forces and hydrodynamic interactions. The experimentally measured aggregation rate coefficient is 3.5 × 10−18 m3 s−1 for the sulfate latex particles and 4.4 × 10−18 m3 s−1 for the amidine latex. These values are smaller than the ones predicted by DLVO theory, and the remaining discrepancies probably originate from inaccuracies of the hydrodynamic resistance function at small separations.
The similarity between bare colloidal particles and particles coated with a saturated PE layer was observed for various other systems, including negatively charged sulfate latex particles in the presence of LPEI, positively charged amidine latex particles in the presence of PSS or PAA, and hematite particles with alginate.142,227 A similar behaviour was also reported for latex particles with poly(methacrylic acid) grafted to their surface.228 This similarity is further supported by direct force measurements, which indicates that interactions between surfaces coated with saturated PE layers are well described by DLVO theory as illustrated in Fig. 9b. These findings clearly demonstrate that forces acting between the charged surfaces and those coated with a saturated PE layer are similar and that they can be understood within DLVO theory. This similarity is due to the very thin and compact nature of the adsorbed PE films.
Particles with an adsorbed saturated PE layer often have higher CCCs.141,142 Particles with grafted PEs on their surface have CCCs in monovalent salts even above 1 M.228,229 Saturated adsorbed PE films typically feature high surface charge densities, which will cause the CCC to shift to high salt concentrations. However, the systems shown in Fig. 13b are characterized by stability ratios larger than unity in the fast aggregation regime, pointing to a more stable suspension than the one predicted theoretically, even at high ionic strengths. Stabilization at high salt levels was also observed in the presence of neutral polymers or for grafted PE brushes.221,228,229 This effect is sometimes referred to as electrosteric stabilization.199,221,228 This additional stabilization cannot be rationalized within DLVO theory, but reflects additional repulsive steric forces. However, such effects are not very important in charged particle suspensions in the presence of oppositely charged PEs.141,142,230
4.2 Aggregation involving unsaturated polyelectrolyte layers
Stability ratios pass through a characteristic minimum with increasing PE doses. Since unsaturated PE layers undergo a charge reversal with increasing the mass of adsorbed PE, this dependence can be rationalized by DLVO theory. Adsorbed unsaturated layers are often laterally heterogeneous, but sometimes they can be more homogeneous. Let us now discuss the aggregation rates of particles in suspension in such situations. We will first focus on the simpler case of homogeneous layers and discuss heterogeneous layers later.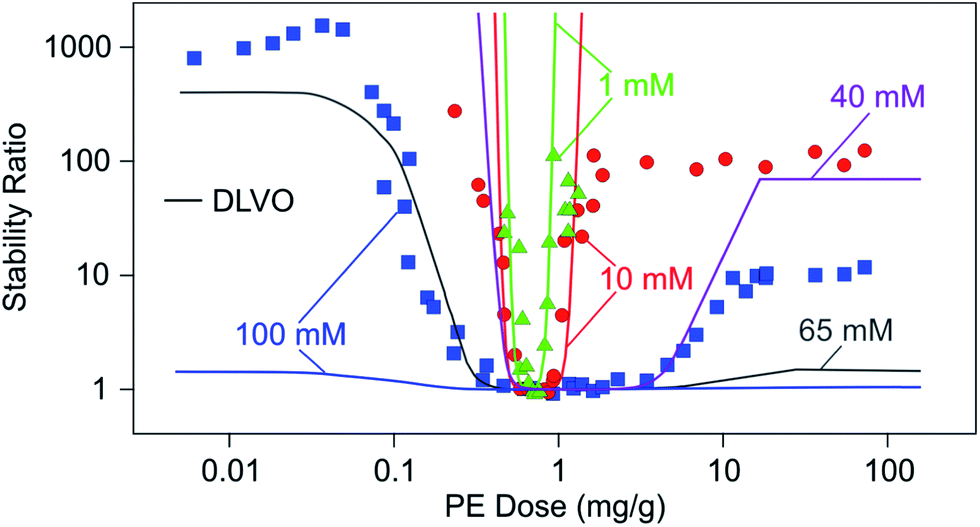 | ||
| Fig. 14 Stability ratios of sulfate latex particles versus the dose of LPEI at different ionic strengths adjusted by a monovalent electrolyte and at pH 4. Solid lines are calculations with DLVO theory.150 Note that only calculated curves are shown for 40 mM and 65 mM. These calculations illustrate that DLVO theory reproduces the overall dependence correctly, albeit not at the appropriate salt concentration. | ||
Let us compare these results with predictions of DLVO theory, whereby the surface potentials were estimated from electrophoresis. At low salt concentrations, DLVO theory reproduces the experimental data well. The likely reason why DLVO theory works in this case is that the adsorbed LPEI film is laterally homogeneous. Force measurements shown in Fig. 11 also suggest that the film is homogeneous on length scales of at least 10 nm. This number is in agreement with the present stability data, since DLVO predictions break down for salt concentrations near and above 10 mM. While the minimum is described reasonably well, the plateau at high LPEI doses is no longer located properly. While the DLVO theory is capable of predicting the overall shape of the stability curve at higher salt concentrations qualitatively, it fails to do so quantitatively. The predicted widths of the instability region and the values of the stability plateaus do not agree with experiments. At higher salt levels, the system is more stable than what is predicted by DLVO theory, possibly due to steric forces.
Another experimental finding confirming the homogeneity of adsorbed LPEI layers is presented in Fig. 15a. This graph shows experimental stability data for different molecular masses.150 No significant trend with the molecular mass can be established and a good agreement with DLVO theory is observed.
