
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel ortho-OPE metallofoldamers: binding-induced folding promoted by nucleating Ag(I)–alkyne interactions†
Ana
Martín-Lasanta
a,
Luis
Álvarez de Cienfuegos
a,
Alice
Johnson
b,
Delia
Miguel
a,
Antonio J.
Mota
c,
Angel
Orte
 d,
Maria Jose
Ruedas-Rama
d,
Maria
Ribagorda
e,
Diego J.
Cárdenas
e,
M.
Carmen Carreño
e,
Antonio M.
Echavarren
b and
Juan M.
Cuerva
*a
d,
Maria Jose
Ruedas-Rama
d,
Maria
Ribagorda
e,
Diego J.
Cárdenas
e,
M.
Carmen Carreño
e,
Antonio M.
Echavarren
b and
Juan M.
Cuerva
*a
aDepartment of Organic Chemistry, Faculty of Science, University of Granada, Granada, Spain. E-mail: jmcuerva@ugr.es
bInstitute of Chemical Research of Catalonia (ICIQ), Tarragona, Spain
cInorganic Chemistry, Faculty of Science, University of Granada, Granada, Spain
dDepartment of Physical Chemistry, Faculty of Pharmacy, University of Granada, Granada, Spain
eDepartment of Organic Chemistry, Universidad Autónoma de Madrid, Madrid, Spain
First published on 29th July 2014
Abstract
We have developed a new family of ortho-oligophenylene ethynylene (o-OPE) metallofoldamers. The folding of these helicates is induced by nucleating carbon–metal interactions between Ag(I) cations and the alkynes of the inner core of the o-OPEs. These o-OPEs form metal–organic assemblies where at least three alkyne moieties are held in close proximity to form novel Ag(I)-complexes with the metal ion lodged into the helical cavity. NMR titration experiments and photokinetic studies have provided quantitative data about the thermodynamic and kinetic features of such binding/folding phenomena. X-ray diffraction and DFT studies have been performed to extract structural information on how the Ag(I) cation is accommodated into the cavity. The great simplicity and versatility of these new metallofoldamers open up the possibility to develop novel structures with applications in material science and/or in asymmetric catalysis.
Introduction
Generating synthetic macromolecules with spatial control is of great interest in view of their prospective applications in catalysis, chemical sensing and material science.1,2 Some of the most successful progress in this area has been provided by the foldamer concept. In supramolecular chemistry, a foldamer is defined as a synthetic chain molecule that adopts a conformationally ordered state in solution by a collection of non-covalent interactions between non-adjacent monomer units. Because coordination bonds are relatively strong among the non-covalent interactions, metallohelicates3 based on oligopyridines and metallofoldamers4 based on salen-type multidentate ligands have been extensively investigated. In most of such cases, the metal coordination sphere is inherently helical (templated approach). Examples in which the metal coordination sphere is not inherently helical, but instead causes a series of cooperative, non-covalent interactions that ultimately result in a folded structure (nucleated approach) are much less common.4 Moreover, the possibility to induce the folding process taking advantage of the coordination capabilities of carbon-based π-donor ligands such as alkynes to carbophilic metals has not been described to date.Herein, we demonstrate the utility of Ag(I)–alkyne bonds to prepare new metal–organic folded assemblies. Inspiration for the work developed here comes from an original idea of Nelson and Moore,5 who proposed that the inner void of folded OPEs should be potentially able to coordinate metals.6 Coinage metal–alkyne interactions are believed to efficiently initiate and mediate several catalytic reactions.7 In contrast to the well-documented chemistry of Ag(I) interacting with heteroatoms, the literature involving structurally characterized η2-alkyne–Ag(I) complexes is rather limited,8 especially in the case of monomeric complexes with more than one alkyne moiety bound to the same Ag(I) metal center.9 Youngs9 and Komatsu9et al. reported respectively the syntheses of (I)2·Ag(I) and II·Ag(I) (Scheme 1), which represent the first structurally characterized complexes involving more than one triple bond interacting with Ag(I). Tetrabenzo[16]dehydroannulene II was found to have a cavity size suitable for the incorporation of one Ag(I) cation, while tribenzo[12]dehydroannulene ligand I was too small and thus formed a sandwich-type complex (I)2·Ag(I).
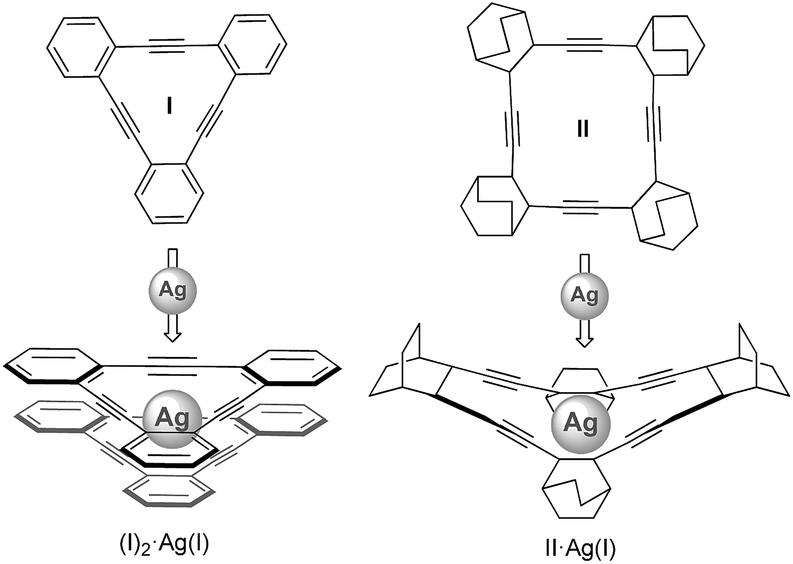 | ||
| Scheme 1 Structure of the ligands and silver complexes reported by Youngs and Komatsu. | ||
In this context, we recently reported a new strategy to covalently lock flexible o-OPEs10 into well-defined single loops (III, Scheme 2), even in a chiral way, by means of an organic staple.11,12 Due to the structural similarity between such stapled π-conjugated systems and benzocyclynes I and II, we envisioned that such a family of o-OPE macrocycles could be used to allocate a carbophilic metal such as Ag(I) into the helical cavity (metallocavitands).13 Furthermore, the Ag(I)–alkyne interaction could induce the helical folding of all-carbon polymers such as acyclic o-OPEs resulting in a new class of metallofoldamers.4
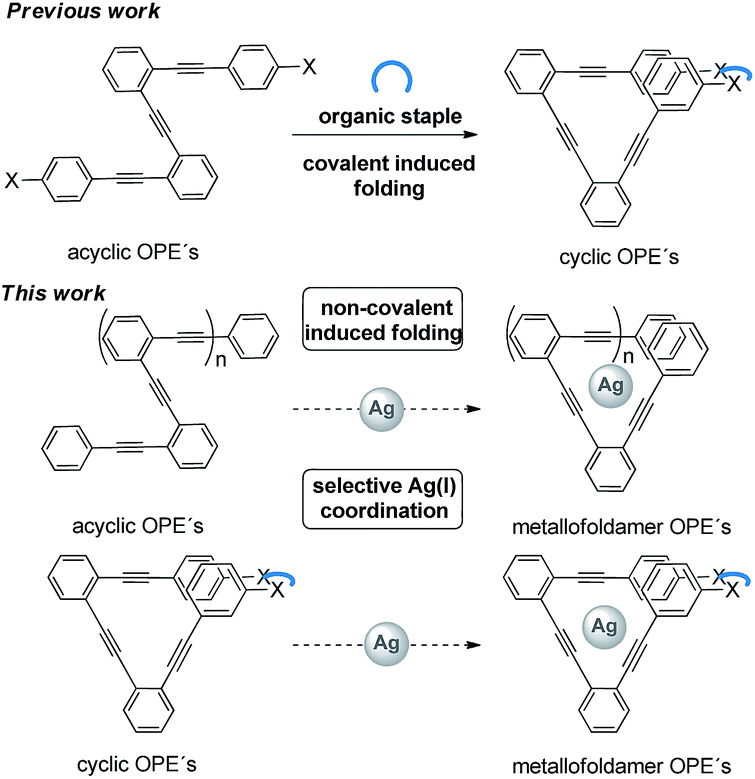 | ||
| Scheme 2 Schematic chemical structures of the o-OPE-based ligands and Ag(I) complexes synthesized. | ||
Continuing our recent interest in all conjugated carbon-based materials,11,14 here we report an in-depth experimental and theoretical study on the ability of Ag(I) cations to form complexes with cyclic and acyclic ortho-oligophenylene ethynylenes (o-OPEs), resulting in a new class of metallofoldamers. We have found that this weak interaction efficiently acts as a nucleating event according to the helix–coil model15 in acyclic ortho-OPEs with more than three triple bonds.
