
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNuclear resonance vibrational spectroscopy reveals the FeS cluster composition and active site vibrational properties of an O2-tolerant NAD+-reducing [NiFe] hydrogenase†
Lars
Lauterbach‡
*ab,
Hongxin
Wang‡
bc,
Marius
Horch‡
a,
Leland B.
Gee
b,
Yoshitaka
Yoda
d,
Yoshihito
Tanaka
e,
Ingo
Zebger
a,
Oliver
Lenz
a and
Stephen P.
Cramer
*bc
aInstitute of Chemistry, Technische Universität Berlin, Straße des 17. Juni 135, 10623 Berlin, Germany. E-mail: lars.lauterbach@tu-berlin.de
bDepartment of Chemistry, University of California, One Shields Ave, Davis, CA 95616, USA. E-mail: spcramer@lbl.gov
cPhysical Biosciences Division, Lawrence Berkeley National Laboratory, One Cyclotron Road, Berkeley, CA 94720, USA
dJASRI, SPring-8, 1-1-1 Kouto, Mikazuki-cho, Sayo-gun, Hyogo 679-5198, Japan
eRIKEN, SPring-8, 1-1-1 Kouto, Mikazuki-cho, Sayo-gun, Hyogo 679-5198, Japan
First published on 30th October 2014
Abstract
Hydrogenases are complex metalloenzymes that catalyze the reversible splitting of molecular hydrogen into protons and electrons essentially without overpotential. The NAD+-reducing soluble hydrogenase (SH) from Ralstonia eutropha is capable of H2 conversion even in the presence of usually toxic dioxygen. The molecular details of the underlying reactions are largely unknown, mainly because of limited knowledge of the structure and function of the various metal cofactors present in the enzyme. Here, all iron-containing cofactors of the SH were investigated by 57Fe specific nuclear resonance vibrational spectroscopy (NRVS). Our data provide experimental evidence for one [2Fe2S] center and four [4Fe4S] clusters, which is consistent with the amino acid sequence composition. Only the [2Fe2S] cluster and one of the four [4Fe4S] clusters were reduced upon incubation of the SH with NADH. This finding explains the discrepancy between the large number of FeS clusters and the small amount of FeS cluster-related signals as detected by electron paramagnetic resonance spectroscopic analysis of several NAD+-reducing hydrogenases. For the first time, Fe–CO and Fe–CN modes derived from the [NiFe] active site could be distinguished by NRVS through selective 13C labeling of the CO ligand. This strategy also revealed the molecular coordinates that dominate the individual Fe–CO modes. The present approach explores the complex vibrational signature of the Fe–S clusters and the hydrogenase active site, thereby showing that NRVS represents a powerful tool for the elucidation of complex biocatalysts containing multiple cofactors.
Introduction
Hydrogenases are nature's catalysts for the reversible cleavage of molecular hydrogen into protons and electrons. In the case of [NiFe] hydrogenase, the active site nickel is coordinated to the protein matrix by four cysteinyl thiolates, two of which serve as bridging ligands to the iron (Fig. 1). The iron is further coordinated by one carbonyl and two cyanide ligands.1 While most [NiFe] hydrogenases are reversibly inactivated by O2, the soluble, NAD+-reducing [NiFe] hydrogenase (SH) from Ralstonia eutropha H16, which couples the reversible oxidation of H2 to the reduction of NAD+, remains highly active even under ambient O2 concentration.2,3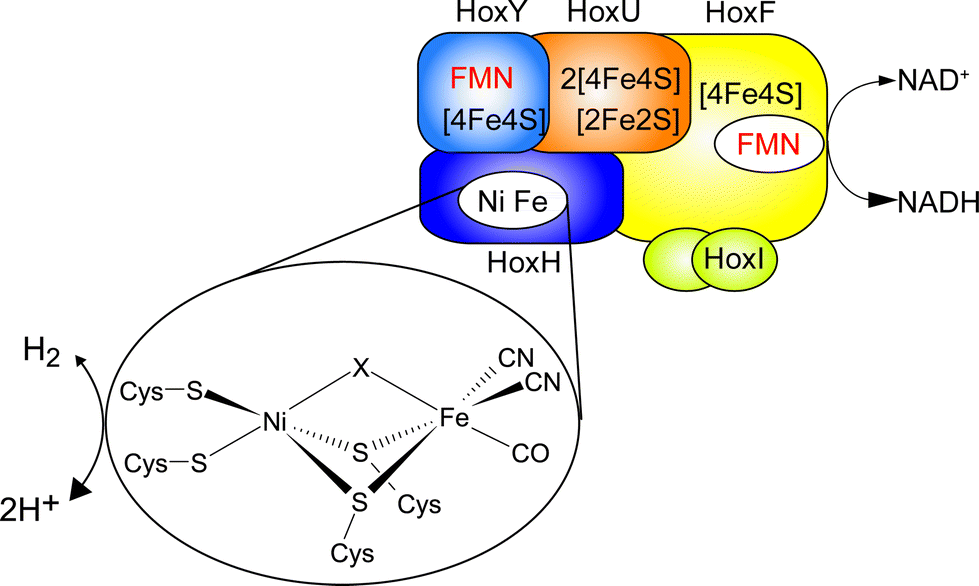 | ||
| Fig. 1 Schematic overview of the cofactor composition of the SH. The [NiFe] active site is shown in the inset, with X denoting the site of hydride or inhibitor binding. | ||