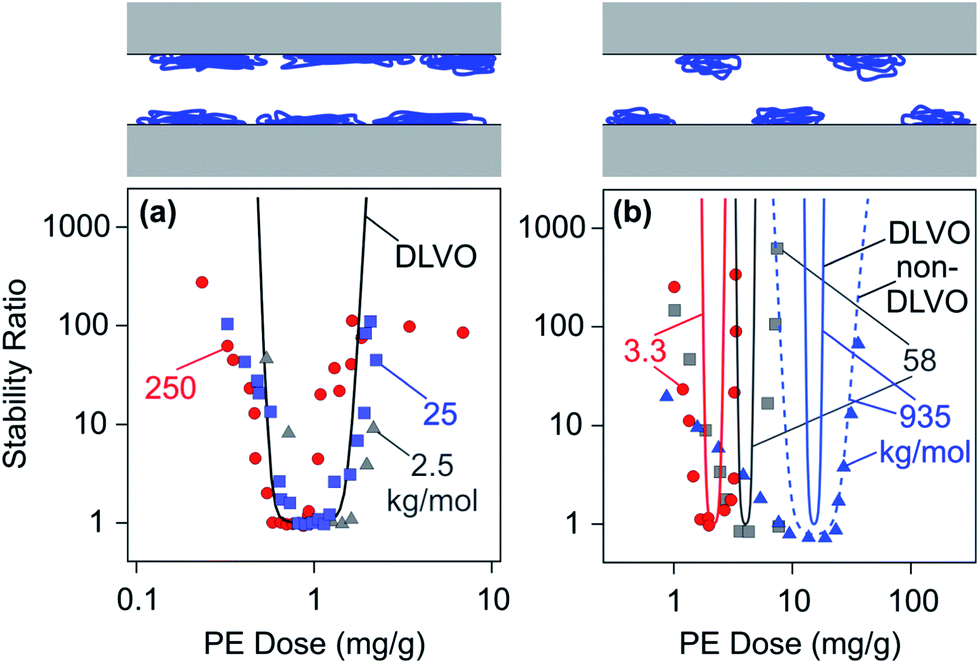 | ||
| Fig. 15 Dependence of stability ratios of sulfate latex particles on the PE dose for different molecular masses in monovalent electrolyte solutions at pH 4.0 and comparison with DLVO theory. (a) LPEI in an electrolyte of 10 mM150 and (b) dendritic PAMAM dendrimers at 1 mM.155 The dashed line illustrates the effect of additional non-DLVO patch–charge attractions. The schemes on the top illustrate the homogeneous LPEI and heterogeneous PAMAM layers. | ||
There are numerous other reports confirming that the aggregation near the charge reversal is rapid and that it slows down away from this point. They involve a wide range of PEs together with positively49,140,144,148,232 and negatively charged particles.31,49,50,56,110,142,145,155 However, the plateaus in the stability ratios at high and low PE dose are difficult to observe experimentally, and therefore they are often missing. Nevertheless, the existence of these plateaus has been clearly demonstrated in some systems.141,142,148,150
With increasing salt concentration, the fast aggregation regime widens and the dependence of the rate coefficient on the PE dose weakens in the slow regime. This salt dependence is generic and was reported for negatively charged latex and silica particles in the presence of cationic PEs31,49,110,142,155 and for positively charged latex particles with PSS or PAA.140,148
Fig. 15b shows aggregation rates of sulfate latex particles in the presence of dendritic PAMAM of different molecular masses.155 When the molecular mass is small, the dependence of the stability ratio on the PE dose is relatively well described by DLVO theory. For large molecular masses, however, the experimentally observed stability ratios are substantially smaller than the predicted ones. Attractive patch–charge interactions between heterogeneous surfaces are likely to be responsible for this reduction. When one approximates these interactions with eqn (19), the experimentally observed trend can be captured relatively well. The higher the molecular mass, the larger the size of the patches, and this increase leads to a larger range of attractive non-DLVO forces; see eqn (20). The stability measurements were carried out at a salt concentration of 1 mM, which corresponds to a Debye length of 10 nm. The nearest-neighbour distances between the dendrimers are below this value for the molecular mass of 3.3 kg mol−1, and thus the film should be considered homogeneous. The shift of the minimum in the stability plot shown in Fig. 15b reflects the shift in the charge neutralization point. This effect is discussed in Section 2.3 and is related to the compact architecture of dendritic PAMAM.
The role of patch–charge heterogeneities is typically manifested in the stability plots by widening of the fast regime and weaker dependence in the slow regime. These trends could also be well predicted by Monte Carlo simulations, where the dendritic PAMAM were modelled as charged hard spheres interacting with screened Coulomb potential.233
Similar dependencies on the molecular mass were observed for amidine latex particles in the presence of PSS or PAA,140,148 for cationic PEs and sulfate latex,56 or silica particles.234,235 However, no dependence of the stability ratio on the molecular mass is observed for LPEI, as illustrated in Fig. 15a.150 This observation reflects the homogeneity of the adsorbed film at length scales above 10 nm.
Another characteristic effect of patch–charge heterogeneities can be observed in the fast aggregation regime near the charge neutralization point. Fig. 16a shows stability ratios versus salt concentration at the charge neutralization point for dendritic PAMAM of different molecular masses. One observes that the stability ratio decreases with decreasing salt concentration and that this effect becomes increasingly pronounced with increasing molecular mass. This trend can be interpreted with the increasing strength of the patch–charge interactions. Calculations of the stability ratio by including the non-DLVO patch–charge contribution given in eqn (19) capture the salt dependence rather well. Therefore, we interpret this enhancement as originating from patch–charge attractions. Due to their electrostatic nature, these interactions are screened at higher salt concentrations. Since these patches increase in size with increasing molecular mass, this effect also becomes more important under these conditions. Fig. 16b illustrates that this enhancement in the stability ratio at the charge neutralization point can be also observed for different linear PEs. This trend was reported for sulfate latex particles neutralized with PVA, BPEI, or poly(aminoethyl methacrylate)49,56,143 or amidine particles with PSS or PAA.140,148 Adsorbed LPEI layers do not show this enhancement due to their lateral homogeneity.150
 | ||
| Fig. 16 The dependence of the stability ratios on the salt concentration at the charge neutralization point. Solid lines are calculations including non-DLVO contributions from patch–charge interactions. (a) Sulfate latex particles at pH 4.0 in the presence of dendritic PAMAM155 and (b) amidine particles in the presence of linear PSS and PAA and sulfate latex in the presence of BPEI and PVA.49,140,143,148 | ||
The question to what extent bridging forces are relevant in the aggregation process of charged particles involving oppositely charged PEs still remains open. The observed trends in the available experimental data are qualitatively consistent with DLVO theory and patch–charge attractions. While the effect of patch–charge attractions can be modelled with an exponential force profile, this treatment is approximate due to inherent lateral heterogeneity of the surface. At this point, no quantitative theory is capable of predicting aggregation rate constants from the respective surface charge distributions. Direct force measurements discussed in Section 3 confirm that bridging events are rare at low salt concentrations, and under these conditions bridging forces will be unimportant. At higher salt concentrations, however, bridging events can be frequently observed with the AFM, and therefore bridging forces could play a more important role. In this regime, however, the prevailing attractive interactions induce fast aggregation, and the corresponding rate depends only weakly on the strength of the attractive forces. In some systems, enhanced aggregation rates in the fast regime in the presence of alginate and multivalent cations were interpreted in terms of gelation, but they might also represent a signature of bridging.236,237 However, analogous effects were not reported for other PEs so far.
5 Outlook
Polyelectrolytes (PEs) adsorb irreversibly to oppositely charged substrates until saturation, which results in thin monolayers, which have the opposite charge than the substrate. Due to the irreversible nature of the adsorption process, the addition of PEs to colloidal suspensions at smaller doses leads to the formation of unsaturated layers, and thereby charge reversal can be induced.The principal forces acting between saturated layers are repulsive due to double layer forces and the corresponding particle suspensions are stable. For unsaturated layers near the charge reversal point, the interaction forces are attractive and the suspensions become unstable. Both phenomena are in agreement with DLVO theory, which can even describe interactions between homogeneous films quantitatively. For laterally heterogeneous films, attractive patch–charge interactions induce additional attractive forces leading to faster aggregation than predicted by DLVO theory.47
In spite of this reasonable level of understanding, we are still far away from being able to quantitatively predict interaction forces and colloidal stability solely from properties of the PEs and of the substrates. The charge reversal point for unsaturated layers can be estimated by assuming stoichiometric charge neutralization, even though numerous PEs adsorb in a super-stoichiometric way due to counterion co-adsorption.31 However, a proper way to address the extent of this co-adsorption process is currently unknown. Similarly, there are a number of uncertainties as to how to reliably estimate electric surface potentials of surfaces with adsorbed PEs.