Results and discussion
A set of acyclic and cyclic o-OPEs (1–16, Chart 1) was synthesized using Sonogashira cross-coupling protocols16 and the macrocyclic compounds were obtained following the stapling methodology developed in our lab.11 Cyclic o-OPE 1 and acyclic 6 were selected as models for the coordination experiments with metal salts (Na(I), Li(I), Zn(II) and Ag(I)) bearing the weakly coordinating tetrafluoroborate counterion.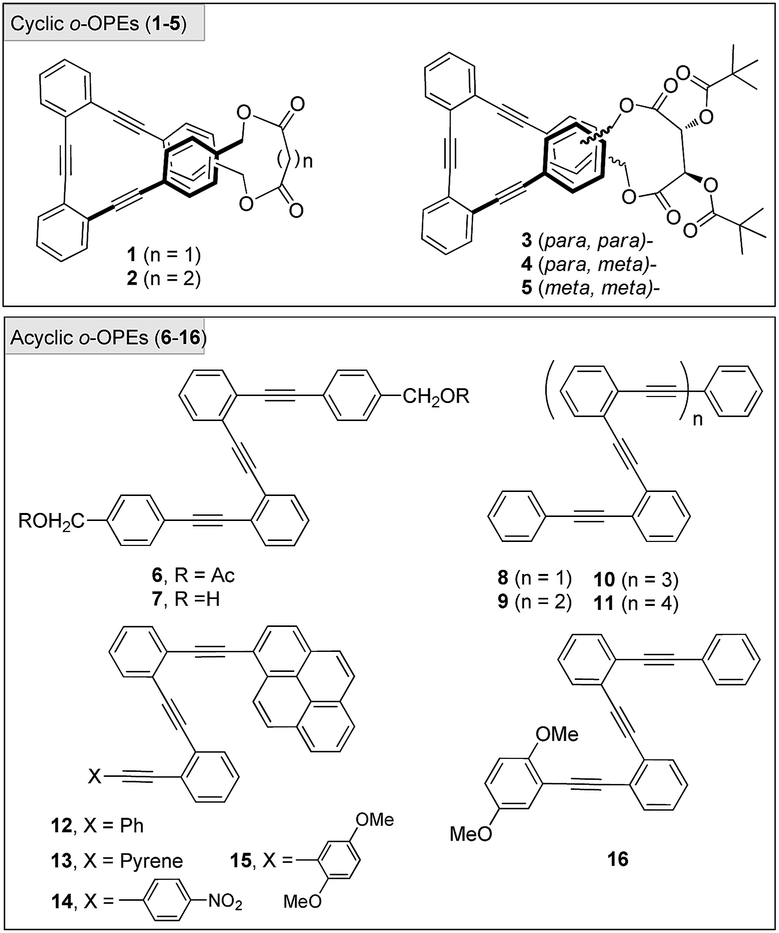 | ||
| Chart 1 Chemical structure of o-OPEs 1–16. | ||
Initial 1H NMR samples (CD2Cl2) were treated with an excess of metal salts under air and at room temperature. Analysis of the 1H NMR spectra before and after metal addition revealed that the Na(I), Li(I) and Zn(II) salts did not induce any appreciable change in the proton spectra, whereas the samples treated with Ag(I) exhibited significant changes in the aromatic region, which evidenced the formation of a new organometallic species (Fig. 1). Interestingly, the addition of Ag(I) led to an almost identical spectra for the complex formed with the acyclic ligand 6 and cyclic o-OPE 1, which unequivocally presents a folded arrangement in solution. Moreover, the upfield shift of the terminal aromatic hydrogens in 6 (Fig. 1d, grey-labeled protons) pointed to the stacking of both rings consistent with a metallo-induced folding phenomenon in solution.
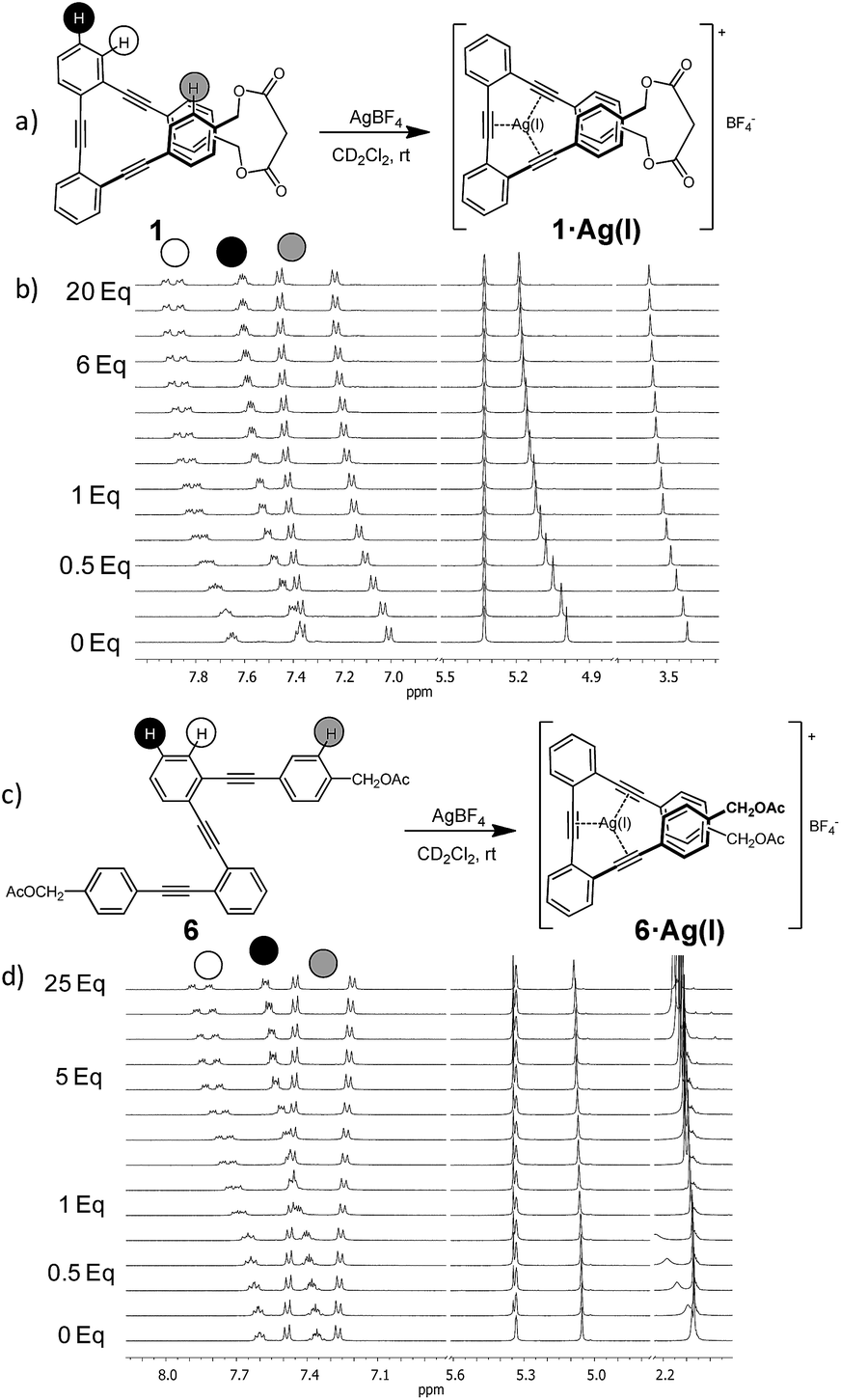 | ||
| Fig. 1 (a and c) Schematic representations of the coordination reaction, (b and d) 1H NMR titration of the ligand with AgBF4 indicating the diagnostic shifts of proton signals (CD2Cl2, 500 MHz, 298 K). Only the representative parts of the spectra are shown. | ||
Since Ag(I) ions can coordinate π-donors such as arenes and alkynes, two kinds of folded structures could be formed by coordination involving the superimposed aromatic rings9,17 or the triple bonds. In order to clarify the type of coordination governing our complexes, a careful analysis of 1H and 13C NMR spectra of o-OPE 1·Ag(I) and 6·Ag(I) was accomplished. The deshielding of the two non-equivalent ortho-H of the inner phenyl groups (Fig. 1b and d, white-labeled hydrogens) indicates a loss of electron density of the triple bonds. These data together with the significant changes (2–5 ppm) observed in the 13C chemical shifts of the alkynes (Fig. S1†) strongly suggested the Ag(I)–alkyne coordination.18 This evidence led us to conclude that Ag(I) was able to bind the three alkynes of the o-OPEs. In light of these results, we decided to study the stability of the Ag(I)–alkyne interaction and its relationship with the nature of the o-OPEs.
