
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric synthesis of N,O-heterocycles via enantioselective iridium-catalysed intramolecular allylic amidation†
Depeng
Zhao
,
Martín
Fañanás-Mastral
,
Mu-Chieh
Chang
,
Edwin
Otten
and
Ben L.
Feringa
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl; Web: http://www.benferinga.com Fax: +31 50 363 4278; Tel: +31 50 3634296
First published on 21st July 2014
Abstract
Chiral N,O-heterocycles were synthesized in high yields and excellent enantioselectivity up to 97% ee via iridium-catalysed intramolecular allylic substitution with nucleophilic attack by the amide oxygen atom. The resulting benzoxazine derivatives were further transformed into challenging chiral N,O-ketals bearing both a tertiary and a quaternary center with excellent diastereoselectivities.
Introduction
Chiral N,O-heterocycles are important structural motifs found in a variety of natural products which exhibit significant biological activities including anti-tumor, antifungal and antibacterial properties.1 Among these heterocycles, oxazolines, oxazines, benzoxazines and their derivatives are of particular importance. Efavirenz (Sustiva), which features a 4H-3,1-benzoxazin-2-one core, is a non-nucleoside reverse transcriptase inhibitor and is used for the treatment of human immunodeficiency virus (HIV) type 1.2 4H-3,1-benzoxazin-4-one derivatives are able to inhibit C1r serine protease.2b Furthermore, N,O-heterocycles play a prominent role in asymmetric synthesis as they are core structures of several chiral ligands used in asymmetric catalysis.3 The preparation of this class of heterocycles and their derivatives usually relies on the use of chiral amino alcohols.2,4 However, direct catalytic asymmetric syntheses of these chiral heterocycles remain challenging. As far as we know, it was not until very recently that the first catalytic methods were reported, which are all based on asymmetric halocyclization of amides (Scheme 1a).5 In order to expand chiral space in heterocyclic chemistry, the development of complementary asymmetric routes to N,O-heterocycles with diverse functional groups is highly desirable.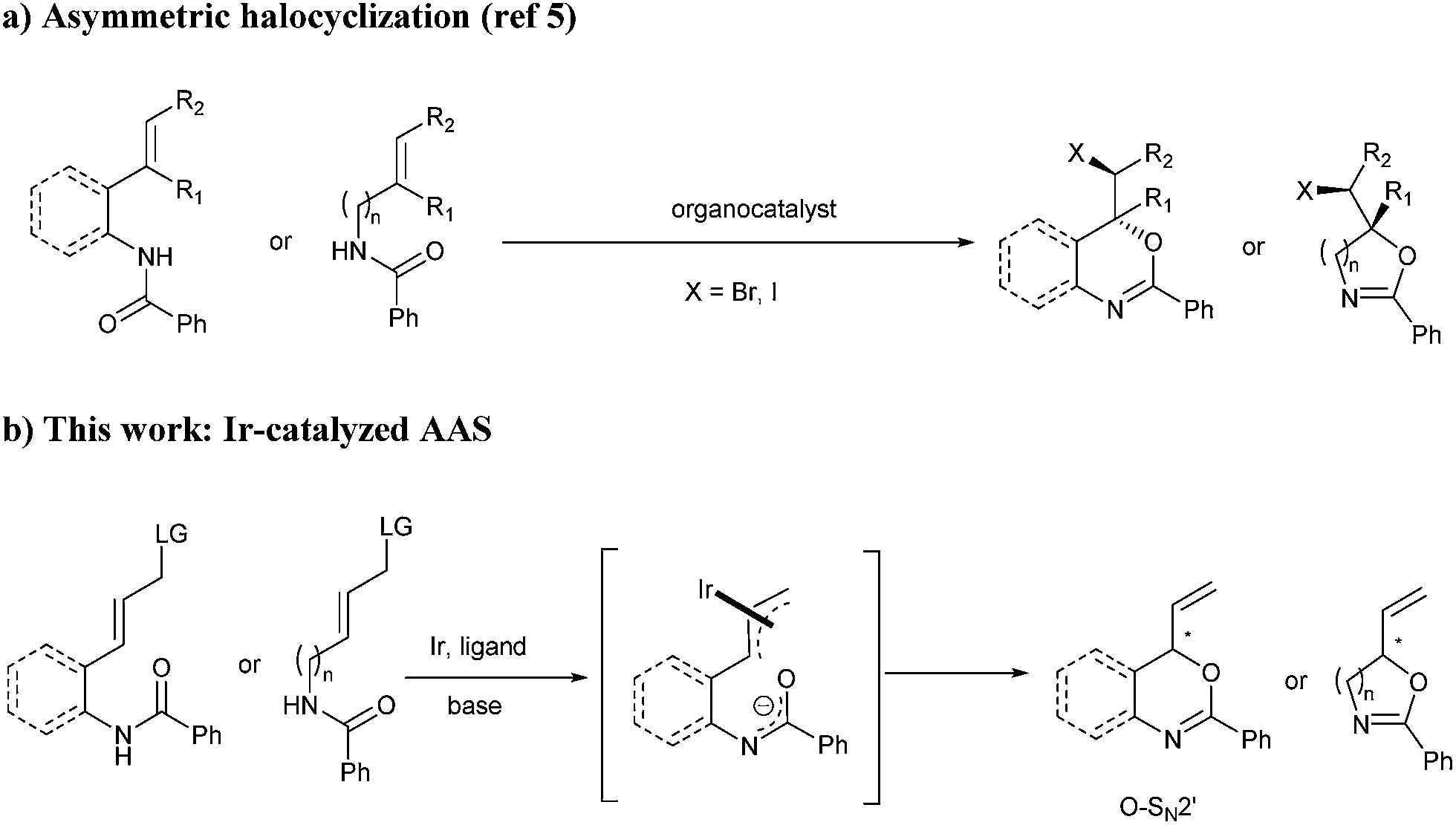 | ||
| Scheme 1 Protocols for catalytic asymmetric synthesis of N,O-heterocycles. | ||