Better characterization of the lateral surface structure of PE-coated surfaces and of the resulting surface charge heterogeneities represents an important need to progress further. At this point, we have little knowledge concerning the type of surface charge heterogeneities, the respective length scales, and when such layers might be considered as homogeneous. Most promising are AFM imaging techniques,134 but obtaining high-resolution maps of surface potentials represents a challenge. Such maps can be interpreted in terms of radial distribution functions, as recently carried out with computer simulation results,41 but corresponding experimental results are only available for dendritic PAMAM.34,133 We further lack reliable models to estimate the interaction forces involving heterogeneously charged surfaces. In particular, such models must go beyond the current simplistic regular lattice arrangements,186,242 and the question of random, liquid-like structures must be addressed.
The relevance of forces that are well established for neutral polymers, such as steric repulsion, bridging attraction, and depletion interactions, should be revisited for PEs in more detail. Based on the above discussion, bridging forces appear irrelevant at low salt levels and for highly charged PEs. With increasing salt levels and decreasing charge densities, the PEs start to resemble neutral polymers, and thus steric and bridging forces will start to play a more important role.109,178 However, it is unclear to us under what conditions this transition happens and what the respective mechanisms are.
On the other hand, depletion interactions have been established to play an important role at elevated PE concentrations. While depletion forces induced by non-adsorbing PEs have been studied in detail,44,201 we have little information concerning such forces for PEs in the presence of oppositely charged substrates. An adsorbed saturated PE layer will make the surface effectively non-adsorbing for additional PE molecules, and thus the depletion interactions in these systems might well resemble the non-adsorbing case.
We hope that these questions will be addressed in the future by combining experimental techniques, computer simulations, and theory. These efforts are expected to lead to a more detailed picture of PE adsorption processes and the resulting interaction forces between substrates, and finally should give rise to reliable predictive tools that could be used to design optimal systems of PEs and substrates for the processes in question.
List of abbreviations
The corresponding structural formulae of all PEs discussed are given in Fig. 1.| AFM | Atomic force microscope |
| BPEI | Branched poly(ethylene imine) |
| CCC | Critical coagulation concentration |
| DLS | Dynamic light scattering |
| DLVO | Derjaguin, Landau, Verwey, and Overbeek |
| LPEI | Linear poly(ethylene imine) |
| PAA | Poly(acrylic acid) |
| PAMAM | Poly(amido amine) |
| PDDA | Poly(diallyldimethyl ammonium) |
| PE | Polyelectrolyte |
| PLL | Poly(L-lysine) |
| PSS | Poly(styrene sulfonate) |
| PVA | Poly(vinyl amine) |
| PVP | Poly(vinyl pyridine) |
| RSA | Random sequential adsorption |
| SFA | Surface forces apparatus |
| SCF | Self-consistent field. |
Acknowledgements
We acknowledge useful discussions with Zbigniew Adamczyk, Vincent Ball, Lars Forsmann, Bo Jönsson, Christophe Labbez, Ger Koper, Robert Meszaros, Raffaele Mezzenga, Georg Papastavrou, Ionel Popa, Maria Santore, Imre Varga, and Corinne Vebert. We further thank Sergiy Minko for providing the high-resolution images appearing in Fig. 6c and d and Zbigniew Adamczyk for his permission to reproduce the data in Fig. 8d. This research was supported by the Swiss National Science Foundation, University of Geneva, State Secretariat for Education, Research and Innovation within the COST Action D43, and the EU Framework Program.References
- B. Bolto and J. Gregory, Water Res., 2007, 41, 2301–2324 CrossRef CAS PubMed.
- D. Horn and F. Linhart, Retention aids, Blackie Academic and Professional, London, 2nd edn, 1996 Search PubMed.
- N. Tobori and T. Amari, Colloids Surf., A, 2003, 215, 163–171 CrossRef CAS.
- A. M. Howe, R. D. Wesley, M. Bertrand, M. Cote and J. Leroy, Langmuir, 2006, 22, 4518–4525 CrossRef CAS PubMed.
- X. N. Chen, R. X. Huang and R. Pelton, Ind. Eng. Chem. Res., 2005, 44, 2078–2085 CrossRef CAS.
- I. Pochard, C. Labbez, A. Nonat, H. Vija and B. Jonsson, Cem. Concr. Res., 2010, 40, 1488–1494 CrossRef CAS PubMed.
- T. Phenrat, N. Saleh, K. Sirk, H. J. Kim, R. D. Tilton and G. V. Lowry, J. Nanopart. Res., 2008, 10, 795–814 CrossRef CAS.
- E. J. Bishop, D. E. Fowler, J. M. Skluzacek, E. Seibel and T. E. Mallouk, Environ. Sci. Technol., 2010, 44, 9069–9074 CrossRef CAS PubMed.
- S. Kim, J. H. So, D. J. Lee and S. M. Yang, J. Colloid Interface Sci., 2008, 319, 48–52 CrossRef CAS PubMed.
- E. Matijevic and S. V. Babu, J. Colloid Interface Sci., 2008, 320, 219–237 CrossRef CAS PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef PubMed.
- V. Salgueirino-Maceira, F. Caruso and L. M. Liz-Marzan, J. Phys. Chem. B, 2003, 107, 10990–10994 CrossRef CAS.
- S. Schwarz, J. E. Wong, J. Bornemann, M. Hodenius, U. Himmelreich, W. Richtering, M. Hoehn, M. Zenke and T. Hieronymus, Nanomed. Nanotechnol. Biol. Med., 2012, 8, 682–691 CrossRef CAS PubMed.
- R. Mezzenga, P. Schurtenberger, A. Burbidge and M. Michel, Nat. Mater., 2005, 4, 729–740 CrossRef CAS PubMed.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- D. G. Shchukin, G. B. Sukhorukov and H. Möhwald, Angew. Chem., Int. Ed., 2003, 42, 4472–4475 CrossRef CAS PubMed.
- A. Fery, F. Dubreuil and H. Möhwald, New J. Phys., 2004, 6, 18 CrossRef.
- T. Radeva and M. Grozeva, J. Colloid Interface Sci., 2005, 287, 415–421 CrossRef CAS PubMed.
- J. Ruhe, M. Ballauff, M. Biesalski, P. Dziezok, F. Grohn, D. Johannsmann, N. Houbenov, N. Hugenberg, R. Konradi, S. Minko, M. Motornov, R. R. Netz, M. Schmidt, C. Seidel, M. Stamm, T. Stephan, D. Usov and H. N. Zhang, Adv. Polym. Sci., 2004, 165, 79–150 CrossRef.
- M. Ballauff and O. Borisov, Curr. Opin. Colloid Interface Sci., 2006, 11, 316–323 CrossRef CAS PubMed.