The SH is therefore a promising enzyme for biotechnological applications, e.g. nucleotide cofactor regeneration.4 The O2 tolerance of the SH relies on its capability to continuously reduce O2 to water and hydrogen peroxide, allowing for H2 oxidation in the presence of oxygen.3 Sequence analysis indicates that the [NiFe] active center of this enzyme is connected to the NAD+-binding site by one [2Fe2S] center and four [4Fe4S] clusters as well as two flavin mononucleotide (FMN) molecules (Fig. 1).5 While the FMN cofactors have been confirmed experimentally, only signals of the [2Fe2S] cluster and one [4Fe4S] cluster5,6 have been detected by electron paramagnetic resonance (EPR) spectroscopy, and similar results have been obtained for other NAD+-reducing [NiFe] hydrogenases.7,8 Until now, the reason for the discrepancy between the number of potential binding motifs and EPR-detected FeS clusters was unclear and experimental evidence for the proposed set of clusters was missing.
Nuclear resonance vibrational spectroscopy (NRVS) is a synchrotron-based vibrational spectroscopic technique that selectively probes iron-specific normal modes and the related molecular coordinates. Therefore it is an ideal technique for the investigation of iron-containing metalloproteins in terms of both structure and dynamics. In the case of [NiFe] hydrogenase, NRVS has first been successfully applied in 2013.9 Likewise, resonance Raman (RR) spectroscopy represents a comparable method to gain complementary insights.10,11 In the case of the SH, however, the application of RR is strongly impeded by FMN-derived fluorescence. Moreover, no RR signal was found for the [NiFe] active site of oxidized SH with several excitation wavelengths (data not shown), demonstrating the necessity of NRVS for the characterization of this particular enzyme.
Experimental
Sample preparation
Ralstonia eutropha was cultured and SH was purified according to ref. 3 except that 18 μM 57FeCl2 was used in the minimal medium. For labelling of the SH with 13CO, 5000 ppm 13CO gas was added into a 16 L desiccator, which contained a R. eutropha culture overproducing the SH and atmospheric gas. The NADH-treated purified SH-samples were prepared by 30 min anaerobic incubation of 1 mM SH with 50 mM NADH. The concentration of oxidized SH samples was 1 mM.NRVS measurements
Nuclear resonance vibrational spectroscopy measurements were conducted at SPring-8 BL09XU and BL19LXU. A liquid N2-cooled Si (1, 1, 1) high heat load monochromator was used to produce an incident beam with ∼1 eV resolution, followed by a high resolution monochromator with an asymmetrically cut Ge (3, 3, 1) and two nested Si (9, 7, 5) crystals to achieve a ∼0.8 meV energy resolution at 14.4125 keV. A 4-channel avalanche photo diode detector array was used to detect the X-ray K-fluorescence from internal conversion and the nuclear gamma ray emission by 57Fe atoms. The detectors were placed as close as possible to the surface of the sample. All measurements were performed at 10 K in the cryostat base. The real sample temperature, as obtained from the spectral analysis, was 30–60 K.To enhance the S/N ratio in the Fe–CO/CN range of the spectra, sectional measurements of these regions were performed. While for the usual scans the time for every point was 5 s, in the case of sectional scans the acquisition time for every data point in the region of interest, i.e. from 400 to 680 cm−1, was 10–20 s and the rest was 1–3 s depending on measurements.
The raw NRVS data were analyzed by the PHOENIX software12 to extract the single-phonon iron partial vibrational density of states (57Fe PVDOS). The resonance peak position for each scan was aligned in the PHOENIX analysis, while the energy scale was calibrated with an external reference ([NEt4][FeCl4]) periodically.