Binding affinity of the Ag(I) cation to o-OPEs
Quantitative binding affinities and stoichiometries were calculated for 1–16 (CD2Cl2–acetone-d6 = 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)19 by 1H NMR titration experiments (Fig. 2 and 3 and the ESI†) using Dynafit code (Table 1).20 Characteristic deshielding of the ortho-H of the inner phenyl groups and 13C NMR chemical shifts of the alkynes attributed to the Ag(I)–alkyne interaction could be unambiguously identified in all of the cases.21 Only one set of signals was observed throughout the titration experiments suggesting stable complexation under fast exchange conditions.22 All compounds, containing up to six alkynes, presented 1:1 ligand–Ag(I) stoichiometries.23
:1)19 by 1H NMR titration experiments (Fig. 2 and 3 and the ESI†) using Dynafit code (Table 1).20 Characteristic deshielding of the ortho-H of the inner phenyl groups and 13C NMR chemical shifts of the alkynes attributed to the Ag(I)–alkyne interaction could be unambiguously identified in all of the cases.21 Only one set of signals was observed throughout the titration experiments suggesting stable complexation under fast exchange conditions.22 All compounds, containing up to six alkynes, presented 1:1 ligand–Ag(I) stoichiometries.23
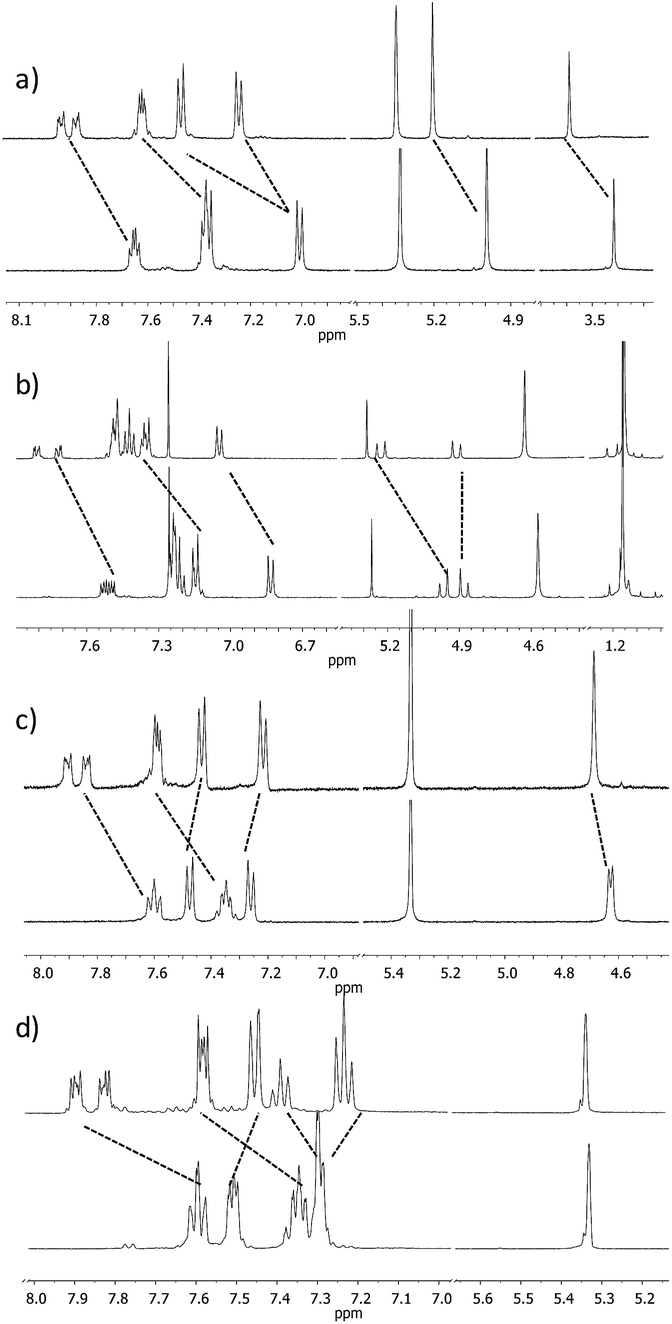 | ||
| Fig. 2
1H NMR spectra of ligand (a) 1 (6 mM), (b) 3 (17 mM), (c) 7 (8 mM), and (d) 8 (14 mM) with an excess of AgBF4 (CD2Cl2–acetone-d6 = 9:1 mixture, 500 MHz, 298 K). Only the representative parts of the spectra are shown. | ||
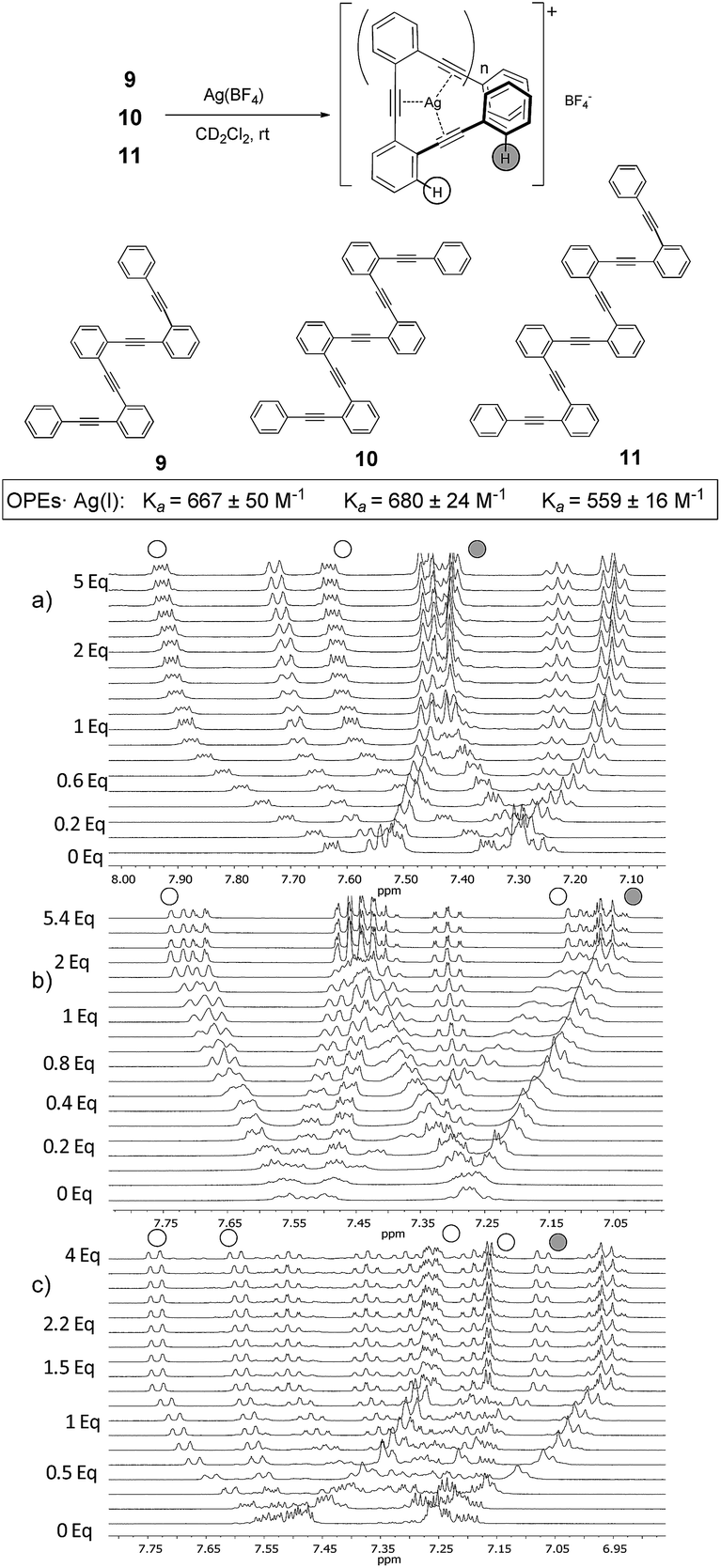 | ||
| Fig. 3
1H-NMR titrations of ligand (a) 9 (10 mM), (b) 10 (20 mM) and (c) 11 (10 mM) with AgBF4 (CD2Cl2–acetone-d6 = 9:1 mixture, 500 MHz, 298 K). | ||
| o-OPEs | K a (M−1) | o-OPEs | K a (M−1) |
|---|---|---|---|
|
a Measured in CD2Cl2–acetone-d6 = 9:1 mixture at 298 K.
|
|||
| 1 | 1001 ± 180 | 9 | 667 ± 50 |
| 2 | 852 ± 260 | 10 | 680 ± 24 |
| 3 | 86 ± 7 | 11 | 559 ± 16 |
| 4 | 53 ± 2 | 12 | 22 ± 5 |
| 5 | 318 ± 25 | 13 | 71 ± 17 |
| 6 | 63 ± 6 | 14 | 7.3 ± 0.3 |
| 7 | 739 ± 140 | 15 | 187 ± 84 |
| 8 | 49 ± 2 | 16 | >105 |
From the 1H NMR data, a higher binding constant was obtained for the cyclic o-OPE 1 (Ka = 1001 ± 180 M−1, Fig. 2a) in comparison to bis(acetylated) acyclic 6, (Ka = 63 ± 6 M−1). This observation is consistent with the existence of a binding site of suitable size that requires a less unfavorable entropic coordination (macrocyclic effect).25 An unexpectedly high binding constant was also obtained for the di-hydroxylated acyclic o-OPE 7 (Ka = 739 ± 140 M−1, Fig. 2c) in comparison to the unsubstituted acyclic 8 analog (Ka = 49 ± 2 M−1, Fig. 2d). Previous DFT calculations revealed that in compound 7 an intramolecular hydrogen bond formed between the two benzyl alcohol groups which further stabilized the folded conformation.11 Hence, the significantly higher value obtained for 7 could reasonably stem from a synergetic effect played by the intramolecular hydrogen bond and the Ag(I) ion acting simultaneously as driving forces in the folding event.26
The succinoyl macrocycle 2 gave a slightly smaller binding constant (Ka = 852 ± 260 M−1) than macrocycle 1. Surprisingly, the para,para- (Fig. 2b) and para,meta-substituted chiral tartrate derivates 3 and 4 presented binding constants one order of magnitude smaller (Ka = 86 ± 7 M−1 and Ka = 53 ± 2 M−1, respectively) than those obtained for ligands 1 and 2. This lower binding affinity could be related to the major rigidity imposed by the chiral tartrate staple11 and, hence, altering the optimal conformation of the alkynes for the coordination (and the corresponding mismatch of the cavity size). Consistent with this hypothesis was the higher value (Ka = 318 ± 25 M−1) measured for the less rigid meta,meta-substituted chiral tartrate derivate 5.
In order to explore if the binding/folding phenomenon could be extended to o-OPEs with more than three alkyne units, we titrated o-OPEs 9 (n = 2, four alkynes), 10 (n = 3, five alkynes) and 11 (n = 4, six alkynes) (Fig. 3).27 Again, a 1:1 stoichiometry was observed for all cases in spite of the increased number of alkyne potential ligands. The 1H NMR spectra recorded during the titration of o-OPE 9 (n = 2) (Fig. 3a) showed analogous changes than the titration experiment of o-OPE 8 (n = 1) (Fig. 2d). Thus, the two ortho-H of the outer phenyl group appear upfield and the two ortho-H of the inner phenyl group are deshielded (Fig. 3). These common features strongly suggest the presence of folded structures. Moreover, the comparison of the binding constant values obtained for 1:1 complexes of o-OPE 8·Ag(I) (Ka = 49 ± 2 M−1) and o-OPE 9·Ag(I) (Ka = 667 ± 50 M−1) revealed that the ability of the alkynes to roll-around the metal is modest in terms of free energy (−2.3 kcal mol−1 and −3.8 kcal mol−1 respectively) but increases significantly with the presence of a new alkyne.