- S. Moya, O. Azzaroni, T. Farhan, V. L. Osborne and W. T. S. Huck, Angew. Chem., Int. Ed., 2005, 44, 4578–4581 CrossRef CAS PubMed.
- J. L. Dalsin, L. Lin, S. Tosatti, J. Voros, M. Textor and P. B. Messersmith, Langmuir, 2005, 21, 640–646 CrossRef CAS PubMed.
- M. Elzbieciak-Wodka, M. Kolasinska-Sojka, D. Wodka, P. Nowak and P. Warszynski, J. Electroanal. Chem., 2011, 661, 162–170 CrossRef CAS PubMed.
- M. A. Cohen Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef CAS PubMed.
- N. T. Qazvini, S. Bolisetty, J. Adamcik and R. Mezzenga, Biomacromolecules, 2012, 13, 2136–2147 CrossRef CAS PubMed.
- N. G. Hoogeveen, M. A. Cohen Stuart and G. J. Fleer, J. Colloid Interface Sci., 1996, 182, 133–145 CrossRef CAS.
- N. G. Hoogeveen, M. A. Cohen Stuart and G. J. Fleer, J. Colloid Interface Sci., 1996, 182, 146–157 CrossRef CAS.
- M. Porus, P. Maroni and M. Borkovec, Langmuir, 2012, 28, 5642–5651 CrossRef CAS PubMed.
- J. Blaakmeer, M. R. Bohmer, M. A. Cohen Stuart and G. J. Fleer, Macromolecules, 1990, 23, 2301–2309 CrossRef CAS.
- R. Rehmet and E. Killmann, Colloids Surf., A, 1999, 149, 323–328 CrossRef CAS.
- J. Kleimann, C. Gehin-Delval, H. Auweter and M. Borkovec, Langmuir, 2005, 21, 3688–3698 CrossRef CAS PubMed.
- E. Seyrek, J. Hierrezuelo, A. Sadeghpour, I. Szilagyi and M. Borkovec, Phys. Chem. Chem. Phys., 2011, 13, 12716–12719 RSC.
- Z. Adamczyk, K. Sadlej, E. Wajnryb, M. Nattich, M. L. Ekiel-Jezewska and J. Blawzdziewicz, Adv. Colloid Interface Sci., 2010, 153, 1–29 CrossRef CAS PubMed.
- B. P. Cahill, G. Papastavrou, G. J. M. Koper and M. Borkovec, Langmuir, 2008, 24, 465–473 CrossRef CAS PubMed.
- G. J. Fleer, M. A. Cohen Stuart, J. M. H. M. Scheutjens, T. Cosgrove and B. Vincent, Polymers at Interfaces, Chapman and Hall, London, 1993 Search PubMed.
- C. Holm, J. F. Joanny, K. Kremer, R. R. Netz, P. Reineker, C. Seidel, T. A. Vilgis and R. G. Winkler, Adv. Polym. Sci., 2004, 166, 67–111 CrossRef CAS.
- R. R. Netz and D. Andelman, Phys. Rep., 2003, 380, 1–95 CrossRef CAS.
- A. V. Dobrynin, Curr. Opin. Colloid Interface Sci., 2008, 13, 376–388 CrossRef CAS PubMed.
- C. Y. Kong and M. Muthukumar, J. Chem. Phys., 1998, 109, 1522–1527 CrossRef CAS PubMed.
- J. Forsman, Langmuir, 2012, 28, 5138–5150 CrossRef CAS PubMed.
- J. Y. Carrillo and A. V. Dobrynin, Langmuir, 2007, 23, 2472–2482 CrossRef CAS PubMed.
- M. A. G. Dahlgren, A. Waltermo, E. Blomberg, P. M. Claesson, L. Sjostrom, T. Akesson and B. Jonsson, J. Phys. Chem., 1993, 97, 11769–11775 CrossRef CAS.
- P. M. Claesson, E. Poptoshev, E. Blomberg and A. Dedinaite, Adv. Colloid Interface Sci., 2005, 114, 173–187 CrossRef PubMed.
- S. Biggs, D. C. Prieve and R. R. Dagastine, Langmuir, 2005, 21, 5421–5428 CrossRef CAS PubMed.
- E. S. Pagac, R. D. Tilton and D. C. Prieve, Langmuir, 1998, 14, 5106–5112 CrossRef CAS.
- X. C. Xing, G. Q. Sun, Z. F. Li and T. Ngai, Langmuir, 2012, 28, 16022–16028 CrossRef CAS PubMed.
- I. Popa, G. Gillies, G. Papastavrou and M. Borkovec, J. Phys. Chem. B, 2009, 113, 8458–8461 CrossRef CAS PubMed.
- M. Finessi, P. Sinha, I. Szilagyi, I. Popa, P. Maroni and M. Borkovec, J. Phys. Chem. B, 2011, 115, 9098–9105 CrossRef CAS PubMed.
- F. Bouyer, A. Robben, W. L. Yu and M. Borkovec, Langmuir, 2001, 17, 5225–5231 CrossRef CAS.
- M. Ashmore, J. Hearn and F. Karpowicz, Langmuir, 2001, 17, 1069–1073 CrossRef CAS.
- Y. K. Leong, P. J. Scales, T. W. Healy and D. V. Boger, Colloids Surf., A, 1995, 95, 43–52 CrossRef CAS.
- B. Derjaguin and L. D. Landau, Acta Physicochim. URSS, 1941, 14, 633–662 Search PubMed.
- E. J. W. Verwey and J. T. G. Overbeek, Theory of Stability of Lyophobic Colloids, Elsevier, Amsterdam, 1948 Search PubMed.
- W. B. Russel, D. A. Saville and W. R. Schowalter, Colloidal Dispersions, Cambridge University Press, Cambridge, 1989 Search PubMed.
- V. K. La Mer and T. W. Healy, Rev. Pure Appl. Chem., 1963, 13, 112–133 CAS.
- J. Gregory, J. Colloid Interface Sci., 1973, 42, 448–456 CrossRef CAS.
- G. Olanya, J. Iruthayaraj, E. Poptoshev, R. Makuska, A. Vareikis and P. M. Claesson, Langmuir, 2008, 24, 5341–5349 CrossRef CAS PubMed.
- A. Naderi, G. Olanya, R. Makuska and P. M. Claesson, J. Colloid Interface Sci., 2008, 323, 223–228 CrossRef CAS PubMed.
- Y. Guo, J. van Beek, B. Zhang, M. Colussi, P. Walde, A. Zhang, M. Kröger, A. Halperin and A. D. Schlüter, J. Am. Chem. Soc., 2009, 131, 11841–11854 CrossRef CAS PubMed.
- J. J. Ramsden, Chem. Soc. Rev., 1995, 24, 73–78 RSC.
- J. R. Lu, X. B. Zhao and M. Yaseen, Curr. Opin. Colloid Interface Sci., 2007, 12, 9–16 CrossRef CAS PubMed.
- Z. Adamczyk, Curr. Opin. Colloid Interface Sci., 2012, 17, 173–186 CrossRef CAS PubMed.