To produce an empirical NRVS spectral simulation, Rhodobacter capsulatus (Rc) [2Fe2S] ferredoxin NRVS13 was used as a representative model for [2Fe2S] clusters, while the Pyrococcus furiosus (Pf) [4Fe4S] ferredoxin NRVS was used as a model for [4Fe4S] clusters (both in their oxidized (ox) and reduced (red) states).14 These spectra were first auto-normalized to 3, representing spectra averaged for one iron (subscript “norm”). For [4Fe4S] clusters, the Pf ferredoxin NRVS was then multiplied by 4 to get a weighed NRVS spectrum considering that the cluster has 4 irons, while for [2Fe2S] clusters, the Rc ferredoxin NRVS was multiplied by 2 for the same reason. Such spectra were then multiplied by the numbers of the same type of clusters included in the SH. The total sum was calculated and divided by 18 in order to account the number of considered Fe ions. For example:
| SHox = [1 × 2 × Rc ferredoxin(ox)norm + 4 × 4 × Pf ferredoxin (ox)norm]/18, |
| SHred = [0.5 × 2 × Rc ferredoxin (ox)norm + 0.5 × 2 × Rc ferredoxin (red)norm + 3 × 4 × Pf ferredoxin (ox)norm + 1 × 4 × Pf ferredoxin (red)norm]/18. |
To evaluate integral intensities in the spectral range of FeS cluster normal modes (0–450 cm−1), spectra were first re-normalized with respect to the integral intensities of the Fe–CO/CN centered modes (450–650 cm−1), assuming that these signals are comparable for different [NiFe] hydrogenases. Using this procedure, spectra were corrected for preparation and acquisition related differences, such that integral FeS intensities of the normalized spectra could be directly compared. The FeS integral intensities were then determined as a marker for the relative amount of Fe ions involved in FeS clusters of different [NiFe] hydrogenases. The whole procedure was performed by using the Bruker OPUS software.
IR measurements
Infrared (IR) spectra with a spectral resolution of 2 cm−1 were recorded on a Bruker Tensor 27 spectrometer, equipped with a liquid nitrogen-cooled mercury cadmium telluride (MCT) detector. The sample compartment was purged with dried air and the sample was held in a temperature-controlled (283 K) gas-tight IR-cell for liquid samples (volume ∼7 μL, optical path length = 50 μm), equipped with CaF2 windows. Spectra were processed and baseline-corrected by using the Bruker OPUS software.Results and discussion
FeS cluster composition
In this communication, we report a combined NRVS and IR spectroscopic characterization of SH in its as-isolated (oxidized) and NADH-reduced state. The partial vibrational density of states (PVDOS) for oxidized and NADH-reduced SH as detected by NRVS is presented in Fig. 2A. As 16 out of 19 expected Fe atoms in this protein are located in [4Fe4S] centers (Fig. 1), the spectra are most likely dominated by contributions from these cofactors. Consistently, the entire pattern between 0 and 400 cm−1 is very similar to the NRVS of the [4Fe4S] cluster in Pf ferredoxin and not far from that of the “standard” [NiFe]-hydrogenase from Desulfovibrio vulgaris (Dv) Miyazaki F, which contains the [NiFe] site, one [3Fe4S] center and two [4Fe4S] clusters.14,9 However, in contrast to Dv hydrogenase, integral intensities in this spectral range are strongly increased by a factor of about 1.5, if the two spectra are normalized with respect to the Fe–CO/CN intensities of the active site (Fig. 3). This observation is in line with the higher amount of FeS clusters in the SH (18 Fe ions) as compared to “standard” [NiFe] hydrogenases (11 Fe ions). Thus, the present study provides the first spectroscopic evidence for the large number of FeS clusters in the SH.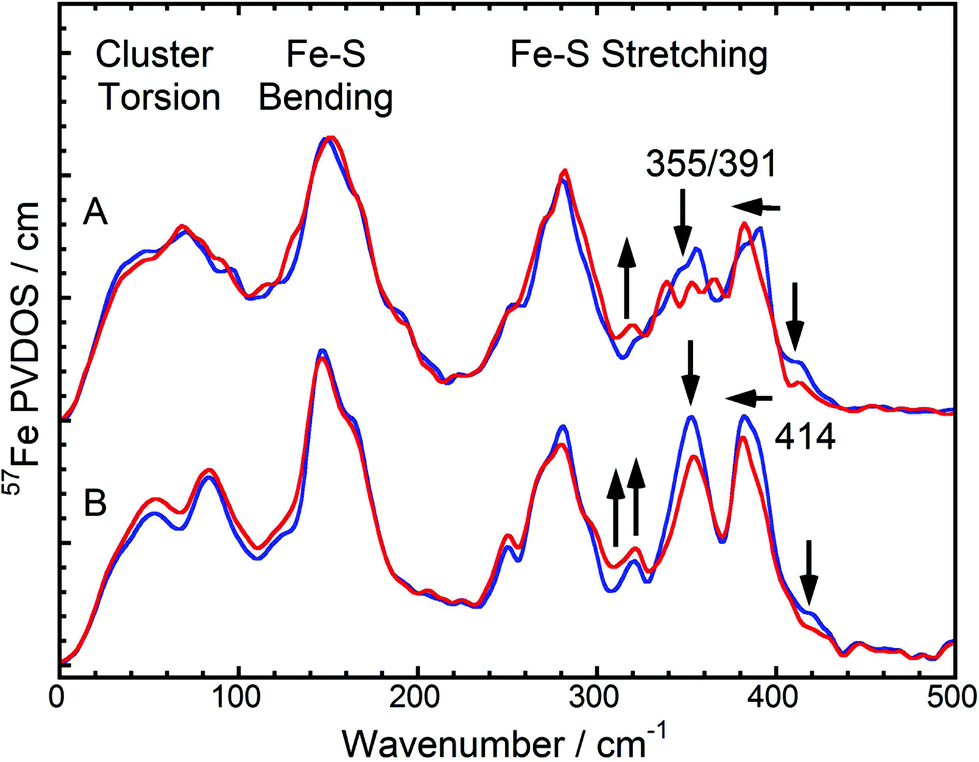 | ||