The affinity constant obtained for the next ligands of the series containing five and six alkynes revealed similar binding constants to o-OPE 9 (compound 10 (n = 2), Ka = 680 ± 24 M−1, compound 11 (n = 3), Ka = 559 ± 16 M−1, Fig. 3b and c). Every additional alkyne should increase the ligand affinity toward Ag(I), however the similar value measured for ligands 9–11 suggests that the Ag(I) can coordinate efficiently to only four alkyne groups.
X-ray crystallographic data and theoretical calculations
We also investigated the structure and the coordination features of the Ag(I)–o-OPE complexes in solid state. We were able to obtain suitable crystals to determine the X-ray structures of the Ag(I) complexes of 1 and 8 (Fig. 4).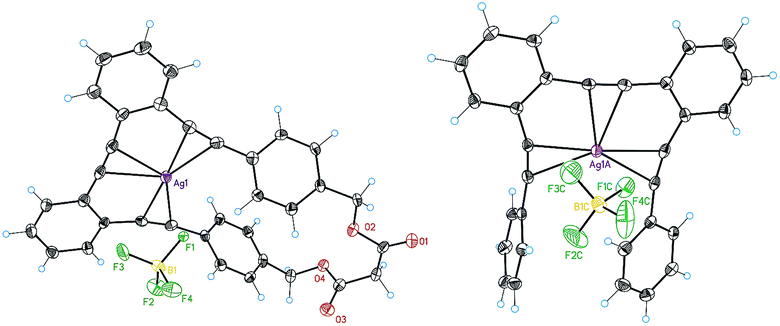 | ||
| Fig. 4 X-ray structures of 1·Ag(I) and 8·Ag(I). | ||
In agreement with the NMR evidence in solution, in both complexes one silver ion was located in the cavity of the o-OPE backbone and simultaneously coordinated to the three alkynes in the solid-state (mean distance Ag(I)–alkyne 2.3 Å).28 In both adducts, the o-OPE moiety was not arranged into a helical geometry around the metals but into a slightly distorted trigonal-bipyramidal geometry (torsion angles 8.8° for complex 8·Ag(I) and 6.4° for complex 1·Ag(I)). In the crystal structure of 1·Ag(I) (Fig. 4), the two apical positions of the distorted trigonal-bipyramidal geometry were occupied by an oxygen atom from the malonyl group of a neighboring complex (ca. 2.57 Å) and a BF4− anion (ca. 2.61 Å). In 8·Ag(I), the silver atom instead of binding an oxygen atom, lay at an average distance of 2.99 Å from one of the benzene rings of the neighboring complex. DFT calculations, using the M06 functional with the 6-31G* basis set for the ligands and the LANL2DZ relativistic effective core potential basis set for the Ag(I) ion21 of the expected cationic structure 8·Ag(I) (Fig. 5a), showed the same slightly distorted trigonal geometry (but with a larger torsion angle of 14.6°), which is presumably the one adopted in solution.
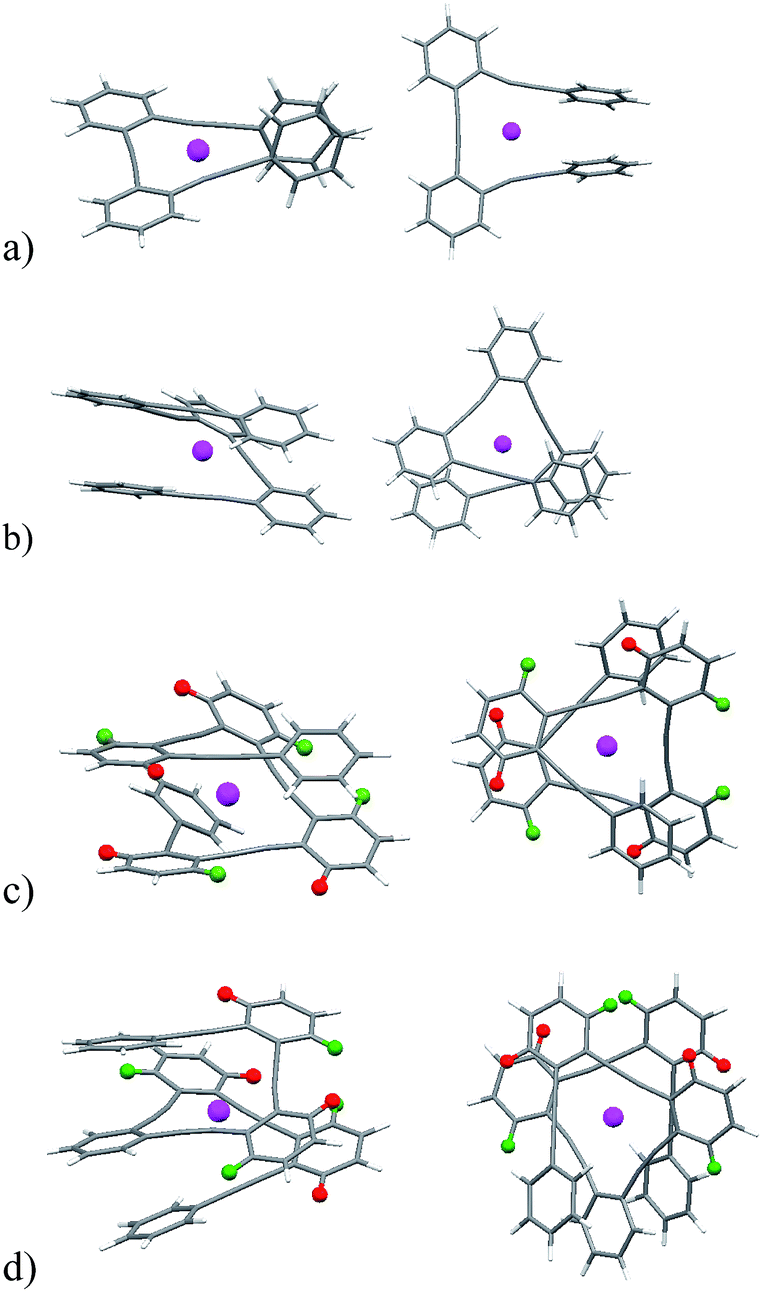 | ||
| Fig. 5 DFT calculated structures for 8·Ag(I), 9·Ag(I), 10·Ag(I), and 11·Ag(I), side views (left) and top views (right). | ||
Although for higher oligomers we were not able to obtain crystal structures, the agreement between experimental and calculated structures of 8·Ag(I) encouraged us to explore the soft accommodating nature of the Ag(I) ion29,30 into o-OPEs 9, 10 and 11 by theoretical calculations (Fig. 5b–d). In the case of 9·Ag(I), 10·Ag(I), and 11·Ag(I) a pseudo-tetrahedral coordination was obtained with the silver cation simultaneously coordinated to the four alkynes. In complexes 10·Ag(I) and 11·Ag(I) the additional alkynes were loosely bound, although a completely folded conformation can be inferred from the NMR data. Both structures are symmetric and, remarkably, the terminal benzene rings are significantly shielded owing to the presence of a superposed aromatic ring. Moreover, the ortho-H of the inner phenyl groups are non-equivalent, with one of them shielded (red hydrogen atoms, Fig. 5c and d) and the other deshielded (green hydrogen atoms, Fig. 5c and d). This spectroscopic behavior can only be explained by taking into account the predicted folded structure (Fig. 5c and d) in which one of such protons is placed over a phenyl ring and the other outside of the anisotropy cone. Interestingly, all of these complexes with four to six alkynes have a helical arrangement, thus inducing chirality around the metal center.31 These results also point toward the potential formation of larger o-OPE metallofoldamers (each o-OPE comprising 4n alkynes chelating n Ag(I) atoms) with two potentially conductive substructures: the metallic core and the all-conjugated carbon backbone.
Photokinetic studies of o-OPEs 12–15
The NMR data and DFT-based theoretical calculations described above provided thermodynamic and structural data, as well as evidence to support the formation of folded structures in solution owing to Ag(I)-coordination features. Nevertheless, an unequivocal demonstration of their existence can only emerge from responses that could only exist in the folded state of the o-OPE. Furthermore, the structural effects of the Ag(I) coordination on the electronic energy levels of the ligands, as well as the kinetics of the random-coil to helix transition are interesting issues to elucidate. For this purpose, we prepared four fluorescent o-OPEs 12–15 bearing one terminal pyrene group and four different rings on the opposite end (Chart 1). Compound 13 was chosen to study the rate constant of pyrenyl-excimer formation.Excimers are short-lived dimeric species formed in the electronic excited state by some compounds. The study is based on the effect of dynamic phenomena, such as quenching or excimer formation, on the fluorescence emission spectra and fluorescence lifetime of pyrene derivatives. In systems with conformational flexibility, metal-ion-mediated conformational changes have been frequently tested using excimer formation kinetics data.32o-OPE 12 was prepared as a model compound, and o-OPEs 14 and 15 were prepared to evaluate the effect of tuning the electronic properties with electron-poor and electron-rich groups.
Despite the wide π-surface of the pyrene moiety, which could potentially interfere with the Ag(I)–alkyne coordination in compounds 12–15, 1H NMR titrations and 13C NMR data evidenced the folding by coordination with the alkynes (Fig. 6 and ESI†). Moreover, the binding constant values calculated had the same order of magnitude as o-OPE 8 (Ka = 22 ± 5 M−1 for 12 and Ka = 71 ± 17 M−1 for 13). Then, we investigated the spectroscopic features of both compounds (Fig. S4†) and the metal binding effect in solution by UV-vis and fluorescence spectroscopy (Fig. 7, S5 and S6†). The most interesting results were obtained from the fluorescence measurements.