- V. Ball, A. Bentaleb, J. Hemmerle, J. C. Voegel and P. Schaaf, Langmuir, 1996, 12, 1614–1621 CrossRef CAS.
- P. M. Claesson, E. Blomberg, J. C. Froberg, T. Nylander and T. Arnebrant, Adv. Colloid Interface Sci., 1995, 57, 161–227 CrossRef CAS.
- R. A. Silva, M. D. Urzua, D. F. S. Petri and P. L. Dubin, Langmuir, 2010, 26, 14032–14038 CrossRef CAS PubMed.
- Z. Adamczyk, J. Barbasz and M. Ciesla, Langmuir, 2011, 27, 6868–6878 CrossRef CAS PubMed.
- A. Mecke, I. Lee, J. R. Baker, M. M. B. Holl and B. G. Orr, Eur. Phys. J. E, 2004, 14, 7–16 CrossRef CAS PubMed.
- S. Block and C. A. Helm, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 030801 CrossRef.
- L. Muresan, P. Maroni, I. Popa, M. Porus, R. Longtin, G. Papastavrou and M. Borkovec, Macromolecules, 2011, 44, 5069–5071 CrossRef CAS.
- M. Porus, P. Maroni and M. Borkovec, Langmuir, 2012, 28, 17506–17516 CrossRef CAS PubMed.
- Y. Samoshina, T. Nylander, V. Shubin, R. Bauer and K. Eskilsson, Langmuir, 2005, 21, 5872–5881 CrossRef CAS PubMed.
- S. M. Notley, S. Biggs, V. S. J. Craig and L. Wagberg, Phys. Chem. Chem. Phys., 2004, 6, 2379–2386 RSC.
- H. G. Pedersen and L. Bergstrom, J. Am. Ceram. Soc., 1999, 82, 1137–1145 CrossRef CAS.
- A. Gromer, M. Rawiso and M. Maaloum, Langmuir, 2008, 24, 8950–8953 CrossRef CAS PubMed.
- T. Abraham, Polymer, 2002, 43, 849–855 CrossRef CAS.
- J. Gregory and S. Barany, Adv. Colloid Interface Sci., 2011, 169, 1–12 CrossRef CAS PubMed.
- C. Porcel, P. Lavalle, V. Ball, G. Decher, B. Senger, J. C. Voegel and P. Schaaf, Langmuir, 2006, 22, 4376–4383 CrossRef CAS PubMed.
- E. Laarz, A. Meurk, J. A. Yanez and L. Bergstrom, J. Am. Ceram. Soc., 2001, 84, 1675–1682 CrossRef CAS.
- A. P. Ngankam and P. R. Van Tassel, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1140–1145 CrossRef CAS PubMed.
- J. Penfold, I. Tucker, R. K. Thomas, D. J. F. Taylor, X. L. Zhang, C. Bell, C. Breward and P. Howell, Langmuir, 2007, 23, 3128–3136 CrossRef CAS PubMed.
- L. Muresan, P. Sinha, P. Maroni and M. Borkovec, Colloids Surf., A, 2011, 390, 225–230 CrossRef CAS PubMed.
- H. Walter, C. Harrats, P. Muller-Buschbaum, R. Jerome and M. Stamm, Langmuir, 1999, 15, 1260–1267 CrossRef CAS.
- Z. Adamczyk, M. Nattich and J. Barbasz, Adv. Colloid Interface Sci., 2009, 147–148, 2–17 CrossRef CAS PubMed.
- N. Kozlova and M. M. Santore, Langmuir, 2006, 22, 1135–1142 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, Environ. Sci. Technol., 2008, 42, 7607–7614 CrossRef CAS.
- S. Abalde-Cela, S. Ho, B. Rodriguez-Gonzalez, M. A. Correa-Duarte, R. A. Alvarez-Puebla, L. M. Liz-Marzan and N. A. Kotov, Angew. Chem., Int. Ed., 2009, 48, 5326–5329 CrossRef CAS PubMed.
- D. Sebok, T. Szabo and I. Dekany, Appl. Surf. Sci., 2009, 255, 6953–6962 CrossRef CAS PubMed.
- R. R. Netz and J. F. Joanny, Macromolecules, 1999, 32, 9013–9025 CrossRef CAS.
- G. J. M. Koper and M. Borkovec, Polymer, 2010, 51, 5649–5662 CrossRef CAS PubMed.
- N. Volk, D. Vollmer, M. Schmidt, W. Oppermann and K. Huber, Adv. Polym. Sci., 2004, 166, 29–65 CrossRef CAS.
- I. Popa, B. P. Cahill, P. Maroni, G. Papastavrou and M. Borkovec, J. Colloid Interface Sci., 2007, 309, 28–35 CrossRef CAS PubMed.
- N. P. Huang, R. Michel, J. Voros, M. Textor, R. Hofer, A. Rossi, D. L. Elbert, J. A. Hubbell and N. D. Spencer, Langmuir, 2001, 17, 489–498 CrossRef CAS.
- Z. Adamczyk, A. Michna, M. Szaraniec, A. Bratek and J. Barbasz, J. Colloid Interface Sci., 2007, 313, 86–96 CrossRef CAS PubMed.
- I. Varga, A. Mezei, R. Meszaros and P. M. Claesson, Soft Matter, 2011, 7, 10701–10712 RSC.
- R. Longtin, P. Maroni and M. Borkovec, Langmuir, 2009, 25, 2928–2934 CrossRef CAS PubMed.
- R. Kargl, T. Mohan, M. Bracic, M. Kulterer, A. Doliska, K. Stana-Kleinschek and V. Ribitsch, Langmuir, 2012, 28, 11440–11447 CrossRef CAS PubMed.
- M. Jiang, I. Popa, P. Maroni and M. Borkovec, Colloids Surf., A, 2010, 360, 20–25 CrossRef CAS PubMed.
- L. E. Enarsson and L. Wagberg, Langmuir, 2008, 24, 7329–7337 CrossRef CAS PubMed.
- R. Meszaros, I. Varga and T. Gilanyi, Langmuir, 2004, 20, 5026–5029 CrossRef CAS.
- R. C. van Duijvenbode, I. B. Rietveld and G. J. M. Koper, Langmuir, 2000, 16, 7720–7725 CrossRef CAS.
- Z. Adamczyk, L. Szyk and P. Warszynski, J. Colloid Interface Sci., 1999, 209, 350–361 CrossRef CAS PubMed.
- H. Dautzenberg, E. Gornitz and W. Jaeger, Macromol. Chem. Phys., 1998, 199, 1561–1571 CrossRef CAS.
- Y. Tie, C. Calonder and P. R. Van Tassel, J. Colloid Interface Sci., 2003, 268, 1–11 CrossRef CAS.
- J. W. Evans, Rev. Mod. Phys., 1993, 65, 1281–1329 CrossRef CAS.
- Z. Adamczyk, B. Senger, J. C. Voegel and P. Schaaf, J. Chem. Phys., 1999, 110, 3118–3128 CrossRef CAS PubMed.