Asymmetric allylic substitution (AAS) represents a powerful transformation for the assembly of congested chiral architectures.6 In particular, Ir-catalysed AAS features high reactivity and allows stereoselective construction of chiral branched allylic compounds.7 Since the pioneering work of Helmchen8 and Hartwig,9 several iridium-catalysed asymmetric allylic substitution reactions with phosphoramidites10 as chiral ligands have been developed. To date, a variety of nucleophiles including C-,11 N-,12 O-13 and S-nucleophiles14 have been used in this type of reaction. We recently developed the first intramolecular Ir-catalysed asymmetric allylic amidation.15 Through nucleophilic attack by the amide nitrogen atom, chiral tetrahydroisoquinolines and saturated N-heterocycles could be obtained with high enantioselectivities. However, to the best of our knowledge, transition metal-catalysed asymmetric allylic substitution with nucleophilic attack by the amide oxygen atom is unprecedented.16
In view of our continuing interest in developing new catalytic methodologies for the construction of chiral heterocycles,17 we decided to study the asymmetric synthesis of N,O-heterocyclic derivatives. We envisioned that reversing the selectivity of the ambidentate amide nucleophile in the iridium-catalysed intramolecular AAS could provide a versatile method to access chiral oxazolines, oxazines and benzoxazines (Scheme 1b). Herein, we report the successful realization of this catalytic transformation i.e. iridium-catalysed intramolecular allylic substitution with nucleophilic attack by the amide oxygen atom. The resulting N,O-heterocyclic derivatives can be readily obtained in high yields and excellent enantioselectivities (up to 97% ee).
Results and discussion
Our preliminary studies started with the intramolecular reaction of 2-benzamido cinnamyl carbonate 1a with 5 mol% of [Ir(cod)Cl]2 in the presence of 10 mol% of phosphoramidite L1 at 50 °C. We were pleased to find that the anticipated reaction proceeded smoothly to provide the benzoxazine product 2a with 31% ee, without formation of any SN2 by-products resulting from attack by either N or O atoms (Table 1, entry 1).|
|
|||||
|---|---|---|---|---|---|
| Entrya | Ligand | T (°C) | Time (h) | Conv.b (%) | eec,e (%) |
| a Reaction conditions: 5 mol % [Ir(cod)Cl]2, 10 mol % of ligand, 0.2 mmol of 1a, 0.2 mmol of base in 2.0 mL THF (0.1 M) unless otherwise noted. b Determined by 1H NMR analysis of the crude reaction mixture. c Determined by HPLC analysis. d Reaction was performed with 4.0 mL THF (0.05 M). e Negative value indicates that opposite enantiomer was obtained. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. | |||||