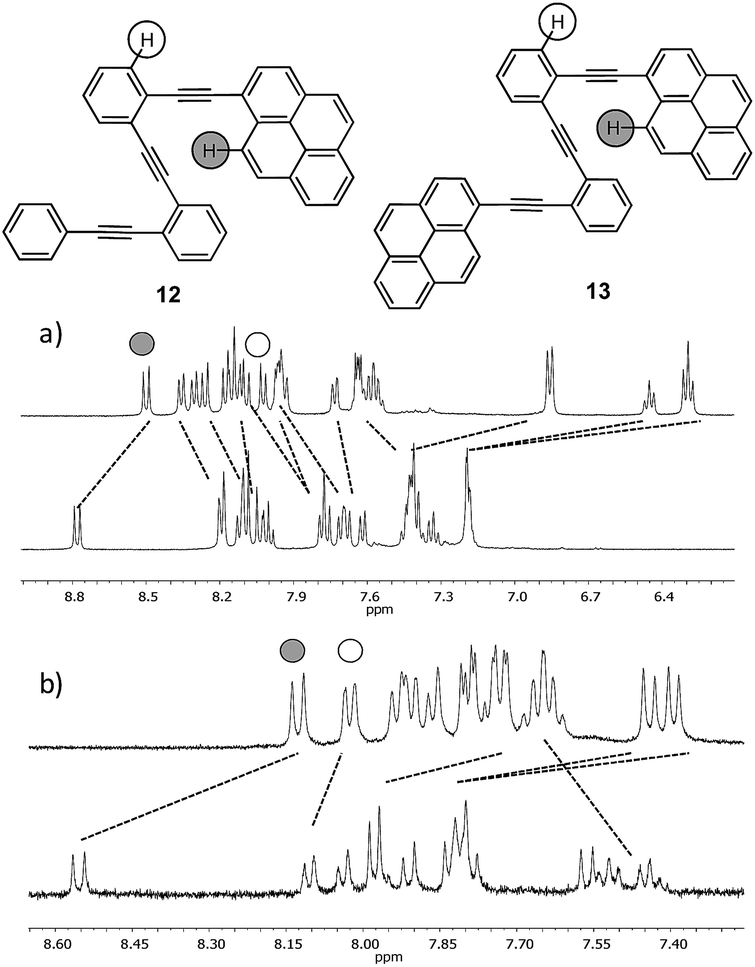 | ||
| Fig. 6
1H NMR spectra of the ligands (a) 12 (7 mM) and (b) 13 (40 mM) with an excess of AgBF4 (CD2Cl2–acetone-d6 = 9:1 mixture, 500 MHz, 298 K). Only the representative parts of the spectra are shown. | ||
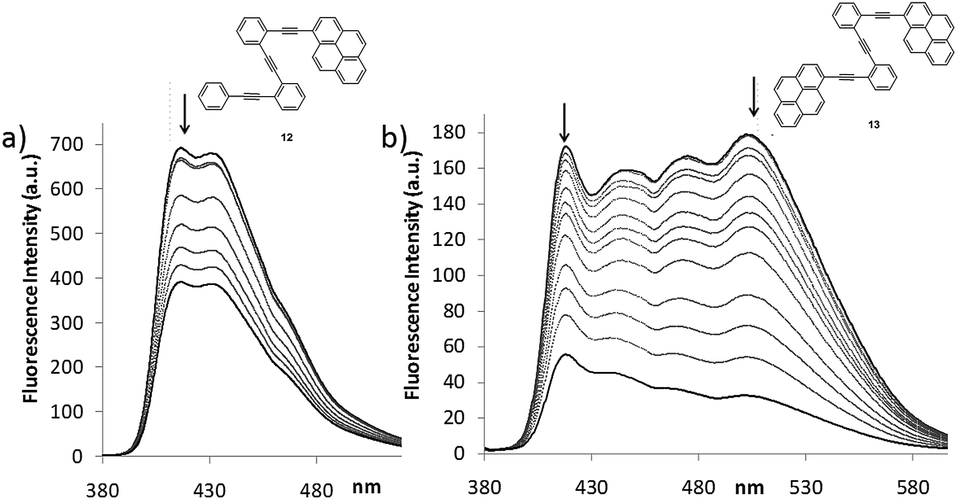 | ||
| Fig. 7 (a) Emission changes observed upon stepwise addition of AgBF4 to a solution of 12 (2.7 × 10−5 M; up to 500 eq.) and (b) 13 (4.4 × 10−5 M; up to 1500 eq.) using λexc = 375 nm in a CH2Cl2–acetone = 9:1 mixture at 298 K. | ||
The emission spectra of 12 and 13 showed significantly different spectral shapes (Fig. 7 and S4†). The emission maxima of 12 occurred at shorter wavelengths (λem from 410 to 450 nm) in comparison to the broad, multi-band emission of 13 (λem from 420 to 520 nm). The formation of intermolecular excimers was ruled out in the working concentration conditions and the long-wavelength band registered for 13 was attributed to the emission of an intramolecularly formed excimer.33 When Ag(I)-titrations of compounds 12 and 13 were recorded, fluorescence quenching was detected in both cases. Nevertheless, a significant ratiometric change was only observed in the titration of 13 (Fig. 7 and S6†).
Time-resolved fluorescence measurements can provide information on the kinetics of excimer formation, and thus on the dynamics of the formation of folded structures. Fluorescence decay traces of 13 at the pyrene monomer emission wavelengths (420 nm, Fig. S7†) and excimer emission wavelengths (560 nm, Fig. S8†) were recorded,21 and global deconvolution analysis was used to extract the fluorescence decay times (Fig. 8). Global analyses of the fluorescence decay traces of 13 in solution showed three different decay times, the shortest (τ3) of 0.713 ± 0.01 ns and two long components of τ2 = 6.37 ± 0.14 and τ1 = 12.36 ± 0.05 ns (Fig. 8a). Importantly, the fluorescence decay traces at the monomer emission wavelengths (420 nm) were effectively mono-exponential with the single τ3 decay time, whereas at the characteristic wavelength of the excimer emission (560 nm) the shortest component is a rise time (it has a negative amplitude), hence indicating the excited-state built up of excimer species. Nevertheless, the sum of all the amplitudes for the excimer trace is not equal to zero, thus indicating the presence of dimers formed in the ground state.34 Analysis of the absolute amplitudes (Fig. 8b) at 560 nm revealed that the amplitude associated with τ2 was almost equal in absolute value to the amplitude associated with τ3. This suggests that the component τ2 corresponds to the emission of the folded species formed dynamically during the excited state, and that τ3 is associated with the kinetics of the excimer formation reaction. On the other hand, prefolded species before excitation were associated with τ1. This is supported by the changes observed in the decay times and amplitudes when Ag(I) was added: the rise-time, τ3, becomes faster (Fig. 8), which involves an Ag(I)-facilitated excimer formation; and the contribution of the ground-state dimer is enhanced, as supported by the growth in τ1 amplitude. In light of these results, we propose the following kinetic model which relates the ground state equilibrium in which compound 13 is involved with the excited-state data (Fig. 9). Unfortunately, this dynamic situation avoids a direct evaluation of the relative ratio between folded and unfolded species.
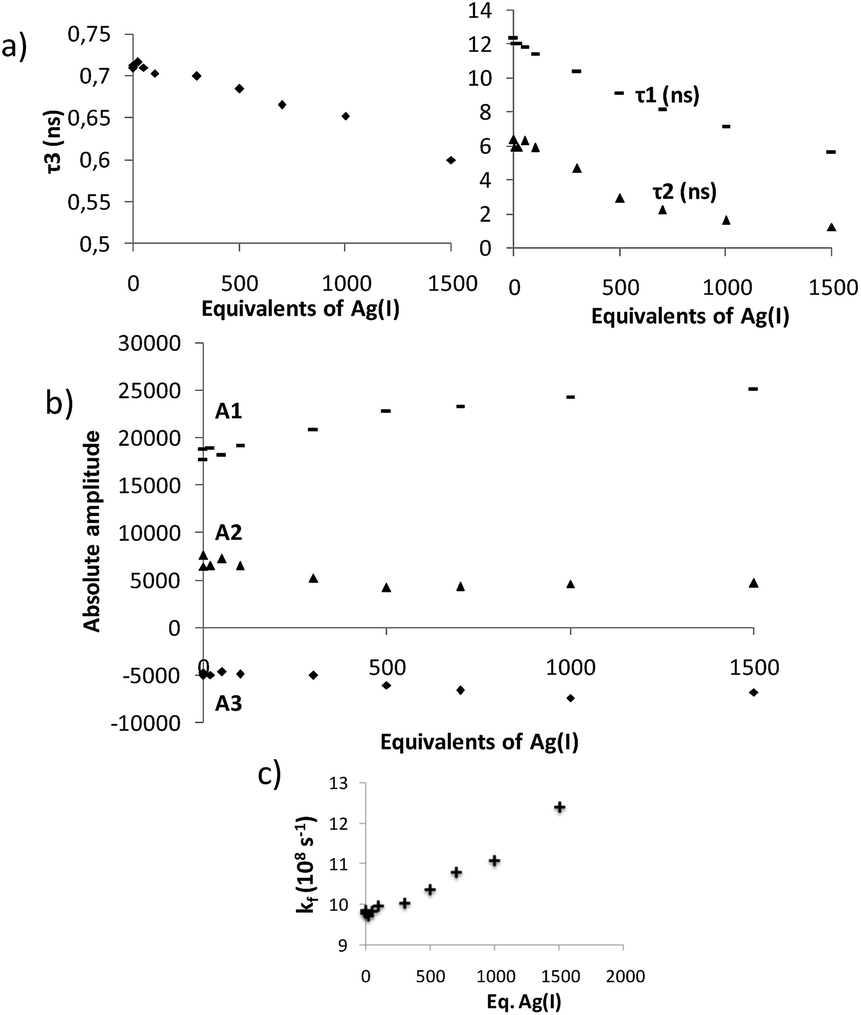 | ||
| Fig. 8 (a) Plots of the global lifetime components τi and (b) absolute amplitudes (at λem = 560 nm) obtained from the fluorescence decays of 13 recorded upon stepwise addition of AgBF4 in a CD2Cl2–acetone-d6 = 9:1 mixture at 298 K. (c) Evolution of the excimer formation rate constant kf due to the Ag(I) coordination. | ||
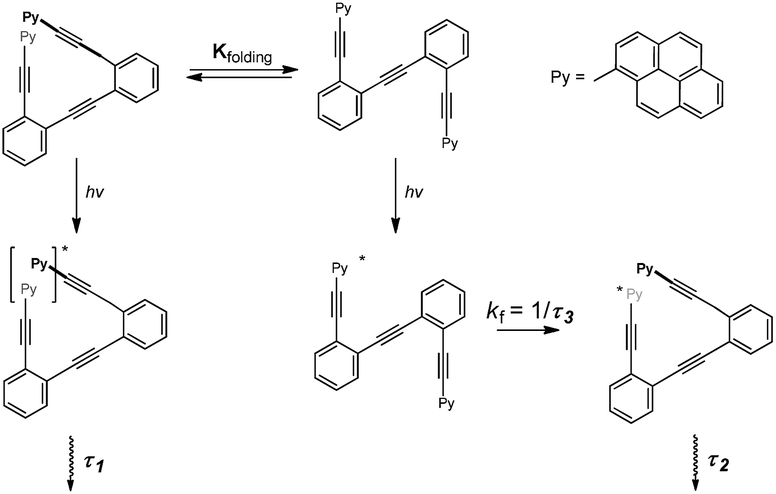 | ||
| Fig. 9 Schematic model which relates the ground state equilibrium of 13 (Kfolding) and the kinetics of the excited-state folding reaction. | ||
Interestingly, compound 12 also presents rich photochemistry. From the global deconvolution analysis two lifetimes could be extracted, τ1 = 2.93 ± 0.03 and τ2 = 1.42 ± 0.06 ns, which were related to two different species in solution. The previously described alkynyl pyrenes only presented one lifetime and, therefore, rotations of the pyrene moiety can be ruled out as the origin of those lifetimes.35 We assigned those lifetimes to the folded and unfolded conformers of compound 12 and, for consistency with compound 13, we assigned the longest one to the folded species. The absence of excimer formation in this case favored the quantification of the two conformers in solution. We performed time-resolved emission spectroscopy (TRES) to obtain the emission spectra associated to each species (Fig. S9†), and quantified a conformational population consisting of a 70:30 folded–unfolded approximate relationship.21
Remarkably, the kinetics of the excited-state folding reaction (kf in Fig. 8c) can be also accessible through the fluorescence decay times. Using compound 12 as a reference, we determined the excited-state folding kinetic constant in the absence of Ag(I) (kf) as (9.8 ± 0.3) × 108 s−1.21,36 This value represents one of the few examples of an experimentally determined kinetic folding constant in OPE-based foldamers.37 As expected, the presence of Ag(I) favors the excited-state folding process, and the kf value increases (Fig. 8c). Although the increase may seem modest when taking into account the number of equivalents of Ag(I) added, it is consistent with the diluted conditions used (10−6 M) and the low binding constant determined for 13 in the 1H NMR experiments.
The effect of tuning the chemical structure of two additional o-OPEs with electron-rich and electron-poor terminal aryl groups (compounds 14 and 15) was subsequently investigated (Fig. 10). A careful examination of the 1H NMR titration spectra revealed that side-chain variation with a nitro group (compound 14) resulted in weaker coordination (Ka = 7.3 ± 0.3 M−1). The strong quenching of the fluorescence due to the electronic communication along the carbon backbone was evidenced by the low fluorescence quantum yield, which characterized this compound in comparison to other pyrene derivatives (ϕ = 0.26 ± 0.03 for 13, 0.009 ± 0.005 for 14, and 0.59 ± 0.07 for 15).21
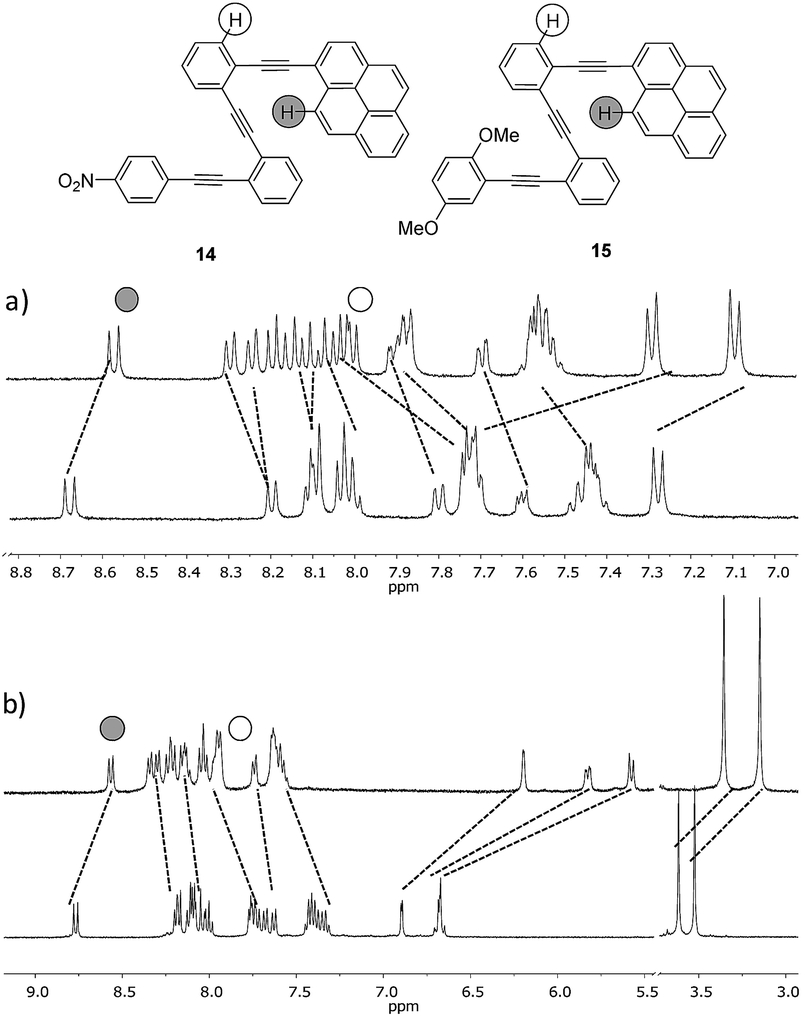 | ||
| Fig. 10 (a) 1H NMR titrations of the ligands 14 and (b) 15 with AgBF4 (CD2Cl2–d6-acetone = 9:1 mixture, 500 MHz, 298 K). | ||
Influence of a new coordination group
Although compound 15, bearing two methoxy groups on the benzene ring, did not show relevant photophysical information, it displayed a completely new phenomenon, evidenced in the 1H-NMR titration owing to methoxy groups taking part in the complexation (Fig. 10b and ESI†).At the beginning of the titration of 15 with up to 0.5 equiv. of Ag(I) the more affected signals were those corresponding to the dimethoxybenzene ring closely followed by the aromatic hydrogens of the inner benzenes. The broadening of the proton signals corresponding to the phenyl ring in a titration range between 0.16 and 1.2 equiv. revealed that a slow exchange process was taking place.21 Moreover, in this range, the protons of the inner aryls experienced more significant shifts from Δδ ≈ 0.11 to 0.23 ppm consistent with a cooperative change. Above 1.2 equiv., no further change was subsequently observed suggesting that the saturation plateau had been reached. A stronger interaction for compound 15 with the silver was also evidenced by the increase of the binding constant (Ka = 187 ± 84 M−1). The coordination of the silver into the foldamer void with a helical conformation was evidenced by the 13C shift of the alkyne signals and by several strong 2D-NOESY cross-peaks.21 Complementary cross-peaks between the methoxy group located at the ortho position (δ = 3.36 ppm) and the more deshielded H-10 proton of the pyrene ring, together with some cross-peaks between the m-MeO group (δ = 3.14 ppm) and other pyrene protons, were consistent with a stable helical conformation in which the o-MeO group was located onto the cavity of the helix probably coordinating the silver ion (Fig. S10†). Hence, compound 15 appeared to be a quite promising ligand to obtain the discrete and stable 1:1 Ag(I)-helix. In order to check if the presence of the pyrene had a crucial role in such behavior, we decided to study its phenyl analog 16. For compound 16, again fast exchange was observed during the 1H NMR titration (Fig. 11). It is noteworthy that when the binding constant was calculated from the NMR data, values higher than 105 M−1 were obtained with good fitting parameters for a 1:1 stoichiometry. NOESY cross-peaks were again observed confirming the helical conformation of 16 in solution (Fig. S11†). The cooperative effect of the alkyne-based coordination and oxygen coordination is remarkable when taking into account the low values of the binding constant for each independent binding site.