- B. Senger, J. C. Voegel and P. Schaaf, Colloids Surf., A, 2000, 165, 255–285 CrossRef CAS.
- I. Popa, G. Papastavrou and M. Borkovec, Phys. Chem. Chem. Phys., 2010, 12, 4863–4871 RSC.
- M. Finessi, PhD, University of Geneva, 2013.
- L. J. Kirwan, P. Maroni, S. H. Behrens, G. Papastavrou and M. Borkovec, J. Phys. Chem. B, 2008, 112, 14609–14619 CrossRef CAS PubMed.
- D. Bauer, H. Buchhammer, A. Fuchs, W. Jaeger, E. Killmann, K. Lunkwitz, R. Rehmet and S. Schwarz, Colloids Surf., A, 1999, 156, 291–305 CrossRef CAS.
- M. R. Bohmer, W. H. A. Heesterbeek, A. Deratani and E. Renard, Colloids Surf., A, 1995, 99, 53–64 CrossRef.
- S. A. Sukhishvili and S. Granick, J. Chem. Phys., 1998, 109, 6861–6868 CrossRef CAS PubMed.
- K. Esumi and M. Goino, Langmuir, 1998, 14, 4466–4470 CrossRef CAS.
- R. C. van Duijvenbode, G. J. M. Koper and M. R. Bohmer, Langmuir, 2000, 16, 7713–7719 CrossRef CAS.
- T. Sennerfors, D. Solberg and F. Tiberg, J. Colloid Interface Sci., 2002, 254, 222–226 CrossRef CAS.
- T. J. Barnes, I. Ametov and C. A. Prestidge, Langmuir, 2008, 24, 12398–12404 CrossRef CAS PubMed.
- V. Shubin, J. Colloid Interface Sci., 1997, 191, 372–377 CrossRef CAS.
- J. Lyklema and L. Deschenes, Adv. Colloid Interface Sci., 2011, 168, 135–148 CrossRef CAS PubMed.
- N. Hansupalak and M. M. Santore, Langmuir, 2003, 19, 7423–7426 CrossRef CAS.
- D. Cakara, C. Chassagne, C. Gehin-Delval and M. Borkovec, Colloids Surf., A, 2007, 294, 174–180 CrossRef CAS PubMed.
- E. Illes and E. Tombacz, Colloids Surf., A, 2003, 230, 99–109 CrossRef CAS PubMed.
- B. Cabot, A. Deratani and A. Foissy, Colloids Surf., A, 1998, 139, 287–297 CrossRef CAS.
- Z. Adamczyk and P. Warszynski, Adv. Colloid Interface Sci., 1996, 63, 41–149 CrossRef CAS.
- M. Semmler, E. K. Mann, J. Ricka and M. Borkovec, Langmuir, 1998, 14, 5127–5132 CrossRef CAS.
- Z. Adamczyk, Adv. Colloid Interface Sci., 2003, 100, 267–347 CrossRef.
- A. V. Dobrynin, A. Deshkovski and M. Rubinstein, Macromolecules, 2001, 34, 3421–3436 CrossRef CAS.
- M. Muthukumar, J. Chem. Phys., 1987, 86, 7230–7235 CrossRef CAS PubMed.
- J. Hierrezuelo, I. Szilagyi, A. Vaccaro and M. Borkovec, Macromolecules, 2010, 43, 9108–9116 CrossRef CAS.
- F. Hook, B. Kasemo, T. Nylander, C. Fant, K. Sott and H. Elwing, Anal. Chem., 2001, 73, 5796–5804 CrossRef CAS.
- A. Vaccaro, J. Hierrezuelo, M. Skarba, P. Galletto, J. Kleimann and M. Borkovec, Langmuir, 2009, 25, 4864–4867 CrossRef CAS PubMed.
- I. Popa, G. Gillies, G. Papastavrou and M. Borkovec, J. Phys. Chem. B, 2010, 114, 3170–3177 CrossRef CAS PubMed.
- Y. Roiter and S. Minko, J. Phys. Chem. B, 2007, 111, 8597–8604 CrossRef CAS PubMed.
- R. Pericet-Camara, G. Papastavrou and M. Borkovec, Langmuir, 2004, 20, 3264–3270 CrossRef CAS.
- S. Minko and Y. Roiter, Curr. Opin. Colloid Interface Sci., 2005, 10, 9–15 CrossRef CAS PubMed.
- L. J. Kirwan, G. Papastavrou, M. Borkovec and S. H. Behrens, Nano Lett., 2004, 4, 149–152 CrossRef CAS.
- F. Valle, M. Favre, P. de los Rios, A. Rosa and G. Dietler, Phys. Rev. Lett., 2005, 95, 158105 CrossRef.
- H. G. M. van de Steeg, M. A. Cohen Stuart, A. de Keizer and B. H. Bijsterbosch, Langmuir, 1992, 8, 2538–2546 CrossRef CAS.
- I. Borukhov, D. Andelman and H. Orland, Macromolecules, 1998, 31, 1665–1671 CrossRef CAS.
- P. Linse, Macromolecules, 1996, 29, 326–336 CrossRef CAS.
- G. Gillies, W. Lin and M. Borkovec, J. Phys. Chem. B, 2007, 111, 8626–8633 CrossRef CAS PubMed.
- J. Hierrezuelo, A. Sadeghpour, I. Szilagyi, A. Vaccaro and M. Borkovec, Langmuir, 2010, 26, 15109–15111 CrossRef CAS PubMed.
- J. Hierrezuelo, A. Vaccaro and M. Borkovec, J. Colloid Interface Sci., 2010, 347, 202–208 CrossRef CAS PubMed.
- W. L. Yu, F. Bouyer and M. Borkovec, J. Colloid Interface Sci., 2001, 241, 392–399 CrossRef CAS.
- E. Illes and E. Tombacz, J. Colloid Interface Sci., 2006, 295, 115–123 CrossRef CAS PubMed.
- S. Sennato, D. Truzzolillo, F. Bordi, F. Sciortino and C. Cametti, Colloids Surf., A, 2009, 343, 34–42 CrossRef CAS PubMed.
- F. Quemeneur, M. Rinaudo, G. Maret and B. Pepin-Donat, Soft Matter, 2010, 6, 4471–4481 RSC.
- A. Feiler, P. Jenkins and J. Ralston, Phys. Chem. Chem. Phys., 2000, 2, 5678–5683 RSC.
- A. Sadeghpour, E. Seyrek, I. Szilagyi, J. Hierrezuelo and M. Borkovec, Langmuir, 2011, 27, 9270–9276 CrossRef CAS PubMed.
- I. Szilagyi, A. Sadeghpour and M. Borkovec, Langmuir, 2012, 28, 6211–6215 CrossRef CAS PubMed.
- I. Szilagyi, D. Rosicka, J. Hierrezuelo and M. Borkovec, J. Colloid Interface Sci., 2011, 360, 580–585 CrossRef CAS PubMed.
- A. Mezei and R. Meszaros, Langmuir, 2006, 22, 7148–7151 CrossRef CAS PubMed.