| 1 | L1 | 50 | 14 | 90 | −31 |
| 2 | L2 | 50 | 14 | 90 | −57 |
| 3 | L3 | 50 | 14 | 75 | −53 |
| 4 | L4 | 50 | 14 | 21 | −7 |
| 5 | L5 | 50 | 14 | 80 | −1 |
| 6 | L6 | 50 | 14 | >95 | 27 |
| 7d | L2 | RT | 16 | 40 | −79 |
| 8 | L7 | 50 | 14 | >95 | 88 |
| 9 | L7 | RT | 40 | >95 | 95 |
| 10 | L8 | RT | 40 | >95 | 91 |
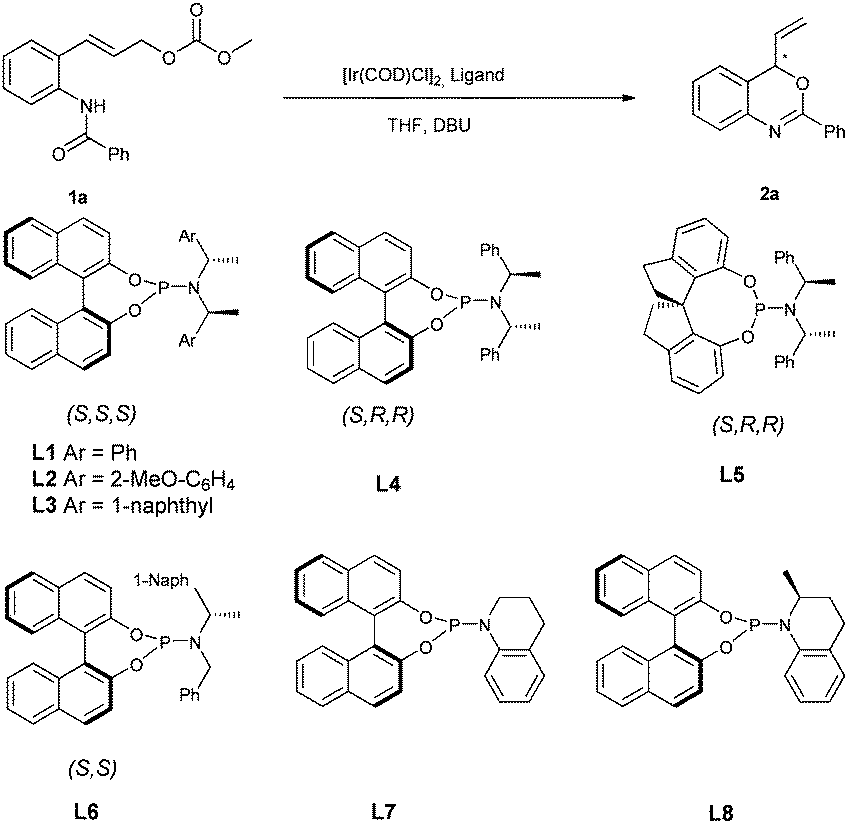
Encouraged by this result, we examined a series of related phosphoramidite ligands L2–L6 (Table 1, entries 2–6). It turned out that the reaction with L2 (ref. 18) at RT led to a promising increase in ee, however, only modest conversion was achieved (entry 7, 79% ee). It has been reported that in allylic substitution with the iridium catalysts derived from commonly used ligands L1 and L2, ortho-substituted cinnamyl substrates in general are not tolerated, with much decreased enantioselectivities observed in many cases.9a,11h,13d,19 The use of tetrahydroquinoline-derived phosphoramidite ligand L7 gave rise to full conversion and furnished the cyclization product with 88% ee at 50 °C and 95% ee at RT (entries 8, 9). Related ligand L8, which features an additional stereogenic center and was reported to give good results in AAS of cinnamyl substrates,20 provided a slightly lower ee in the present transformation (entry 10). It should be noted that the active catalyst generated from L1 or L2 features an Ir–C(sp3) bond in accordance with the structure shown by Hartwig,9 while the active catalyst derived from L7 or L8, introduced by You's group, incorporates an Ir–C(sp2) bond instead.20
With a highly selective catalyst based on ligand L7 in hand, we further optimized the reaction conditions (Table 2). Evaluation of several bases indicated that the reaction was slower with inorganic bases such as Cs2CO3 and K2CO3, probably due to poor solubility (Table 2, entries 2 and 3), whereas the use of DABCO as a base led to a slightly higher ee (97% ee vs. 95% ee) and a similar reaction rate as compared with DBU (entries 1 and 4). With DABCO as the optimal base, the reaction was then performed with 2.5 mol% of [Ir(cod)Cl]2 at room temperature (entry 5). However, the reaction turned out to be very slow and did not reach full conversion under these conditions. Gratifyingly, when the reaction was performed at 35 °C with 5 mol% (2.5 mol% of [Ir(cod)Cl]2) catalyst, the ee was not affected and the