| Fig. 2 NRVS-derived 57Fe PVDOS in the FeS cluster mode region. (A): Oxidized (blue) and NADH-reduced (red) SH. (B): A weighted sum of one oxidized [2Fe2S] cluster of Rc ferredoxin plus four oxidized [4Fe4S] clusters of Pf ferredoxin (blue) vs. a weighted sum of one 50% reduced / 50% oxidized [2Fe2S] cluster of Rc ferredoxin plus one reduced and three oxidized [4Fe4S] clusters of Pf ferredoxin (red). NRVS data of Rc and Pf ferredoxins have been published previously.13,14 Arrows indicate the changes between the spectra in each pair. | ||
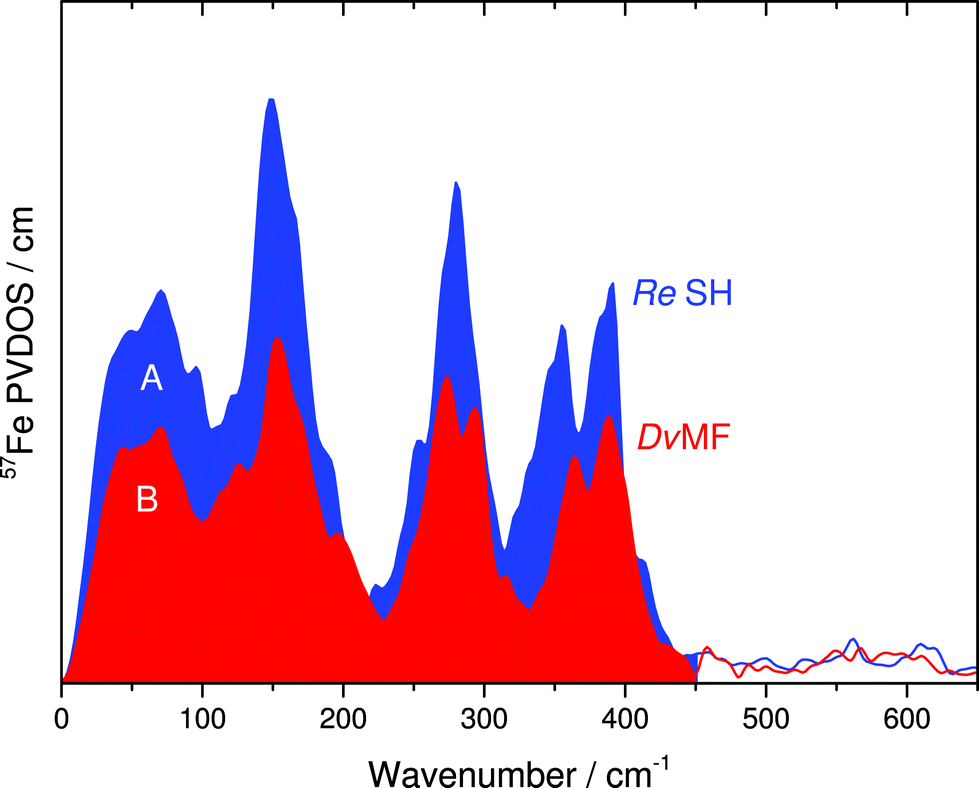 | ||
| Fig. 3 Comparison of integral intensities for the FeS cluster related 57Fe PVDOS in the 0–450 cm−1 range of (A) oxidized SH and (B) oxidized Dv hydrogenase. Spectra were normalized with respect to the integral intensities of the Fe(CO)(CN)2 moiety (450–650 cm−1). | ||
In general, pronounced spectral features of FeS clusters range from (delocalized) torsional modes below 100 cm−1via breathing and bending modes near 150 cm−1 to structurally sensitive stretching vibrations between 250 and about 420 cm−1 (Fig. 2). The oxidized SH shows two intensive bands with peak maxima around 355 cm−1 and 391 cm−1, which reflect normal modes with predominant Fe–S stretching character (Fig. 2A, blue). While both modes have contributions from displacements of the bridging sulfides (Fe–Sb stretching), the band at 355 cm−1 also includes increased movements of terminal cysteines (Fe–St stretching).15 Furthermore, oxidized SH shows a band at 414 cm−1, which is characteristic for [2Fe2S] clusters of ferredoxins (e.g. those from Rc and Aquifex aeolicus (Aa)).13 The Dv hydrogenase does not contain a [2Fe2S] cluster. Thus, no corresponding NRVS signal is observed.9 Remarkably, a weighted sum of one oxidized [2Fe2S] cluster of Rc ferredoxin13 and four oxidized [4Fe4S] clusters of Pf ferredoxin14 provides an ideal fit for the NRVS of as-isolated SH (Fig. 2B, blue and S1C/E†). Including an oxidized [3Fe4S] cluster (Pf ferredoxin)9 into the simulation instead of one of the [4Fe4S] clusters does not improve the agreement with the experimental spectrum (Fig. S1D†). In contrast, the weighted sum spectra would exhibit obvious band splittings at around 150 and 290 cm−1, the latter of which is also observed for [3Fe4S] cluster-containing Dv hydrogenase (Fig. 3B and S1B†). No such splitting is observed for the SH, suggesting that the previously proposed presence of a native [3Fe4S] cluster in enzymes of this type5,16 is unlikely.