- Z. Adamczyk, A. Zembala and A. Michna, J. Colloid Interface Sci., 2006, 303, 353–364 CrossRef CAS PubMed.
- I. Popa, G. Papastavrou and M. Borkovec, Macromolecules, 2010, 43, 1129–1136 CrossRef CAS.
- J. Dejeu, L. Buisson, M. C. Guth, C. Roidor, F. Membrey, D. Charraut and A. Foissy, Colloids Surf., A, 2006, 288, 26–35 CrossRef CAS PubMed.
- W. Lin, P. Galletto and M. Borkovec, Langmuir, 2004, 20, 7465–7473 CrossRef CAS PubMed.
- J. Sabin, C. Vazquez-Vazquez, G. Prieto, F. Bordi and F. Sarmiento, Langmuir, 2012, 28, 10534–10542 CrossRef CAS PubMed.
- J. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 3rd edn, 2011 Search PubMed.
- S. H. Behrens and M. Borkovec, J. Phys. Chem. B, 1999, 103, 2918–2928 CrossRef CAS.
- G. Toikka, R. A. Hayes and J. Ralston, Langmuir, 1996, 12, 3783–3788 CrossRef CAS.
- C. Gutsche, U. F. Keyser, K. Kegler and F. Kremer, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 031403 CrossRef CAS.
- P. G. Hartley, I. Larson and P. J. Scales, Langmuir, 1997, 13, 2207–2214 CrossRef CAS.
- J. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 2nd edn, 1992 Search PubMed.
- P. Sinha, I. Szilagyi, F. J. Montes Ruiz-Cabello, P. Maroni and M. Borkovec, J. Phys. Chem. Lett., 2013, 4, 648–652 CrossRef CAS.
- R. M. Pashley, J. Colloid Interface Sci., 1981, 83, 531–546 CrossRef CAS.
- G. Toikka and R. A. Hayes, J. Colloid Interface Sci., 1997, 191, 102–109 CrossRef CAS.
- G. Vigil, Z. H. Xu, S. Steinberg and J. Israelachvili, J. Colloid Interface Sci., 1994, 165, 367–385 CrossRef CAS.
- P. F. Luckham and J. Klein, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 865–878 RSC.
- P. Berndt, K. Kurihara and T. Kunitake, Langmuir, 1992, 8, 2486–2490 CrossRef CAS.
- R. Pericet-Camara, G. Papastavrou, S. H. Behrens, C. A. Helm and M. Borkovec, J. Colloid Interface Sci., 2006, 296, 496–506 CrossRef CAS PubMed.
- E. Poptoshev and P. M. Claesson, Langmuir, 2002, 18, 2590–2594 CrossRef CAS.
- E. Poptoshev, M. W. Rutland and P. M. Claesson, Langmuir, 2000, 16, 1987–1992 CrossRef CAS.
- P. M. Claesson and B. W. Ninham, Langmuir, 1992, 8, 1406–1412 CrossRef CAS.
- G. Maurdev, L. Meagher, J. Ennis and M. L. Gee, Langmuir, 2001, 34, 4151–4158 CAS.
- C. E. McNamee, M. Matsumoto, P. G. Hartley, P. Mulvaney, Y. Tsujii and M. Nakahara, Langmuir, 2001, 17, 6220–6227 CrossRef CAS.
- A. Szucs, T. Haraszti, I. Dekany and J. H. Fendler, J. Phys. Chem. B, 2001, 105, 10579–10587 CrossRef.
- P. G. Hartley and P. J. Scales, Langmuir, 1998, 14, 6948–6955 CrossRef CAS.
- V. Bosio, F. Dubreuil, G. Bogdanovic and A. Fery, Colloids Surf., A, 2004, 243, 147–155 CrossRef CAS PubMed.
- T. Abraham, D. Christendat, Z. Xu, J. Masliyah, J. F. Gohy and R. Jerome, AIChE J., 2004, 50, 2613–2626 CrossRef CAS.
- R. Podgornik and M. Licer, Curr. Opin. Colloid Interface Sci., 2006, 11, 273–279 CrossRef CAS PubMed.
- H. H. Huang and E. Ruckenstein, Langmuir, 2012, 28, 16300–16305 CrossRef CAS PubMed.
- M. Rief, F. Oesterhelt, B. Heymann and H. E. Gaub, Science, 1997, 275, 1295–1297 CrossRef CAS.
- T. Hugel, M. Grosholz, H. Clausen-Schaumann, A. Pfau, H. Gaub and M. Seitz, Macromolecules, 2001, 34, 1039–1047 CrossRef CAS.
- X. Chatellier, T. J. Senden, J. F. Joanny and J. M. di Meglio, Europhys. Lett., 1998, 41, 303–308 CrossRef CAS.
- G. Papastavrou, L. J. Kirwan and M. Borkovec, Langmuir, 2006, 22, 10880–10884 CrossRef CAS PubMed.
- E. Poptoshev, M. W. Rutland and P. M. Claesson, Langmuir, 1999, 15, 7789–7794 CrossRef CAS.
- S. J. Miklavic, D. Y. C. Chan, L. R. White and T. W. Healy, J. Phys. Chem., 1994, 98, 9022–9032 CrossRef CAS.
- P. Richmond, J. Chem. Soc., Faraday Trans. 2, 1975, 71, 1154–1163 RSC.
- I. Popa, G. Papastavrou, M. Borkovec, M. Trulsson and B. Jonsson, Langmuir, 2009, 25, 12435–12438 CrossRef CAS PubMed.
- J. Swenson, M. V. Smalley and H. L. M. Hatharasinghe, Phys. Rev. Lett., 1998, 81, 5840–5843 CrossRef CAS.
- Y. Zhou, Y. Gan, E. J. Wanless, G. J. Jameson and G. V. Franks, Langmuir, 2008, 24, 10920–10928 CrossRef CAS PubMed.
- I. Borukhov, D. Andelman and H. Orland, J. Phys. Chem. B, 1999, 103, 5042–5057 CrossRef CAS.
- J. Forsman and S. Nordholm, Langmuir, 2012, 28, 4069–4079 CrossRef CAS PubMed.
- M. Turesson, J. Forsman and T. Akesson, Langmuir, 2006, 22, 5734–5741 CrossRef CAS PubMed.
- D. Truzzolillo, F. Bordi, F. Sciortino and S. Sennato, J. Chem. Phys., 2010, 133, 024901 CrossRef CAS PubMed.
- C. E. Woodward, T. Akesson and B. Jonsson, J. Chem. Phys., 1994, 101, 2569–2576 CrossRef CAS PubMed.
- A. Broukhno, B. Jonsson, T. Akesson and P. N. Vorontsov-Velyaminov, J. Chem. Phys., 2000, 113, 5493–5501 CrossRef CAS PubMed.
- R. Podgornik, P. Akesson and B. Jonsson, J. Chem. Phys., 1995, 102, 9423–9434 CrossRef CAS PubMed.
- R. Podgornik, J. Chem. Phys., 2003, 118, 11286–11296 CrossRef CAS PubMed.
- S. Biggs and T. W. Healy, J. Chem. Soc., Faraday Trans., 1994, 90, 3415–3421 RSC.
- S. Biggs, J. L. Burns, Y. D. Yan, G. J. Jameson and P. Jenkins, Langmuir, 2000, 16, 9242–9248 CrossRef CAS.
- A. Sharma, S. N. Tan and J. Y. Walz, J. Colloid Interface Sci., 1997, 191, 236–246 CrossRef CAS.
- A. J. Milling, J. Phys. Chem., 1996, 100, 8986–8993 CrossRef CAS.
- X. J. Gong and T. Ngai, Langmuir, 2013, 29, 5974–5981 CrossRef CAS PubMed.
- D. Kleshchanok, R. Tuinier and P. R. Lang, J. Phys.: Condens. Matter, 2008, 20, 073101 CrossRef.
- Y. Zeng and R. von Klitzing, Langmuir, 2012, 28, 6313–6321 CrossRef CAS PubMed.
- M. Elimelech, J. Gregory, X. Jia and R. A. Williams, Particle Deposition and Aggregation: Measurement, Modeling, and Simulation, Butterworth-Heinemann Ltd., Oxford, 1995 Search PubMed.
- P. Sandkuhler, M. Lattuada, H. Wu, J. Sefcik and M. Morbidelli, Adv. Colloid Interface Sci., 2005, 113, 65–83 CrossRef PubMed.
- C. Gogelein, G. Nagele, J. Buitenhuis, R. Tuinier and J. K. G. Dhont, J. Chem. Phys., 2009, 130, 204905 CrossRef PubMed.
- M. Lattuada, H. Wu and M. Morbidelli, Langmuir, 2004, 20, 4355–4362 CrossRef CAS.
- K. Masschaele, J. Fransaer and J. Vermant, Soft Matter, 2011, 7, 7717–7726 RSC.
- E. P. Honig, G. J. Roebersen and P. H. Wiersema, J. Colloid Interface Sci., 1971, 36, 97–102 CrossRef CAS.
- S. H. Behrens, M. Borkovec and P. Schurtenberger, Langmuir, 1998, 14, 1951–1954 CrossRef CAS.
- M. Kobayashi, M. Skarba, P. Galletto, D. Cakara and M. Borkovec, J. Colloid Interface Sci., 2005, 292, 139–147 CrossRef CAS PubMed.
- M. Schudel, S. H. Behrens, H. Holthoff, R. Kretzschmar and M. Borkovec, J. Colloid Interface Sci., 1997, 196, 241–253 CrossRef CAS PubMed.
- H. Holthoff, S. U. Egelhaaf, M. Borkovec, P. Schurtenberger and H. Sticher, Langmuir, 1996, 12, 5541–5549 CrossRef CAS.
- W. Lin, M. Kobayashi, M. Skarba, C. Mu, P. Galletto and M. Borkovec, Langmuir, 2006, 22, 1038–1047 CrossRef CAS PubMed.
- H. Holthoff, A. Schmitt, A. Fernandez-Barbero, M. Borkovec, M. A. Cabrerizo-Vilchez, P. Schurtenberger and R. Hidalgo-Alvarez, J. Colloid Interface Sci., 1997, 192, 463–470 CrossRef CAS.
- S. H. Xu and Z. W. Sun, Soft Matter, 2011, 7, 11298–11308 RSC.
- D. R. E. Snoswell, J. M. Duan, D. Fornasiero and J. Ralston, J. Colloid Interface Sci., 2005, 286, 526–535 CrossRef CAS PubMed.
- H. Kihira, N. Ryde and E. Matijevic, J. Chem. Soc., Faraday Trans., 1992, 88, 2379–2386 RSC.
- M. B. Einarson and J. C. Berg, J. Colloid Interface Sci., 1993, 155, 165–172 CrossRef CAS.
- C. Schneider, M. Hanisch, B. Wedel, A. Jusufi and M. Ballauff, J. Colloid Interface Sci., 2011, 358, 62–67 CrossRef CAS PubMed.
- S. H. Behrens, D. I. Christl, R. Emmerzael, P. Schurtenberger and M. Borkovec, Langmuir, 2000, 16, 2566–2575 CrossRef CAS.
- T. Hiemstra and W. H. van Riemsdijk, Langmuir, 1999, 15, 8045–8051 CrossRef CAS.
- S. Y. Shulepov and G. Frens, J. Colloid Interface Sci., 1996, 182, 388–394 CrossRef CAS.
- J. D. Feick and D. Velegol, Langmuir, 2002, 18, 3454–3458 CrossRef CAS.
- K. L. Chen, S. E. Mylon and M. Elimelech, Environ. Sci. Technol., 2006, 40, 1516–1523 CrossRef CAS.
- G. Fritz, V. Schadler, N. Willenbacher and N. J. Wagner, Langmuir, 2002, 18, 6381–6390 CrossRef CAS.
- X. Guo and M. Ballauff, Langmuir, 2000, 16, 8719–8726 CrossRef CAS.
- C. N. Likos, K. A. Vaynberg, H. Lowen and N. J. Wagner, Langmuir, 2000, 16, 4100–4108 CrossRef CAS.
- R. G. Smits, G. J. M. Koper and M. Mandel, J. Phys. Chem., 1993, 97, 5745–5751 CrossRef CAS.
- H. W. Walker and S. B. Grant, Colloids Surf., A, 1996, 119, 229–239 CrossRef CAS.
- M. Trulsson, J. Forsman, T. Akesson and B. Jonsson, Langmuir, 2009, 25, 6106–6112 CrossRef CAS PubMed.
- S. Schwarz, K. Lunkwitz, B. Kessler, U. Spiegler, E. Killmann and W. Jaeger, Colloids Surf., A, 2000, 163, 17–27 CrossRef CAS.
- S. Schwarz, W. Jaeger, B. R. Paulke, S. Bratskaya, N. Smolka and J. Bohrisch, J. Phys. Chem. B, 2007, 111, 8649–8654 CrossRef CAS PubMed.
- T. Abe, S. Kobayashi and M. Kobayashi, Colloids Surf., A, 2011, 379, 21–26 CrossRef CAS PubMed.
- K. L. Chen, S. E. Mylon and M. Elimelech, Langmuir, 2007, 23, 5920–5928 CrossRef CAS PubMed.
- J. E. Seebergh and J. C. Berg, Langmuir, 1994, 10, 454–463 CrossRef CAS.
- A. Sharma, S. N. Tan and J. Y. Walz, J. Colloid Interface Sci., 1997, 190, 392–407 CrossRef CAS.
- M. J. Snowden, S. M. Clegg, P. A. Williams and I. D. Robb, J. Chem. Soc., Faraday Trans., 1991, 87, 2201–2207 RSC.
- S. Rawson, K. Ryan and B. Vincent, Colloids Surf., 1988, 34, 89–93 CrossRef CAS.
- P. Richmond, J. Chem. Soc., Faraday Trans. 2, 1974, 70, 1066–1073 RSC.
| This journal is © The Royal Society of Chemistry 2014 |