reaction was complete when 3 equiv. of DABCO were used (Table 2, entries 6 and 7). Importantly, the enantioselectivity could be maintained and the reaction was completed in a shorter time when carried out at 50 °C (Table 2, entry 8, 81% yield, 97% ee). As shown in Table 2, the enantioselectivity and reaction rate can both be enhanced with additional equivalents of DABCO. It seems that this catalytic conversion tolerates higher temperature when DABCO is used as the base. In contrast, the use of DBU as the base under the same conditions led to 2a with only 89% ee (Table 2, entry 9).
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | [Ir(cod)Cl]2 (mol%) | T (°C) | Base (equiv.) | Time (h) | Conv.b (%) | eec (%) |
| a Reaction was performed on a 0.2 mmol scale in 2.0 mL THF. b Determined by 1H NMR analysis of the crude reaction mixture. c Determined by HPLC analysis. d Value in parenthesis is the yield of isolated 2a. DABCO = 1,4-diazabicyclo[2.2.2]octane. ND = Not determined. | ||||||
| 1 | 5% | RT | DBU (1) | 40 | >95 | 95 |
| 2 | 5% | RT | Cs2CO3 (1) | 40 | 75 | 94 |
| 3 | 5% | RT | K2CO3 (1) | 40 | 87 | 96 |
| 4 | 5% | RT | DABCO (1) | 40 | >95 | 97 |
| 5 | 2.5% | RT | DABCO (1) | 24 | 31 | ND |
| 6 | 2.5% | 35 | DABCO (1) | 48 | 90 | 96 |
| 7 | 2.5% | 35 | DABCO (3) | 48 | >95 | 97 |
| 8 | 2.5% | 50 | DABCO (3) | 24 | >95 (81)d | 97 |
| 9 | 2.5% | 50 | DBU (3) | 24 | >95 | 89 |
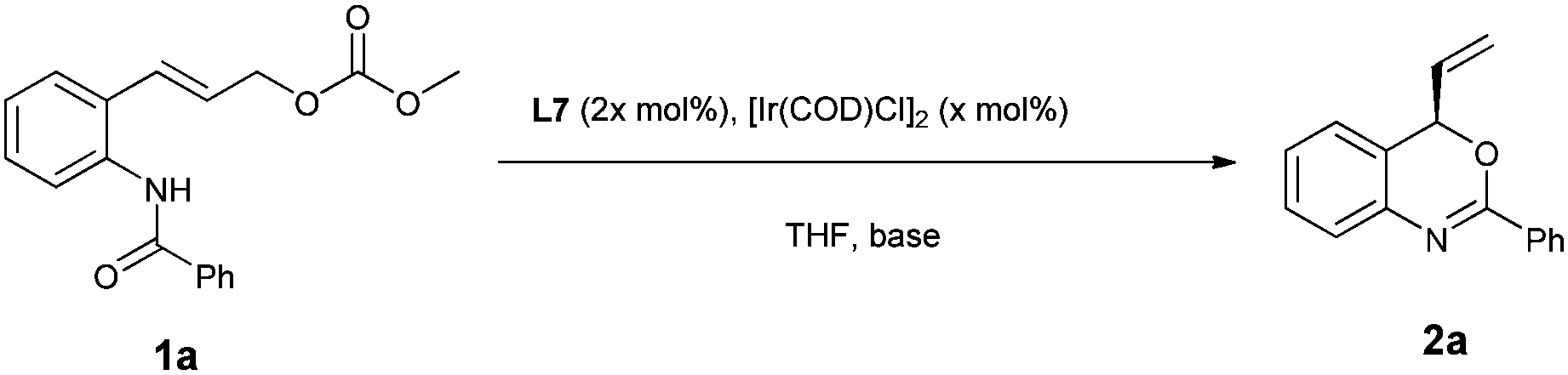
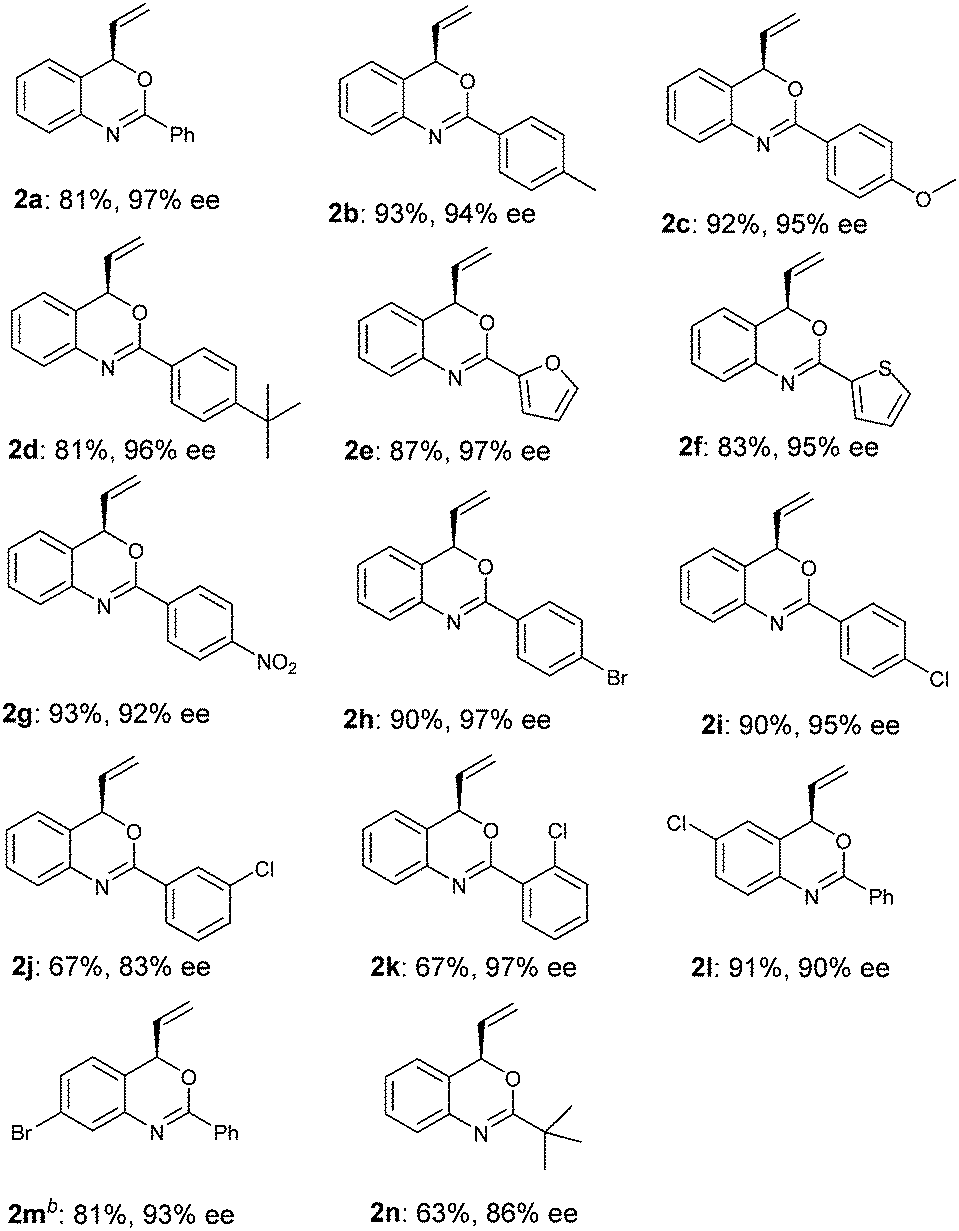
Under the optimized reaction conditions (Table 2, entry 8), we explored the substrate scope of this new catalytic allylic cyclization (Table 3). High tolerance for substituents at the para-position of the benzamide aryl group was observed, irrespective of the electronic and steric properties (Table 3, 2b–2d and 2g–2i). Substrates 1j and 1k with substituents at the meta- and ortho- position of the benzamide aryl group were also tested, and it was shown that 2j with meta-Cl gave a reduced ee, while ortho-Cl substituted substrate 1k provided 2k with 97% ee. Heteroarylamides 1e and 1f were also suitable substrates for the present cyclization reaction. Benzoxazine 2n with a t-butyl substituent at the 2-position could be obtained under standard reaction conditions in 63% yield and 86% ee. Notably, substrates 1l and 1m with substituents at the C4 and C5 position of the cinnamyl carbonate gave the desired products 2l and 2m in good yields and enantioselectivities.