Upon reduction of the SH with NADH, a modified band pattern is observed for the spectral region between 300 and 400 cm−1, reflecting (partial) reduction of FeS clusters. Moreover, the band at 414 cm−1, assigned to the oxidized [2Fe2S] cluster, decreases in intensity by around 50% (Fig. 2A, red and S1E†). This observation indicates partial reduction of the [2Fe2S] cluster. Consistently, a lowered intensity has also been observed for the corresponding band upon reduction of [2Fe2S] cluster-containing ferredoxins from Rc and Aa.13 In general, band positions and intensities between 0 and 300 cm−1 show only marginal changes after reduction of the SH while drastic changes have been observed for Pf ferredoxin and Dv hydrogenase.13,9 This indicates that not all FeS clusters in SH were reduced under the experimental conditions chosen. Indeed, a weighted sum of a 50% reduced / 50% oxidized [2Fe2S] cluster (from Rc ferredoxin) plus one reduced and three oxidized [4Fe4S] clusters (from Pf ferredoxin) provides the best fit to the experimental spectrum of NADH-reduced SH (Fig. 2, red and S1C/E†). This finding is consistent with previous EPR spectroscopic studies, revealing signals for the reduced (paramagnetic) states of the [2Fe2S] cluster and in total only one of the four [4Fe4S] clusters.5,6 According to our data, this latter observation can be clearly related to incomplete reduction of FeS clusters, as also observed for respiratory Complex I, whose water-soluble domain is homologous to the SH.17 Consequently, pronounced antiferromagnetic coupling, high-spin ground states, or very fast spin relaxation can now be excluded as alternative explanations for the absence of EPR-detectable clusters. In summary, we have probed for the first time the entirety of FeS clusters of an NAD+-reducing [NiFe] hydrogenase in its oxidized and reduced states, thereby disentangling the discrepancy between predicted and EPR-detectable FeS clusters in this enzyme type.5
While the NRVS of oxidized and reduced SH are well simulated by the corresponding weighted sums of one Rc ferredoxin-derived [2Fe2S] cluster and four [4Fe4S] clusters of Pf ferredoxin, clear deviations can still be observed in the structural marker region between 300 and 400 cm−1 (Fig. 2). Relative intensities of bands in this region have been proposed as structural markers for Fe–S–C–C dihedral angles. These dihedral angles affect contributions of S–C–C bending coordinates to normal modes with predominant Fe–S stretching character, thereby influencing iron-displacement and NRVS intensities.9 In this respect, the balanced doublets in the weighted sum spectra suggest little contributions from S–C–C bending coordinates, i.e. Fe–S–C–C dihedral angles close to 90°. The SH NRVS exhibits a pattern that is similar to Pf ferredoxin and the weighted sum spectrum (Fig. 2), indicating that most Fe–S–C–C dihedral angles are close to 90° in SH, too. However, an intensity drop of the low-frequency feature around 355 cm−1 is indicative of the co-existence of [4Fe4S] clusters with Fe–S–C–C dihedral angles approaching 0/180° (Fig. 2A).14
There is no crystal structure available for the SH; however, dihedral angles might be similar to those in Complex I. Thus, the SH-related NRVS might be well explained by the atomic coordinates derived from the crystal structure of Complex I. Indeed 14 out of 20 (70%) Fe–S–C–C dihedrals in Complex I approach 90° (±24°) while the remaining six (30%) assume values close to 0/180° (±31°).18 This finding explains the SH NRVS and supports the structural similarity between Complex I and NAD+-reducing [NiFe] hydrogenases.