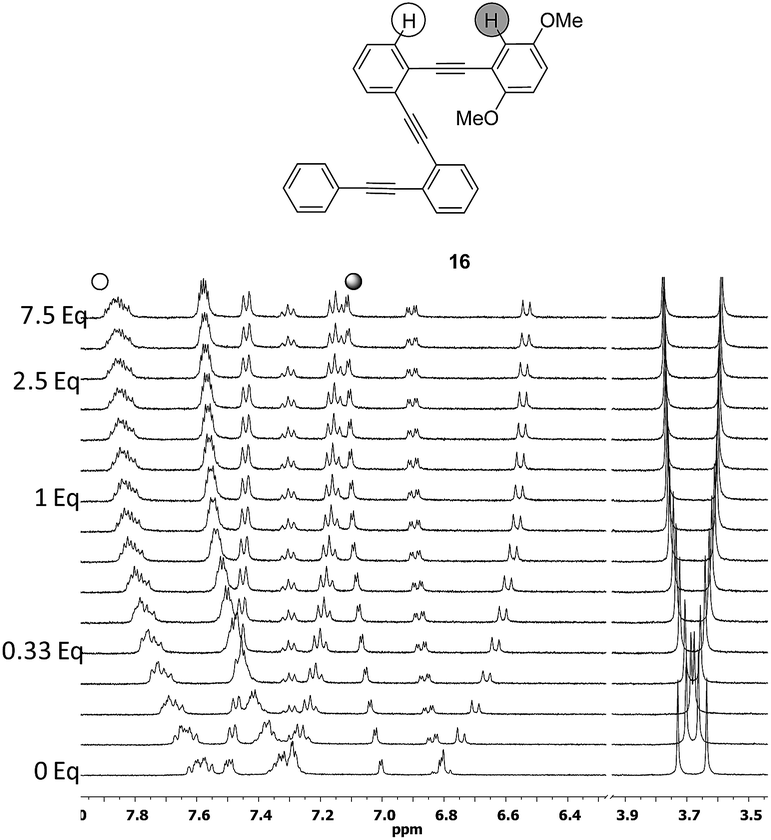 | ||
| Fig. 11
1H NMR titration of 16 with AgBF4 (CD2Cl2–acetone-d6 = 9:1 mixture, 500 MHz, 298 K). | ||
Conclusions
For the first time, the Ag(I)–alkyne interaction has been shown to induce the folding of non-conformationally restricted o-OPE foldamers by simple C–C bond rotations yielding a new class of metallofoldamers. The diverse interactions of silver with the o-OPE defines the binding constant and three-dimensional structure of the foldamer. These interactions can be modulated as a function of size and/or the substituents in the organic structures, allowing foldamers of different structure and strength using Ag(I) as the only promoter. Additionally, the synergetic effect of hydrogen bonding and preorganization of the ligands has been detected to increase the stability of the organometallic products facilitating the binding/folding event. It is noteworthy that the unexpected but promising role displayed by the MeO group in compounds 15 and 16, opens up the opportunity to design a new generation of more robust helicates. The possibility to change the alkoxy chain, e.g. a chiral group, could potentially be applicable to form a single P or M helicate and to transfer the chirality to the metal center.38 Thus, the development of new chiral metal catalysts or chemoselective chemical sensors could start to become a realistic goal.Acknowledgements
This research was funded by the Regional Government of Andalucía (project P09-FQM-4571) and the ICIQ Foundation. DM thanks Regional Government of Andalucía for her contract. AML thanks MICINN for her FPU fellowship. The authors thank the Centro de Servicios de Informática y Redes de Comunicaciones (CSIRC), Universidad de Granada, for providing the computing time.Notes and references
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271 CrossRef CAS PubMed; H. Juwarker, J.-M. Suk and K.-S. Jeong, Chem. Soc. Rev., 2009, 38, 3316 RSC; Foldamers: Structure, Properties and Applications, ed. S. Hecht and I. Huc, Wiley-VCH, Weinheim, Germany, 2007 CrossRef PubMed; D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef PubMed; S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef.
- T. Martinek and F. Fülöp, Chem. Soc. Rev., 2012, 41, 687 RSC; Y.-B. Lim, K.-S. Moon and M. Lee, Chem. Soc. Rev., 2009, 38, 925 RSC; M. C. Petty, Molecular Electronics: From Principles to Practice, Wiley-VCH, Weinheim, Germany, 2008 CrossRef CAS PubMed; E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166 CrossRef CAS PubMed.
- H. Miyake and H. Tsukube, Chem. Soc. Rev., 2012, 41, 6977 RSC; A. Petitjean, L. A. Cuccia, J.-M. Lehn, H. Nierengarten and M. Schmutz, Angew. Chem., Int. Ed., 2002, 41, 1195 CrossRef CAS; C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005 CrossRef PubMed; J.-M. Lehn, Supramolecular Chemistry, Concepts and Perspectives, Wiley-VCH, Weinheim, 1995 Search PubMed.
- Metallofoldamers: Supramolecular Architectures from Helicates to Biomimetics, ed. G. Maayan and M. Albrecht, Wiley-VCH, Weinheim, Germany, 2013 CrossRef CAS PubMed; Z. Dong, J. N. Plampin III, G. P. A. Yap and J. M. Fox, Org. Lett., 2010, 12, 4002 CrossRef CAS PubMed; G. Maayan, Eur. J. Org. Chem., 2009, 5699 CrossRef; S. Akine, Y. Morita, F. Utsuno and T. Nabeshima, Inorg. Chem., 2009, 48, 10670 CrossRef PubMed; Z. Dong, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2007, 129, 11850 CrossRef PubMed; Z. Dong, R. J. Karpowicz, S. Bai, G. P. A. Yap and J.-M. Fox, J. Am. Chem. Soc., 2006, 128, 14242 CrossRef PubMed; F. Zhang, S. Bai, G. P. A. Yap, V. Tarwade and J. M. Fox, J. Am. Chem. Soc., 2005, 127, 10590 CrossRef PubMed; R. B. Prince, T. Okada and J. S. Moore, Angew. Chem., Int. Ed., 1999, 38, 233 CrossRef.
- J. C. Nelson, J. G. Saven, J. S. Moore and P. G. Wolynes, Science, 1997, 277, 1793 CrossRef CAS PubMed.
- For seminal works of meta-oligophenylene ethynylenes (OPEs): R. B. Prince, J. G. Saven, P. G. Wolynes and J. S. Moore, J. Am. Chem. Soc., 1999, 121, 3114 CrossRef CAS; M. S. Gin, T. Yokozawa, R. B. Prince and J. S. Moore, J. Am. Chem. Soc., 1999, 121, 2643 CrossRef; L. Brunsveld, E. W. Meijer, R. B. Prince and J. S. Moore, J. Am. Chem. Soc., 2001, 123, 7978 CrossRef PubMed; S. Lahiri, J. L. Thompson and J. S. Moore, J. Am. Chem. Soc., 2000, 122, 11315 CrossRef; A. Tanatani, M. J. Mio and J. S. Moore, J. Am. Chem. Soc., 2001, 123, 1792 CrossRef PubMed; K. Matsuda, M. T. Stone and J. S. Moore, J. Am. Chem. Soc., 2002, 124, 11836 CrossRef PubMed; A. Tanatani, T. S. Hughes and J. S. Moore, Angew. Chem., Int. Ed., 2002, 41, 325 CrossRef; M. T. Stone, J. M. Fox and J. S. Moore, Org. Lett., 2004, 6, 3317 CrossRef PubMed; R. F. Kelly, B. Rybtchinski, M. T. Stone, J. S. Moore and M. R. Wasielewski, J. Am. Chem. Soc., 2007, 129, 4114 CrossRef PubMed; R. A. Smaldone and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 5444 CrossRef PubMed; R. A. Smaldone and J. S. Moore, Chem.–Eur. J., 2008, 14, 2650 CrossRef PubMed; S. Y.-L. Leung, A. Y.-Y. Tam, C.-H. Tao, H. S. Chow and V. W.-W. Yam, J. Am. Chem. Soc., 2012, 134, 1047 CrossRef PubMed.
- Modern Gold Catalyzed Synthesis; A, ed. S. K. Hashmi and F. D. Toste, Wiley-VCH, Weinheim, Germany, 2012 RSC; M. Bandini, Chem. Soc. Rev., 2011, 40, 1358 RSC; L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933 CrossRef CAS; A. Corma, A. Leyva-Perez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef PubMed; S. Flügge, A. Anoop, R. Goddard, W. Thiel and A. Fürstner, Chem.–Eur. J., 2009, 15, 8558 CrossRef PubMed; Silver in Organic Chemistry, ed. M. Harmata, Wiley-VCH, Weinheim, Germany, 2010 CrossRef PubMed; M. Naodovic and Y. Yamamoto, Chem. Rev., 2008, 108, 3132 CrossRef PubMed; J.-M. Weibel, A. Blanc and P. Pale, Chem. Rev., 2008, 108, 3149 CrossRef PubMed; Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef PubMed; E. Jimenez-Nunez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef PubMed; N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef PubMed; U.-H. Letinois, J.-M. Weibel and P. Pale, Chem. Soc. Rev., 2007, 36, 759 RSC.
- H. Lang, T. Stein, S. Back and G. Rheinwald, J. Organomet. Chem., 2004, 689, 2690 CrossRef CAS; H. Lang, N. Mansilla, R. Claus, T. Rüffer and G. Rheinwald, Inorg. Chim. Acta, 2001, 373, 93 CrossRef; S. M. Cortez and R. G. Raptis, Coord. Chem. Rev., 1997, 162, 495 CrossRef; H. Lang, K. Köhler and S. Blau, Coord. Chem. Rev., 1995, 143, 113 CrossRef.
- A. Das, C. Dash, M. A. Celik, M. Yousufuddin, G. Frenking and H. V. R. Dias, Organometallics, 2013, 32, 3135 CrossRef CAS; A. Reisinger, N. Trapp, I. Krossing, S. Altmannshofer, V. Herz, M. Presnitz and W. Scherer, Angew. Chem., Int. Ed., 2007, 46, 8295 CrossRef PubMed; A. Kunze, S. Balalaie, R. Gleiter and F. Rominger, Eur. J. Org. Chem., 2006, 2942 CrossRef; R. Gleiter, T. V. Hirschheydt and F. Rominger, Eur. J. Org. Chem., 2000, 2127 CrossRef; A. Kunze, R. Gleiter and F. Rominger, Chem. Commun., 1999, 171 RSC; T. Nishinaga, T. Kawamura and K. Komatsu, J. Chem. Soc. ( Chem. Commun. , 1998 , 2263 ) Search PubMed; J. D. Ferrara, A. Djebli, C. A. Tessier and W. J. Youngs, J. Am. Chem. Soc., 1988, 110, 647 CrossRef.
- For seminal works of ortho-oligophenylene ethynylenes (OPEs): R. A. Blatchly and G. N. Tew, J. Org. Chem., 2003, 68, 8780 CrossRef CAS PubMed; T. V. Jones, R. A. Blatchly and G. N. Tew, Org. Lett., 2003, 5, 3297 CrossRef PubMed; T. V. Jones, M. M. Slutsky, R. Laos, T. F. A. de Greef and G. N. Tew, J. Am. Chem. Soc., 2005, 127, 17235 CrossRef PubMed; R. A. Blatchly and G. N. Tew, J. Org. Chem., 2003, 68, 8780 CrossRef PubMed; M. M. Slutsky, T. V. Jones and G. N. Tew, J. Org. Chem., 2007, 72, 342 CrossRef PubMed; M.-X. Zhu, W. Lu, N. Zhu and C.-M. Che, Chem.–Eur. J., 2008, 14, 9736 CrossRef PubMed; J. Jiang, M. M. Slutsky, T. V. Jones and G. N. Tew, New J. Chem., 2010, 34, 307 RSC; Y.-T. Shen, N. Zhu, X.-M. Zhang, K. Deng, W. Feng, Q. Yan, S. Lei, D. Zhao, Q.-D. Zeng and C. Wang, Chem.–Eur. J., 2011, 17, 7061 CrossRef PubMed; J. Li, G. Hu, N. Wang, T. Hu, Q. Wen, P. Lu and Y. Wang, J. Org. Chem., 2013, 78, 3001 CrossRef PubMed.
- N. Fuentes, A. Martín-Lasanta, L. Álvarez de Cienfuegos, R. Robles, D. Choquesillo-Lazarte, J. M. García-Ruiz, A. J. Mota, L. Martínez-Fernández, I. Corral, D. J. Cárdenas, M. Ribagorda, M. C. Carreño and J. M. Cuerva, Angew. Chem., Int. Ed., 2012, 51, 13036 CrossRef CAS PubMed.
- For other approaches to covalently lock supramolecular arrangements, see: S. Hecht and A. Khan, Angew. Chem., Int. Ed., 2003, 42, 6021–6024 CrossRef CAS PubMed; R. A. Smaldone, E.-C. Lin and J. S. Moore, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 927 CrossRef; C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891 CrossRef; Y.-K. Kim, P. S. Kutchukian and G. L. Verdine, Org. Lett., 2010, 12, 3046 CrossRef PubMed; H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281 CrossRef; D. J. Yeo, S. L. Warriner and A. J. Wilson, Chem. Commun., 2013, 49, 9131 RSC; M. J. Kim, Y. R. Choi, H. G. Jeon, P. Kang, M.-G. Choi and K.-S. Jeong, Chem. Commun., 2013, 49, 11412 RSC; J. Yu, J. R. Horsley, K. E. Moore, J. G. Shapter and A. D. Abell, Chem. Commun., 2014, 50, 1652 RSC; I.-E. S. Müller, B. Bernet, C. Dengiz, W. B. Schweizer and F. Diederich, Eur. J. Org. Chem., 2014, 941 CrossRef.
- P. D. Frischmann and M. J. MacLachlan, Chem. Soc. Rev., 2013, 42, 871 RSC.
- N. Fuentes, L. Álvarez de Cienfuegos, A. Parra, D. Choquesillo-Lazarte, J. M. García-Ruiz, M. Marcos, E. Buñuel, M. Ribagorda, M. C. Carreño, D. J. Cárdenas and J. M. Cuerva, Chem. Commun., 2011, 47, 1586 RSC; S. Rodríguez-Bolívar, F. M. Gómez-Campos, L. Álvarez de Cienfuegos, N. Fuentes, D. J. Cárdenas, E. Buñuel, J. E. Carceller, A. Parra and J. M. Cuerva, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 125424 CrossRef CAS; N. Fuentes, A. Martín-Lasanta, L. Álvarez de Cienfuegos, M. Ribagorda, A. Parra and J. M. Cuerva, Nanoscale, 2011, 3, 4003 RSC; A. J. Mota, L. Álvarez de Cienfuegos, S. P. Morcillo, N. Fuentes, D. Miguel, S. Rodríguez-Bolívar, F. M. Gómez-Campos, D. J. Cárdenas and J. M. Cuerva, ChemPhysChem, 2012, 13, 3857 CrossRef PubMed; A. Martín-Lasanta, D. Miguel, T. García, J. A. López-Villanueva, S. Rodríguez-Bolivar, F. M. Gómez-Campos, E. Buñuel, D. J. Cárdenas, L. Álvarez de Cienfuegos and J. M. Cuerva, ChemPhysChem, 2012, 13, 860 CrossRef PubMed.
- B. H. Zimm and J. K. Bragg, J. Chem. Phys., 1959, 31, 526 CrossRef CAS.
- R. Chinchilla and C. Najera, Chem. Soc. Rev., 2011, 40, 5084 RSC; R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed; K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef.
- J. E. Gano, G. Subramaniam and R. Birnbaum, J. Org. Chem., 1990, 55, 4760 CrossRef CAS; H. C. Kang, A. W. Hanson, B. Eaton and V. Boelelheide, J. Am. Chem. Soc., 1985, 107, 1979 CrossRef; J. L. Pierre, P. Baret, P. Chautemps and M. Armand, J. Am. Chem. Soc., 1981, 103, 2986 CrossRef.
- A mechanistic study of a silver-catalyzed Sonogashira protocol based on 1H, 13C and 109Ag-NMR data reported by Pale, et al. provided several data to contrast our findings: U. Létinois-Halbes, P. Pale and S. Berger, J. Org. Chem., 2005, 70, 9185–9190 CrossRef PubMed . See Fig. S1 in ESI† for details.
- AgOTf was also tested in the titration experiments as a silver source but the best results were obtained using AgBF4. This fact is related to the lesser coordinating character of BF4− in comparison to the triflate anion.
- P. Kuzmic, Anal. Biochem., 1996, 237, 260 CrossRef CAS PubMed.
- See ESI† for details.
- Titrations of compounds 1, 7 and 8 were repeated several times at different concentrations and the same titration curves were obtained in all of the trials.
- Tolane and bis-alkynyl trimer were also titrated and sequential changes in the chemical shift were recorded indicating that Ag(I)–alkyne coordination occurred. However, the titration curves showed no clear break point over the range investigated, suggesting the coexistence of several complexes with different stoichiometries in solution. In addition, an isomer of compound 9 in which the central 1,3-substituted benzene also led to similar results evidencing that at least three alkynes located in ortho positions (adjacent coordination sites) were required for the metal-assisted folding. See Fig. S2 in ESI† for details.
- Modern NMR instruments can record good quality spectra with sub-millimolar concentrations and therefore NMR might be suitable for measuring association constants up to and even above 106 M−1, although many literature references will state that 105 M−1 is the limit for NMR titration experiments. See: P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305 RSC.
- M. J. Chmielewski, T. Zielinski and J. Jurczak, Pure Appl. Chem., 2007, 79, 1087 CrossRef CAS.
- A mono-hydroxylated o-OPE was synthesized and titrated giving a value of Ka = 16.8 ± 0.8 M−1, closer to the ones obtained for 8. See Fig. S3 in ESI† for details.
- Poly-hydroxylated backbones turned out to be quite insoluble products hence their non-substituted analogs were chosen for the study.
- The Ag(I)–alkyne values are shorter than the ones reported for the II·Ag(I) complex (ca. 2.7–2.8 Å, from ref. 9) but they are quite similar to that measured for [(cyclooctyne)3Ag][PF6] (ca. 2.3–2.4 Å, from ref. 9).
- A. G. Young and L. R. Hanton, Coord. Chem. Rev., 2008, 252, 1346 CrossRef CAS.
- G. F. Swiegers and T. J. Malefetse, Chem. Rev., 2000, 100, 3483 CrossRef CAS PubMed.
- E. C. Constable, Chem. Soc. Rev., 2013, 42, 1637 RSC; Y. Wang, J. Xu, Y. Wang and H. Chen, Chem. Soc. Rev., 2013, 42, 2930 RSC; J. Crassous, Chem. Commun., 2012, 48, 9684 RSC.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260 CrossRef CAS PubMed; S. H. Lee, S. H. Kim, K. S. Kim, J. H. Jung and J. S. Kim, J. Org. Chem., 2005, 70, 9288 CrossRef PubMed; H. Yuasa, N. Miyagawa, T. Izumi, M. Nakatani and I. Hashimoto, Org. Lett., 2004, 6, 1489 CrossRef PubMed; F. M. Winnick, Chem. Rev., 1993, 93, 587 CrossRef.
- The fluorescence emission profiles were invariant with concentration in the range of 10−5 to 10−7 M.
- P. Reynders, W. Kühnle and K. A. Zachariasse, J. Am. Chem. Soc., 1990, 112, 3929 CrossRef CAS.
- See for example: H. Maede, T. Maeda, K. Mizuno, K. Fujimoto, H. Shimizu and M. Inouye, Chem.–Eur. J., 2006, 12, 824 CrossRef CAS PubMed; A. C. Benniston, A. Harriman, D. J. Lawrie and S. A. Rostron, Tetrahedron Lett., 2004, 45, 2503 CrossRef.
- A. M. Maçanita and K. A. Zachariasse, J. Phys. Chem. A, 2011, 115, 3183 CrossRef PubMed.
- W. Y. Yang, R. B. Prince, J. Sabelko, J. S. Moore and M. Grubele, J. Am. Chem. Soc., 2000, 122, 3248 CrossRef CAS; Z. Yu, S. Weidner, T. Risse and S. Hecht, Chem. Sci., 2013, 4, 4156–4167 RSC.
- G. Maayan, M. D. Ward and K. Kirshenbaum, Chem. Commun., 2009, 56–58 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details. Synthesis of all new substrates and complexes. 1H NMR and 13C NMR spectra of new compounds and the corresponding NMR titrations. Photophysical and theoretical data. CCDC 1012031 and 1012032. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc01988a |
| This journal is © The Royal Society of Chemistry 2014 |