|
|
|---|
| a Reaction conditions: 2.5 mol % [Ir(cod)Cl]2, 5.0 mol % of ligand, 0.2 mmol of 1, 0.6 mmol DABCO in 2.0 mL THF at 50 °C for 24 h. Yields of isolated products after column chromatography; enantiomeric excess determined by HPLC or GC analysis (see the ESI†). b The absolute configuration of 2m was determined as R by X-ray crystallographic analysis (Fig. 1).21 |
|
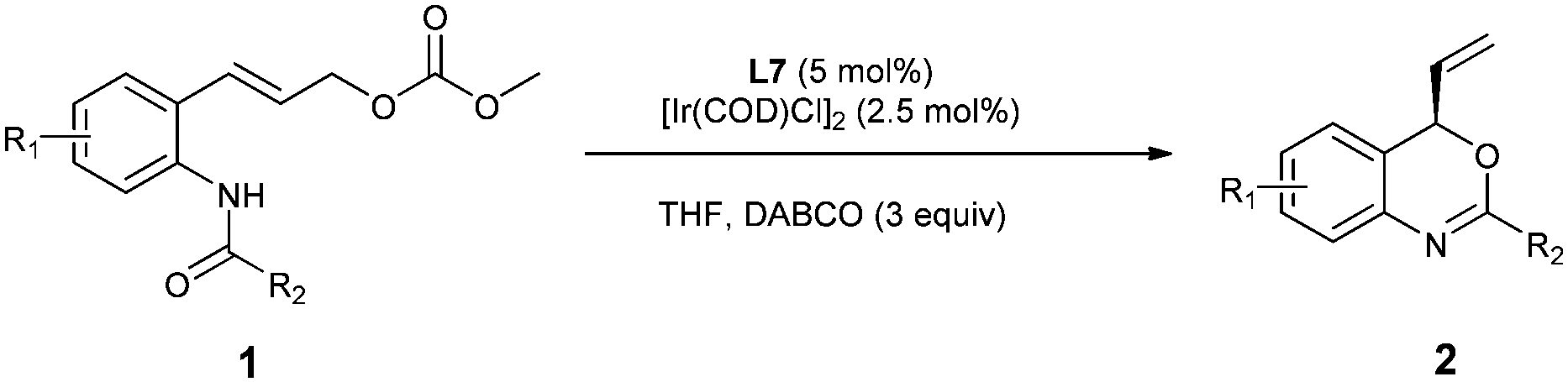
To further demonstrate the versatility of the reaction, we examined some aliphatic substrates to provide the corresponding chiral oxazoline and oxazine. Under the optimized reaction conditions for the enantioselective synthesis of benzoxazines 2, the reaction of aliphatic substrates 1o and 1p gave rise to a complex mixture of products. However, through a slight modification of the reaction conditions, i.e. running the reaction at room temperature and using DBU (0.5 equiv.) as the base instead of DABCO, the corresponding chiral oxazoline 2o and oxazine 2p were obtained with excellent ee (Scheme 2).
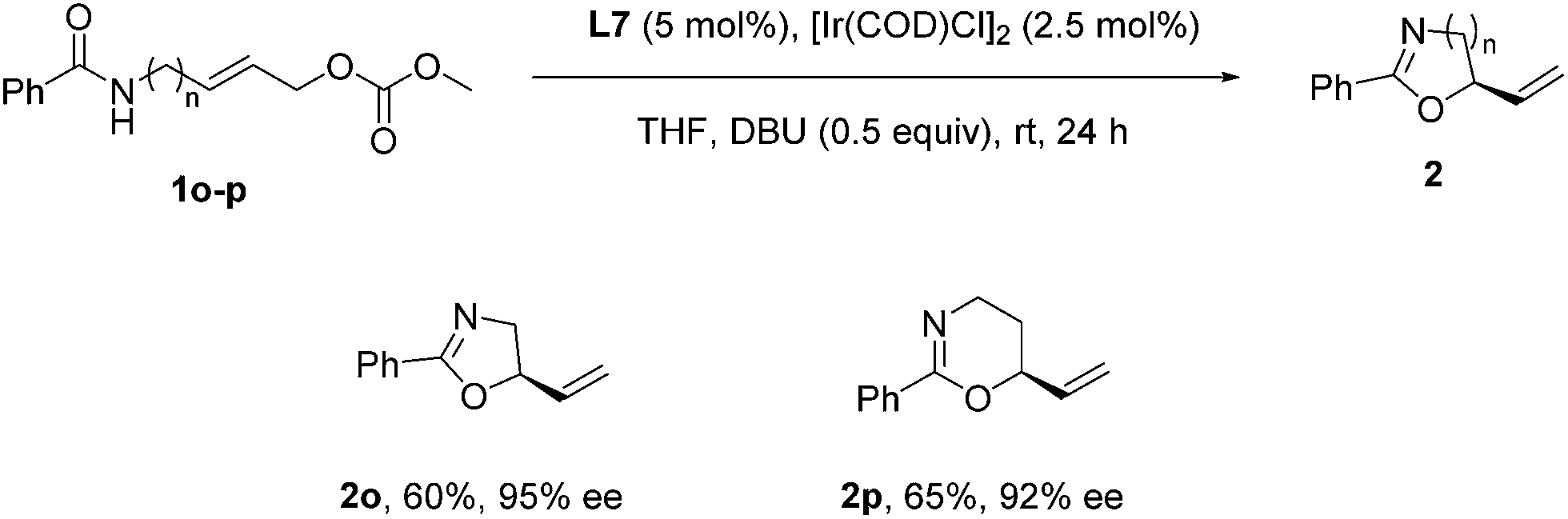 | ||
| Scheme 2 Ir-catalysed synthesis of chiral oxazoline 2o and oxazine 2p. | ||
In order to evaluate the synthetic applicability of this new catalytic method, a number of transformations of the benzoxazine products were carried out (Scheme 3). The allylation products of this transformation feature a terminal double bond which allows for further functionalization. As an example, the vinyl moiety of 2a underwent cross-metathesis with ethyl acrylate in the presence of the Grubbs–Hoveyda II catalyst without compromising the enantiomeric excess of product 3.
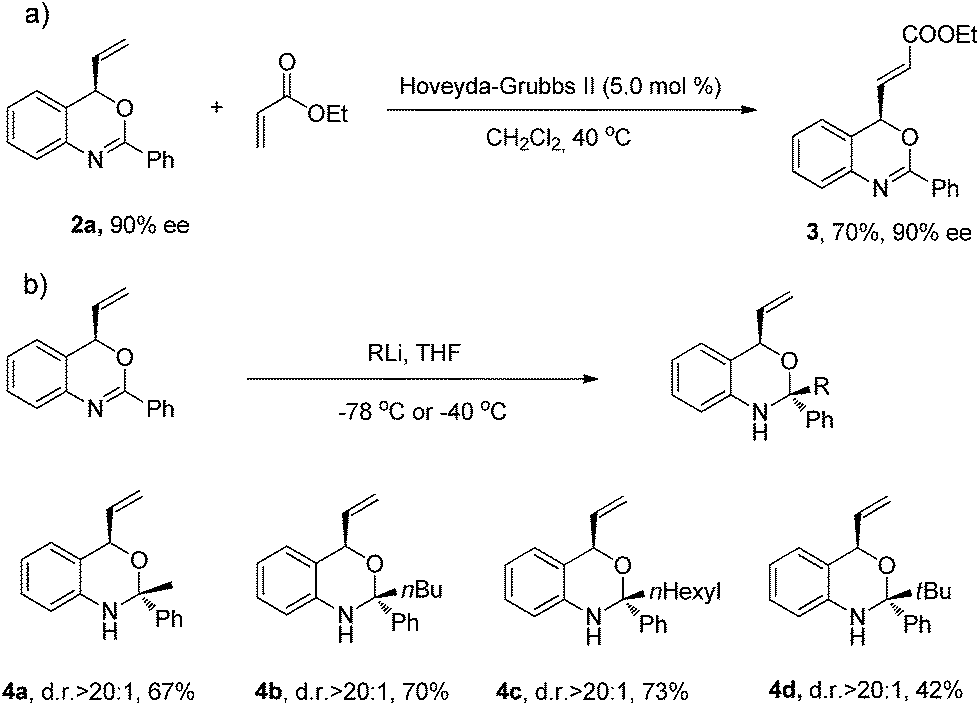 | ||
| Scheme 3 Transformations of benzoxazine 2a. | ||
In view of the synthetic challenges to prepare chiral hemiaminals,22 we also explored the 1,2-addition of organolithium reagents to chiral benzoxazine 2a.23 As shown in Scheme 3, a series of linear and branched organolithium reagents were successfully employed, furnishing the chiral N,O-ketals 4a–4d bearing both a tertiary and a quaternary stereogenic center with excellent diastereoselectivities. The relative configurations of 4a and 4d were determined by NOESY studies.24 Surprisingly, the X-ray analysis of 4a (ref. 21) confirmed that the methyl group is installed cis to the vinyl group (Fig. 1). It is expected that under kinetic control, the alkyl nucleophile would prefer to attack from the opposite side of the vinyl group due to steric hindrance.23,25 While the detailed mechanism is as yet unclear, on the basis of control experiments,26 we propose that there is an equilibrium between the closed acetal form and open imine form of the product formed in the reaction (Scheme 4). Through the ring opening and subsequent closing, the kinetically favored intermediate epimerizes in situ to the thermodynamically more stable intermediate, although alternative pathways cannot be excluded.
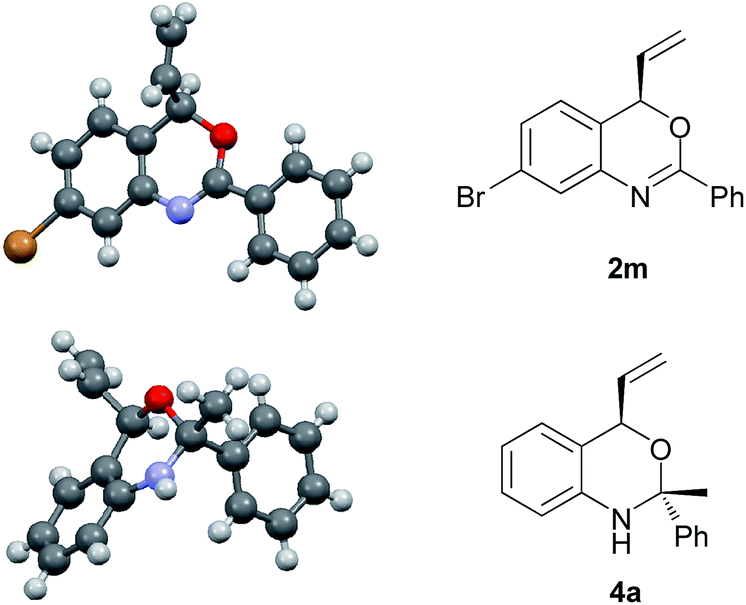 | ||
| Fig. 1 X-ray crystal structures of 2m and 4a. | ||
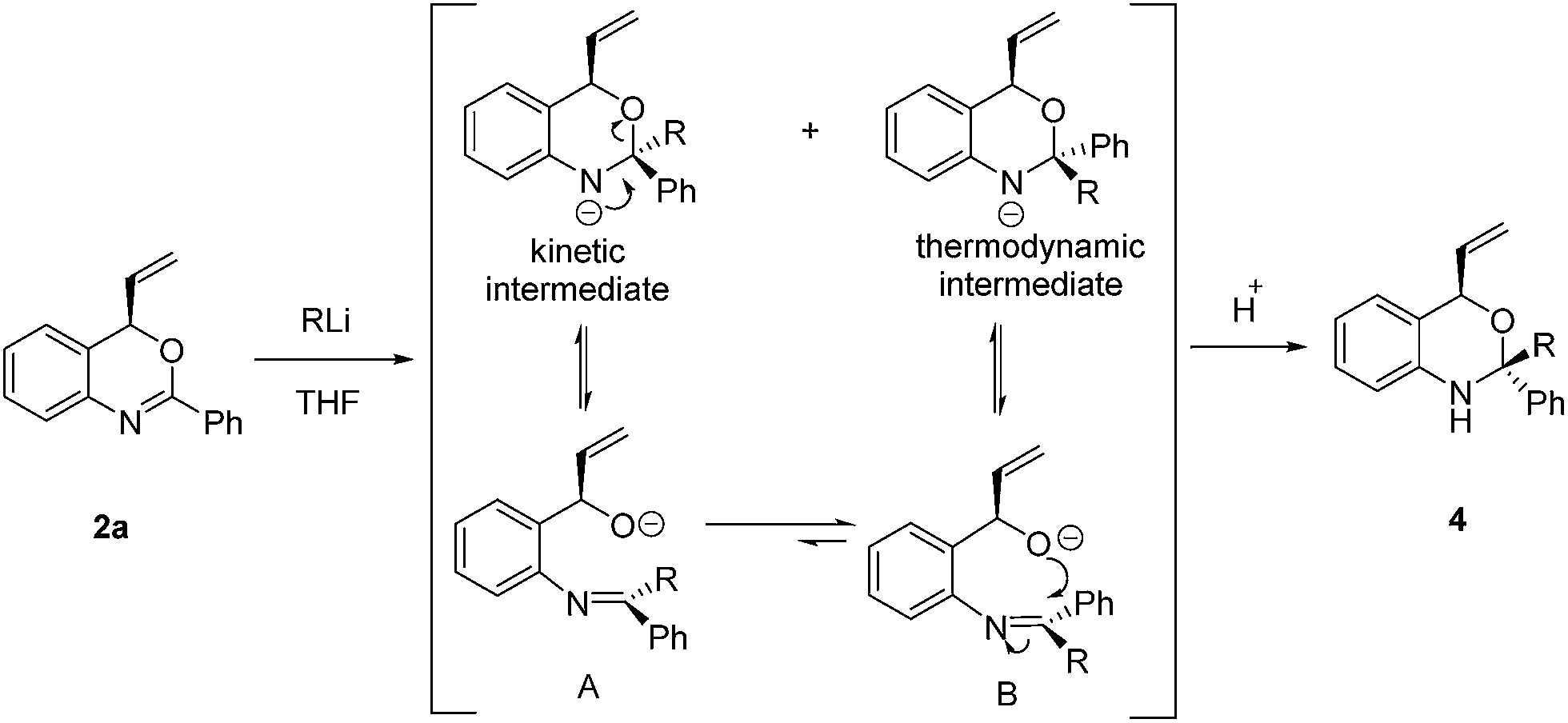 | ||
| Scheme 4 Proposed mechanism for the addition of organolithium reagents to benzoxazines. | ||
Conclusions
In conclusion, we have developed the first enantioselective iridium-catalyzed intramolecular allylic substitution reaction with nucleophilic attack by the amide oxygen atom. A series of N,O-heterocycles, i.e., oxazolines, oxazines and benzoxazine derivatives, could be readily obtained with high enantioselectivities. Furthermore, the resulting benzoxazine derivatives were transformed into chiral N,O-ketals bearing tertiary and quaternary centers with excellent diastereoselectivities. This methodology offers excellent opportunities to explore the “3rd dimension” in common planar N,O-heterocycles.Acknowledgements
The Netherlands Organization for Scientific Research (NWO-CW), the National Research School of Catalysis (NRSC-C), the European Research Council (ERC advanced grant 227897 to BLF), the Royal Netherlands Academy of Arts and Sciences (KNAW) and the Ministry of Education Culture and Science (Gravity program 024.601035) are acknowledged for financial support.Notes and references
-
(a) G. Pattenden, J. Heterocycl. Chem., 1992, 29, 607 CrossRef CAS PubMed
; (b) E. J. Corey, B. Czakó and L. Kürti, Molecules and Medicine, Wiley, Hoboken, 2007 Search PubMed
-
(a) N. Chinkov, A. Warm and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 2957 CrossRef CAS PubMed
-
(a) G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336 CAS
- A. Solladié-Cavallo, P. Lupattelli, C. Bonini, V. Ostuni and N. D. Blasio, J. Org. Chem., 2006, 71, 9891 Search PubMed
-
(a) V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681 CrossRef CAS PubMed
-
(a)
Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, Ed. U. Kazmaier, Springer, 2012 Search PubMed
- For recent reviews, see:
(a) G. Helmchen, A. Dahnz, P. Dübon, M. Schelwies and R. Weihofen, Chem. Commun., 2007, 675 RSC
-
(a) J. P. Janssen and G. Helmchen, Tetrahedron Lett., 1997, 38, 8025 CrossRef CAS
-
(a) T. Ohmura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 15164 CrossRef CAS PubMed
- For reviews, see:
(a) J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486 CrossRef CAS PubMed
- For selected examples, see:
(a) J. Y. Hamilton, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2013, 135, 994 CrossRef CAS PubMed
- For selected examples, see:
(a) M. Lafrance, M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3470 CrossRef CAS PubMed
- For selected examples, see:
(a) C. Fischer, C. Defieber, T. Suzuki and E. M. Carreira, J. Am. Chem. Soc., 2004, 126, 1628 CrossRef CAS PubMed
- For selected examples, see:
(a) M. Ueda and J. F. Hartwig, Org. Lett., 2010, 12, 92 CrossRef CAS PubMed
- J. F. Teichert, M. Fañanás-Mastral and B. L. Feringa, Angew. Chem., Int. Ed., 2011, 50, 688 CrossRef CAS PubMed
- For examples of intramolecular oxazine formation employing chiral allylic substrates, see:
(a) J. E. Joo, K. Y. Lee, V. T. Pham, Y. S. Tian and W. H. Ham, Org. Lett., 2007, 9, 3627 CrossRef CAS PubMed
- For selected examples, see:
(a) A. Rudolph, P. H. Bos, A. Meetsma, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2011, 50, 5834 CrossRef CAS PubMed
- K. Tissot-Croset, D. Polet and A. Alexakis, Angew. Chem., Int. Ed., 2004, 43, 2426 CrossRef CAS PubMed
-
(a) A. Alexakis and D. Polet, Org. Lett., 2004, 6, 3529 CrossRef CAS PubMed
- W.-B. Liu, C. Zheng, C.-X. Zhuo, L.-X. Dai and S.-L. You, J. Am. Chem. Soc., 2012, 134, 4812 CrossRef CAS PubMed
- CCDC 957090 (4a) and 957091 (2m) contain the supplementary crystallographic data for this paper.
-
(a) M. Miyashita, M. Sasaki, I. Hattori, M. Sakai and K. Tanino, Science, 2004, 305, 495 CrossRef CAS PubMed
- For transformations of benzoxazines, see: R. R. Schmidt and B. Beitzke, Chem. Ber., 1983, 116, 2115 Search PubMed
- For details, see the ESI†.
- B. H. Lipshutz, J. A. Kozlowski and R. S. Wilhelm, J. Org. Chem., 1983, 48, 546 CrossRef CAS
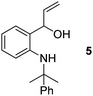
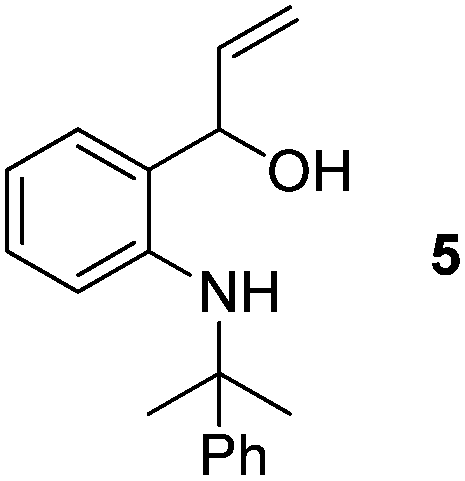
- When the reaction of 2a was performed with 3 equiv. of MeLi at RT, formation of compound 5 was observed. This might proceed via attack of MeLi on the open imine structure
 .
.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data for all new compounds. CCDC 957090 and 957091. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc01940g |
| This journal is © The Royal Society of Chemistry 2014 |