Active site vibrational properties
Despite the lower spectral intensity of the active site (only 1 Fe ion compared to 18 located in FeS clusters), NRVS also allows probing Fe–CO and Fe–CN stretching and bending modes (400–650 cm−1),9 which provide valuable information on structural and electronic properties of the catalytic [NiFe] center (Fig. 4, left and S2†).10,11 A detailed knowledge of the molecular coordinates included in these modes is essential for the understanding of the complex signature of the Fe(CO)(CN)2 moiety.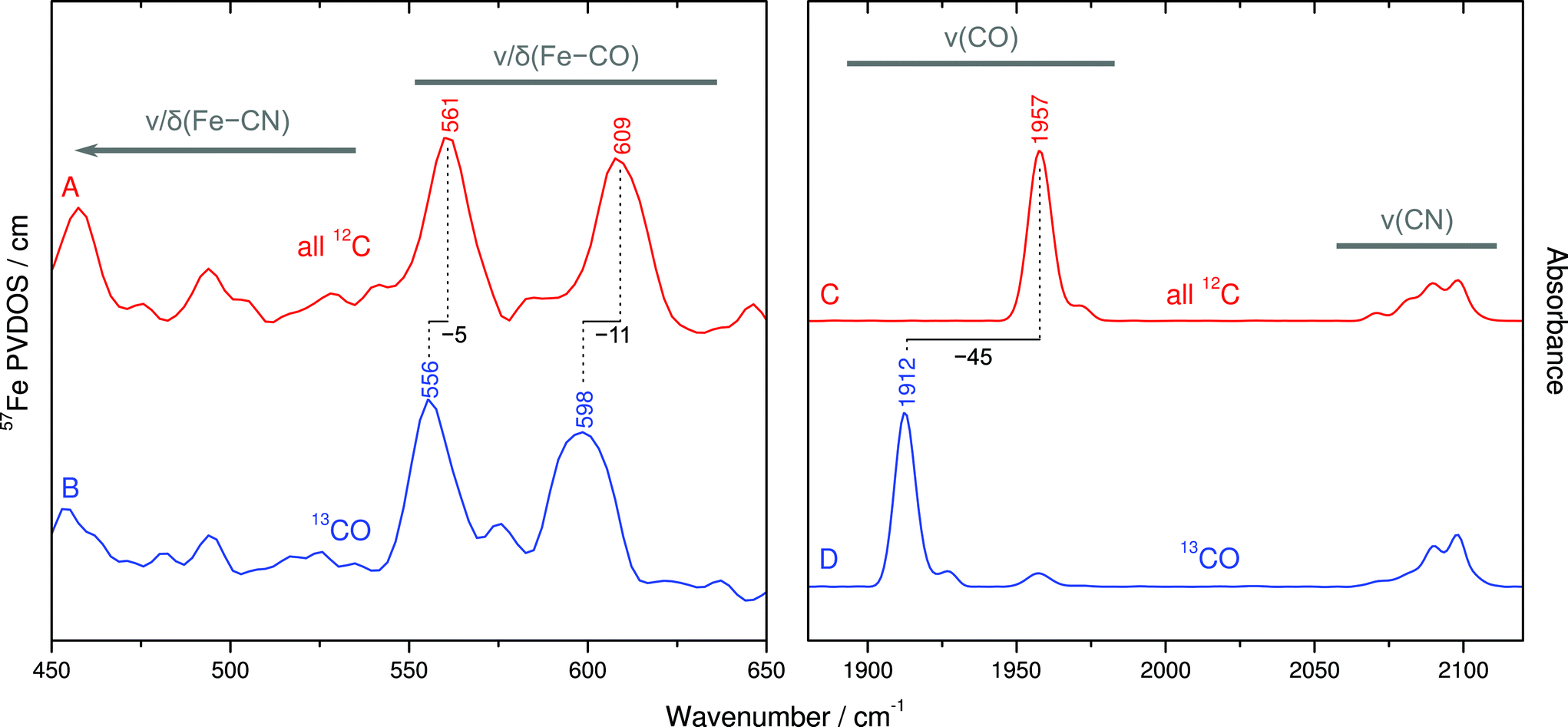 | ||
| Fig. 4 Vibrational signature for the Fe(CO)(CN)2 moiety of the oxidized SH active site. Left panel: Fe–CO/CN metal–ligand modes as revealed by NRVS. Right panel: CO/CN stretching modes probed by IR spectroscopy. A/C: non-labeled SH (red). B/D 13CO-labeled SH (blue). | ||
To disentangle this vibrational manifold, we have used selective 13C labeling of the CO ligand by isolating SH from cells grown in a 13CO-enriched atmosphere. To verify selective 13CO labeling, we have first recorded IR spectra from both unlabeled and 13CO-containing as-isolated SH (Fig. 4C and D). While unlabeled SH exhibits the typical CO stretching band at 1957 cm−1,19 this feature is considerably displaced by −45 cm−1 in case of 13CO-labeled SH, reflecting the larger mass of 13C. Moreover, both spectra exhibit the same set of four CN stretching bands at around 2098, 2090, 2081 and 2072 cm−1, as reported previously.19 These observations demonstrate that the 13CO ligand of the [NiFe] site originates from externally supplied gaseous 13CO, which is consistent with observations previously made for the regulatory hydrogenase from R. eutropha.20
The comparison of NRVS signatures between 550–650 cm−1 obtained from unlabeled and 13CO-labeled SH reveals isotopic shifts of 5–11 cm−1, indicating that major contributions of these bands are due to Fe–CO stretching and bending (Fig. 4A and B). In contrast, bands at lower frequencies are largely unaffected by 13CO labeling, demonstrating their affiliation with normal modes that are dominated by Fe–CN coordinates. In addition, closer inspection of 13CO isotopic shifts may also yield information about the character of individual Fe–CO modes in terms of predominant stretching and bending contributions.21 The larger 13C isotopic shift observed for the band at 609 cm−1 (Δν = −11 cm−1) likely reflects more pronounced Fe–C–O bending contributions, while the smaller 13C effect on the band at 561 cm−1 (Δν = −5 cm−1) is indicative of a predominant Fe–CO stretching character. In line with previous computational studies,10,11 these observations provide the first experimental insights into normal mode compositions (Fe–CO vs. Fe–CN) of the [NiFe] hydrogenase active site. Interestingly, Fe–CO modes of the SH, as probed by NRVS, appear at slightly higher frequencies compared to oxidized Dv hydrogenase, while the IR-traceable CO stretching vibrations are nearly the same. This observation may provide further details on structural and electronic properties of the SH active site.
Conclusions
In conclusion, the first combined NRVS and IR characterization of an oxygen-tolerant [NiFe]-hydrogenase revealed the content and redox behavior of FeS clusters in the NAD+-reducing enzyme from Ralstonia eutropha H16. We demonstrated the presence of one [2Fe2S] center and four [4Fe4S] clusters, which are only partly reduced upon incubation of the enzyme with the native electron donor NADH. This finding unravels the discrepancy between the number of predicted and EPR-detected FeS clusters in SH and related proteins, including Complex I, which underscores the benefit of NRVS for the characterization of complex metalloproteins. Furthermore, our approach provides access to the entire vibrational signature of the Fe(CO)(CN)2 moiety of the hydrogenase active site. For the first time, selective isotope labeling with gaseous 13CO allowed the experimental discrimination between Fe–CO and Fe–CN normal modes. Moreover, bands with predominant Fe–CO stretching and bending character could be identified. In summary, vibrational spectroscopy in conjunction with selective isotope labeling allows detailed insights into molecular coordinates of the active site in hydrogenase. An extension of this approach in terms of other site-specific isotope exchanges will likely provide further valuable information about functional and structural aspects of hydrogen-converting biocatalysts.Acknowledgements
The SH biochemistry and IR work (LL, MH, IZ and OL) were supported by the Deutsche Forschungsgemeinschaft (DFG, Cluster of Excellence UniCat). LL received a short-term EMBO scholarship for performing the work in this study. HW, LG and SPC were supported by National Institutes of Health (GM-65440 to SPC). The NRVS experiments were performed at BL09XU and BL19LXU of SPring8 approved under JASRI and RIKEN proposal numbers 2013B0103, 2014B1032 and 20133892).Notes and references
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS PubMed.
- K. Schneider and H. G. Schlegel, Biochem. J., 1981, 193, 99–107 CAS.
- L. Lauterbach and O. Lenz, J. Am. Chem. Soc., 2013, 135, 17897–17905 CrossRef CAS PubMed.
- L. Lauterbach, O. Lenz and K. A. Vincent, FEBS J., 2013, 280, 3058–3068 CrossRef CAS PubMed.
- M. Horch, L. Lauterbach, O. Lenz, P. Hildebrandt and I. Zebger, FEBS Lett., 2012, 586, 545–556 CrossRef CAS PubMed.
- M. Horch, L. Lauterbach, M. Saggu, P. Hildebrandt, F. Lendzian, R. Bittl, O. Lenz and I. Zebger, Angew. Chem., Int. Ed., 2010, 49, 8026–8029 CrossRef CAS PubMed.
- F. Germer, I. Zebger, M. Saggu, F. Lendzian, R. Schulz and J. Appel, J. Biol. Chem., 2009, 284, 36462–36472 CrossRef CAS PubMed.
- K. Schneider, R. Cammack and H. G. Schlegel, Eur. J. Biochem., 1984, 142, 75–84 CrossRef CAS PubMed.
- S. Kamali, H. Wang, D. Mitra, H. Ogata, W. Lubitz, B. C. Manor, T. B. Rauchfuss, D. Byrne, V. Bonnefoy, F. E. Jenney Jr, M. W. Adams, Y. Yoda, E. Alp, J. Zhao and S. P. Cramer, Angew. Chem., Int. Ed. Engl., 2013, 52, 724–728 CrossRef CAS PubMed.
- M. Horch, J. Schoknecht, M. A. Mroginski, O. Lenz, P. Hildebrandt and I. Zebger, J. Am. Chem. Soc., 2014, 136, 9870–9873 CrossRef CAS PubMed.
- E. Siebert, M. Horch, Y. Rippers, J. Fritsch, S. Frielingsdorf, O. Lenz, F. V. Escobar, F. Siebert, L. Paasche, U. Kuhlmann, F. Lendzian, M. A. Mroginski, I. Zebger and P. Hildebrandt, Angew. Chem., Int. Ed., 2013, 52, 5162–5165 CrossRef CAS PubMed.
- W. Sturhahn, Hyperfine Interact., 2000, 125, 149 CrossRef CAS.
- Y. Xiao, M. L. Tan, T. Ichiye, H. Wang, Y. Guo, M. C. Smith, J. Meyer, W. Sturhahn, E. E. Alp, J. Zhao, Y. Yoda and S. P. Cramer, Biochemistry, 2008, 47, 6612–6627 CrossRef CAS PubMed.
- D. Mitra, V. Pelmenschikov, Y. Guo, D. A. Case, H. Wang, W. Dong, M. L. Tan, T. Ichiye, F. E. Jenney, M. W. Adams, Y. Yoda, J. Zhao and S. P. Cramer, Biochemistry, 2011, 50, 5220–5235 CrossRef CAS PubMed.
- D. Mitra, S. J. George, Y. Guo, S. Kamali, S. Keable, J. W. Peters, V. Pelmenschikov, D. A. Case and S. P. Cramer, J. Am. Chem. Soc., 2013, 135, 2530–2543 CrossRef CAS PubMed.
- C. Zaborosch, M. Köster, E. Bill, K. Schneider, H. G. Schlegel and A. X. Trautwein, Biometals, 1995, 8, 149–162 CrossRef CAS.
- H. R. Bridges, E. Bill and J. Hirst, Biochemistry, 2012, 51, 149–158 CrossRef CAS PubMed.
- L. A. Sazanov and P. Hinchliffe, Science, 2006, 311, 1430–1436 CrossRef CAS PubMed.
- R. P. Happe, W. Roseboom, G. Egert, C. G. Friedrich, C. Massanz, B. Friedrich and S. P. J. Albracht, FEBS Lett., 2000, 466, 259–263 CrossRef CAS.
- I. Bürstel, P. Hummel, E. Siebert, N. Wisitruangsakul, I. Zebger, B. Friedrich and O. Lenz, J. Biol. Chem., 2011, 286, 44937–44944 CrossRef PubMed.
- M. Tsubaki, R. B. Srivastava and N. T. Yu, Biochemistry, 1982, 21, 1132–1140 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Representations of data analyses (Fig. S1 and S2). See DOI: 10.1039/c4sc02982h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |