
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A water-soluble boronate-based fluorescent probe for the selective detection of peroxynitrite and imaging in living cells†
Xiaolong
Sun
a,
Qingling
Xu
b,
Gyoungmi
Kim
b,
Stephen E.
Flower
a,
John P.
Lowe
a,
Juyoung
Yoon
*b,
John S.
Fossey
c,
Xuhong
Qian
d,
Steven D.
Bull
*a and
Tony D.
James
*a
aDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: s.d.bull@bath.ac.uk; t.d.james@bath.ac.uk
bDepartment of Chemistry and Nano Science, Global Top 5 Program, Ewha Womans University, Seoul 120-750, Korea. E-mail: jyoon@ewha.ac.kr
cSchool of Chemistry, University of Birmingham, Birmingham, Edgbaston, West Midlands B15 2TT, UK
dSchool of Pharmacy, East China University of Science and Technology, Meilong Road 130, Shanghai 200237, China
First published on 23rd May 2014
Abstract
A water-soluble boronate-based fluorescent probe was evaluated for the detection of peroxynitrite (ONOO−) in the presence of a monosaccharide. The enhanced fluorescence of the probe when bound with D-fructose was switched off in the presence of peroxynitrite. In contrast, other reactive oxygen/nitrogen species (ROS/RNS) led to only slight fluorescence decreases due to protection by an internal N–B interaction. The interaction of the probe with D-fructose not only strengthens the fluorescence signal, but also protects the boronic acid from oxidation by other ROS/RNS. Therefore, under conditions generating various ROS/RNS, the boronate-based saccharide complex preferentially reacts with peroxynitrite. The probe was used in cell imaging experiments for the detection of endogenous and exogenous peroxynitrite. The sensor displays good “on–off” responses towards peroxynitrite, both in RAW 264.7 cells and HeLa cells.
Introduction
Peroxynitrite (ONOO−) – a combination of nitric oxide and the superoxide radical anion – has been recognized as a strong oxidant in physiological and pathological processes. It was first discovered as a biological endogenous oxidant in 1990.1 Under physiological conditions, peroxynitrite is also a highly reactive molecule with a very short life time (∼10 ms) involved in cell signal transduction2 and apoptosis in HL-60 cells,3 and PC-12 cells.4 Many biomolecules are oxidised and/or nitrated by peroxynitrite-derived radicals, including DNA, tyrosine residues, thiols, and unsaturated fatty-acid-containing phospholipids.5 Peroxynitrite formation has been implicated in Alzheimer's disease, Parkinson's disease, Huntington's disease, and traumatic brain injury.6–8 Recently, peroxynitrite was found as a key trigger of skeletal muscle hypertrophy via activation of calcium signaling.9 Thus, the importance of peroxynitrite has led to researchers seeking effective and applicable approaches for its detection.Synthetic fluorescent probes have the potential to be powerful tools for peroxynitrite detection since they can measure intracellular ONOO− directly.10 Working towards the fluorescence detection of peroxynitrite, Yang et al. developed a range of chemo-sensors in which ONOO− reacts with activated ketones to form dioxiranes,11–13 and Yang and Qian have also designed a three-channel fluorescent probe capable of distinguishing peroxynitrite from hypochlorite.14 Recently, Ai et al. reported a genetically encoded fluorescent probe for the detection of peroxynitrite.15 However, it still remains a great challenge to use small-molecular fluorescent probes to detect ONOO− selectively and sensitively amongst the large number of biologically relevant reactive oxygen and nitrogen species (e.g. H2O2 and ClO−).
We have a long-standing interest in boronic acids for monosaccharide and anion detection,16–18 and have found that boronic acid derivatives rapidly and reversibly interact with saccharides in aqueous media. In previous work, we investigated the reaction of “integrated” and “insulated” boronate-based fluorescent probes with hydrogen peroxide (pKa = 11.6) in the presence of monosaccharides.19 Chang and co-workers have also developed a series of boronate-based derivatives for the fluorescence detection of H2O2 in living systems.20–30
Functional N-substituted-1,8-naphthalimide derivatives have been successfully employed as DNA targeting, anticancer and cellular imaging agents.31,32 Due to its excellent photophysical and photochemical properties, N-substituted-1,8-naphthalimide is widely used as a D–π–A chromophore in the design of fluorescent probes.33,34
From our previous research, we found that boronic ester formation causes an enhanced interaction between the neighboring amine and the boron atom. The enhanced N–B interaction35 (whether direct or via solvent insertion) hinders the reaction between boron and H2O2 in the presence of saccharides resulting in a much slower decrease in fluorescence intensity (cf. a saccharide free system).
It is known that ONOO− (pKa = 6.8) – a strong nucleophile – reacts rapidly and stoichiometrically with aromatic boronate derivatives (κ ∼ 106 M−1 S−1 at pH 7.4, Scheme 1).36–38 From this observation and our previous results, we wondered if a powerful nucleophile such as ONOO− could overcome the protection given to the boron atom by the N–B interaction in the “insulated” probes. Therefore, we reasoned that it would be possible to develop boronate-based “insulated” probes for the selective intracellular mapping of peroxynitrite in the presence of a monosaccharide (D-fructose was chosen as a model saccharide since it has a high binding affinity, ∼4400 mol−1).18
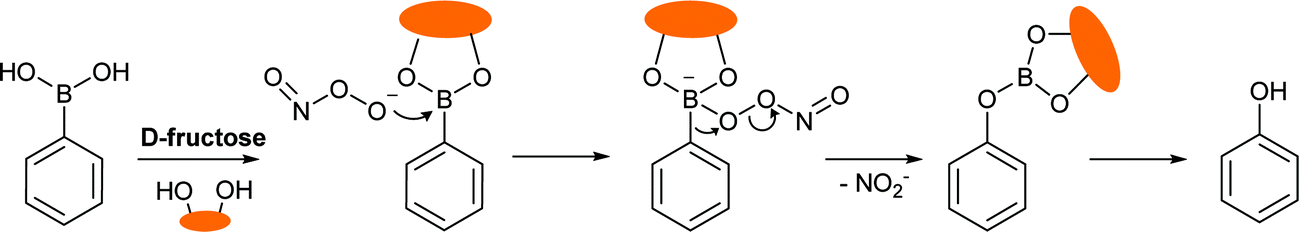 | ||
| Scheme 1 The reaction mechanism between a generic aryl boronic acid/ester and ONOO−. | ||
Results and discussion
Design strategy
Probe 1 (λabs = 440 nm, ε = 9500 M−1 cm−1, Scheme 2) was designed based on a PET fluorescence mechanism, with the introduction of hydroxyethoxyl side chain to not only improve the water-solubility, but also to provide a potential route to develop a polymer bound receptor. Probe 1 was synthesised in three steps starting from 4-bromo-1,8-naphthalic anhydride in 24% overall yield (Scheme S1†).39 The boronic acid moiety is widely recognized as an excellent binding node through which to connect a monosaccharide with the fluorescent probe. The “insulated” system displays an “off–on” response towards D-fructose due to the enhanced N–B interaction (Scheme 2).40 Furthermore, we decided to evaluate the fluorescence properties of the monosaccharide complex towards peroxynitrite in vitro and also using in vivo cellular imaging for endogenous and exogenous derived peroxynitrite.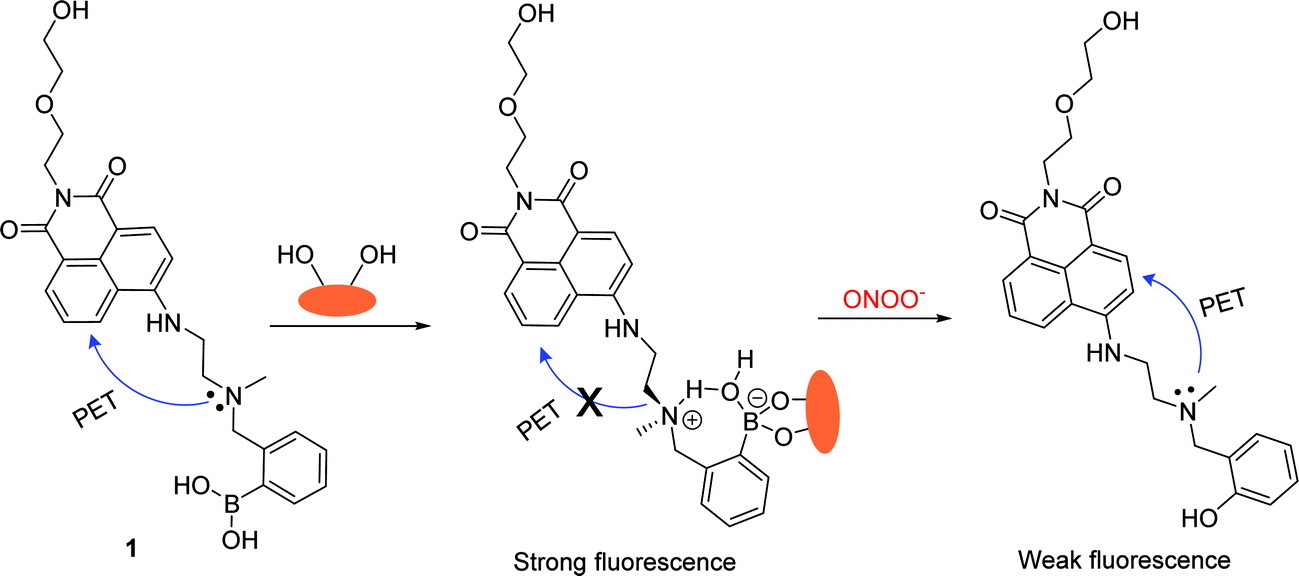 | ||
| Scheme 2 Design strategy of probe 1 for the detection of peroxynitrite in the presence of D-fructose. | ||
We carried out a pH titration to evaluate the effect of pH on the fluorescence of probe 1 (2 μM). As shown by Fig. 1, the fluorescence intensity of the probe 1 decreased at pH values above 8.0, while in the range between 3.0 and 8.0, the pH change had minimal effect on the fluorescence intensity. Thus, the probe can be expected to work well under physiological conditions (pH 7.30, PBS buffer). In the presence of D-fructose (100 mM), the fluorescence of the formed 1–D-fructose complex increases due to the enhanced N–B interaction at different pH values. The fluorescence of the 1–D-fructose complex decreases over a pH range of 3–11.
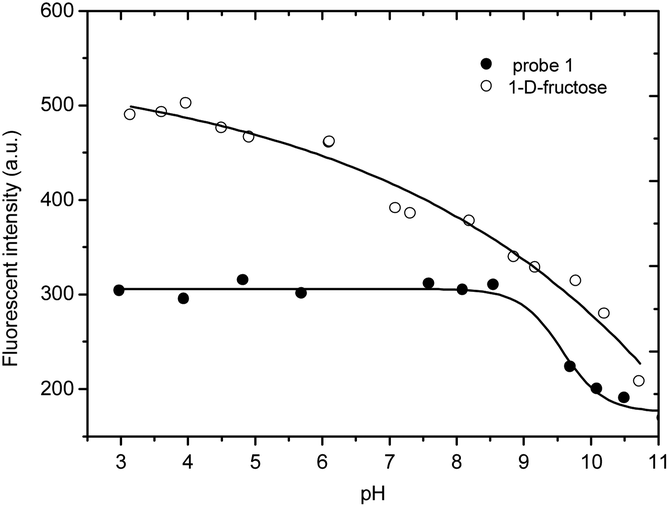 | ||
| Fig. 1 pH titration of probe 1 (2 μM) and 1–D-fructose (probe 1, 2 μM; D-fructose, 100 mM), modulated by utilizing aqueous hydrochloric acid (1.2 N) and sodium hydroxide solution (1 N). | ||
UV-Vis and emission spectra towards ONOO−
In the case of probe 1, a significant “off–on” signal response is seen on binding with D-fructose due to inhibition of the PET mechanism. From Fig. 2a, the maximum fluorescence intensity of probe 1 (2 μM, λem = 525 nm) is increased two-fold in the presence of D-fructose (100 mM) in pH 7.3 buffer solution. However, when the arylboronic ester moiety of probe 1 was transformed into a phenol upon adding peroxynitrite, the fluorescence was further reduced due to the stronger PET from the amine in the boron free system. As can be seen from the dose-dependent titration curve in Fig. 2b, the enhanced fluorescence of the probe 1–D-fructose complex was finally reduced to a F (in the presence of ONOO−)/F0 (in the absence of ONOO−) = ca. 0.10 over a ONOO− concentration range of 0–297 μM. In contrast, for the saccharide free system small amounts of peroxynitrite (60 μM) caused a big change in fluorescence intensity (F/F0 = ca. 0.10, Fig. 2c and d). In the UV-Vis spectra (Fig. S2†), the free boronic acid probe 1 (2 μM) displayed a maximum absorption (A = 0.026) at 440 nm while the binding of D-fructose (100 mM) led to a decrease to A = 0.019 at the maximum absorption wavelength. In the presence of ONOO− (100 μM), a new peak (A = 0.021) at 360 nm developed, ascribable to the formation of phenol.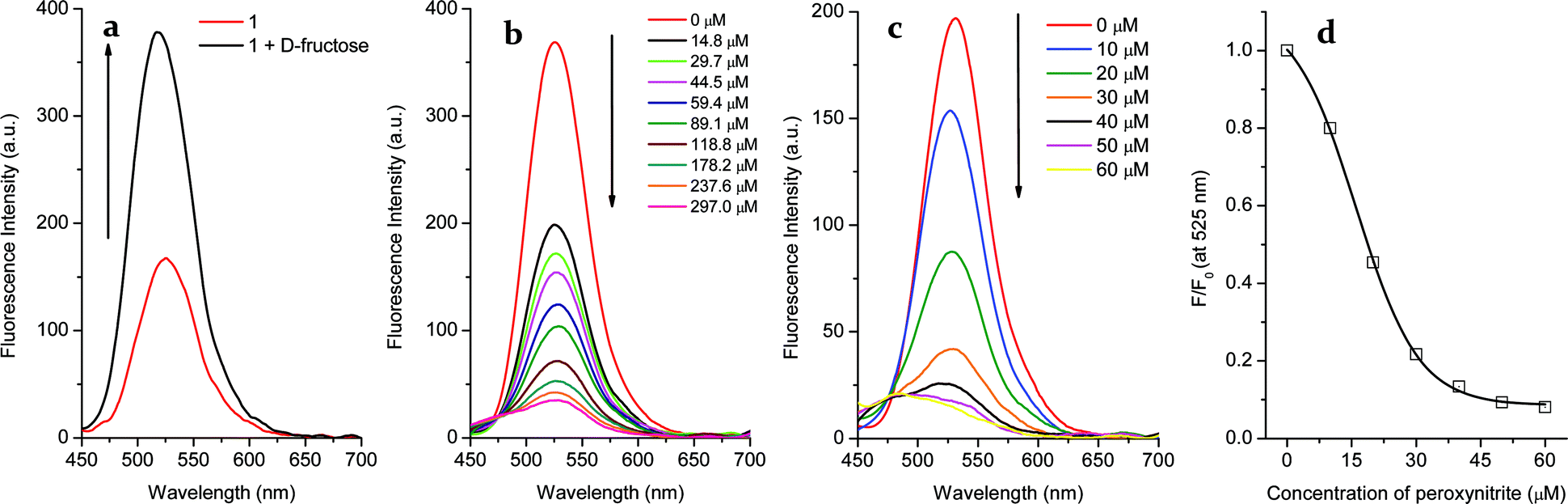 | ||
| Fig. 2 (a) Fluorescence spectra of probe 1 (2 μM) before and after the addition of D-fructose (100 mM). After the addition of D-fructose, the mixtures were stirred for 10 min; (b) probe 1–D-fructose complex (probe 1, 2 μM; D-fructose, 100 mM) at different concentrations of ONOO−. After addition of peroxynitrite, the mixtures were stirred for 5 min; (c) fluorescence spectra of probe 1 (2 μM) in the presence of various concentrations of peroxynitrite in pH 7.3 buffer solution; (d) the non-linear relationship between probe 1 (2 μM) and ONOO− (0–60 μM) in pH 7.3 buffer solution. The spectra were collected after 5 min stirring for each dose. The data were collected in PBS buffer (1/15 M, pH 7.30) with excitation at 410 nm (Ex slit: 5 nm, Em slit: 5 nm). | ||
Thus, the fluorescence of probe 1 is turned on by saccharide binding, since boronic ester formation causes an enhanced interaction between the neighbouring amine and the boron atom.
Emission spectra towards H2O2
Since boronate-based derivatives can be oxidised to phenol by H2O2 and ONOO−, it is very important to discriminate between them by fluorescence tools. Independently, we tested the responses of probe 1 and the 1–D-fructose complex towards hydrogen peroxide (Fig. 3 and S4†). With the free boronic acid system, the fluorescence of probe 1 (2 μM) increased to F/F0 = ca. 1.59 in the presence of hydrogen peroxide (100 μM) over 1 h in pH 7.30 buffer solution (Fig. S4†). When the solution was adjusted to pH 8.10, the fluorescence decreased most probably due to the decomposition of the intermediate to phenol (Fig. 3a). This is different from the process observed for the detection of ONOO− in which the boronic acid was transformed into phenol quickly and directly. However, in the case of the 1–D-fructose complex the fluorescence showed only a slight drop to F/F0 = ca. 0.75, even after the addition of H2O2 (1 mM) over 1 h (Fig. 3b and S7†). Therefore, the probe 1–D-fructose complex does not produce a significant response to H2O2.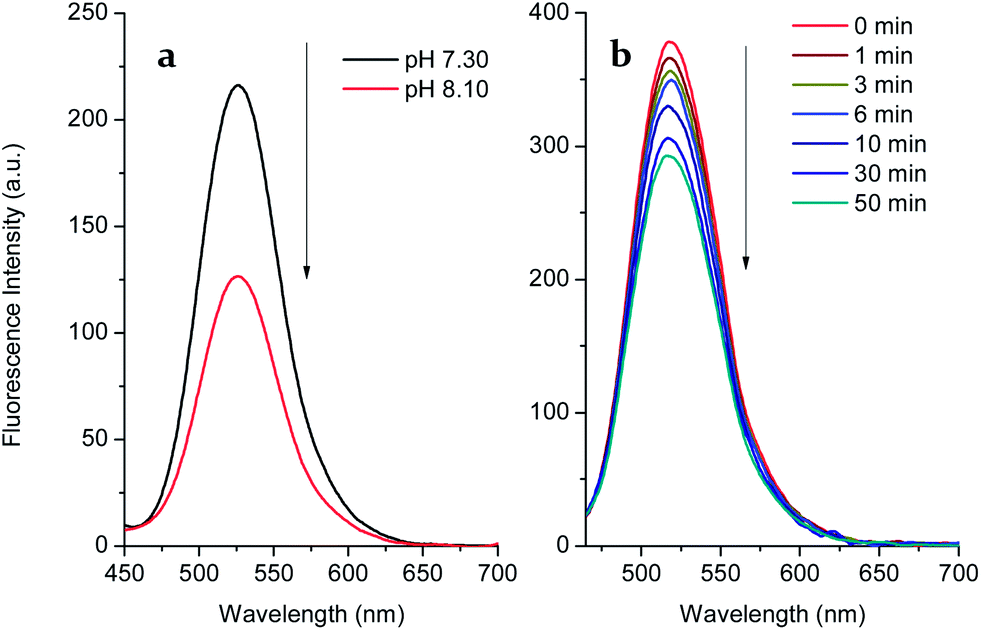 | ||
| Fig. 3 (a) Fluorescence spectra of probe 1 after reaction with H2O2 at pH 7.30 and pH 8.10. The pH was adjusted from 7.30 to 8.10 using aqueous sodium hydroxide (10 N); (b) fluorescence spectra of the probe 1–D-fructose complex (probe 1, 2 μM; D-fructose, 100 mM) in the presence of hydrogen peroxide (1 mM) at pH 7.3 buffer solution. The spectra were collected at different times with excitation at 410 nm (Ex slit: 5.0, Em slit: 5.0). | ||
Thus, the interaction of probe 1 with D-fructose not only strengthens the fluorescence signal, but also protects the boronic acid from oxidation by hydrogen peroxide via the N–B interaction. However, the oxidation reaction between boron and peroxynitrite was still very rapid and was complete within 1 min (Fig. S3†).
The sugar complex reacts stoichiometrically and rapidly with ONOO− to form the phenol product. Therefore, under conditions generating both H2O2 and ONOO−, the probe 1–D-fructose complex preferentially reacts with ONOO−.
Selectivity tests towards ROS/RNS
We also investigated the selectivity of the 1–D-fructose complex towards other reactive oxygen and nitrogen species, such as hypochlorite (−OCl), nitric oxide (NO), nitrite (NO2−), nitrate (NO3−), peroxyl radicals (ROO˙), superoxide (O2−) and hydroxyl radicals (˙OH) in pH 7.30 buffer solution (Fig. 4 and S8†). Among them, only hypochlorite (100 μM) caused a big fluorescence decrease ((F − F0)/F = ca. 0.52) (Fig. 4b). As reported previously,37 aryl boronic acids and esters can be oxidised into phenol by hypochlorite. However, under the same concentrations of −OCl (100 μM) and ONOO− (100 μM), peroxynitrite reacts much more strongly with the 1–D-fructose sensing system ((F − F0)/F = ca. 0.78).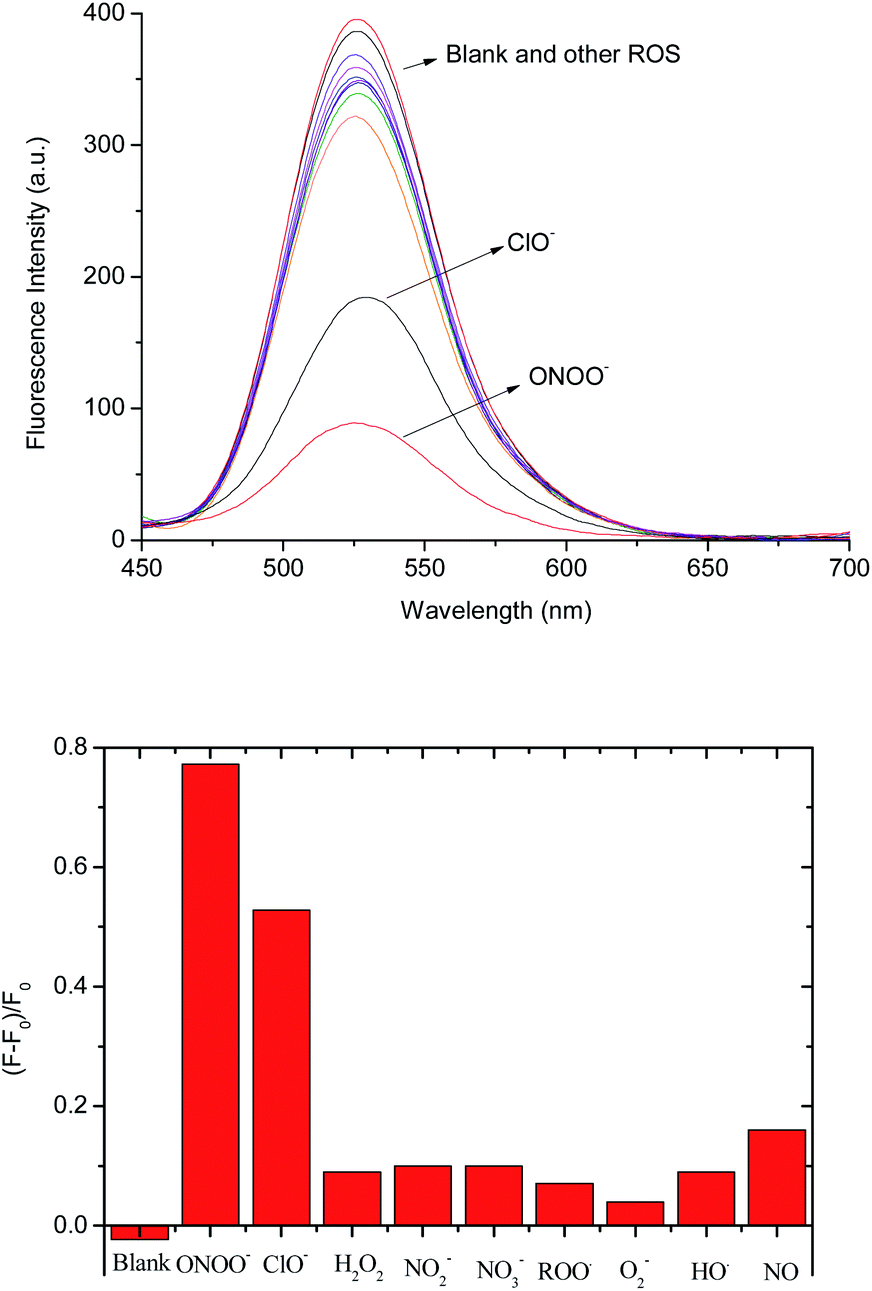 | ||
| Fig. 4 (a) Fluorescence spectra of probe 1–D-fructose (probe 1, 2 μM; D-fructose, 100 mM) in the presence of various ROS/RNS: ONOO− (100 μM, 5 min), −OCl (100 μM, 5 min), H2O2 (100 μM, 5 min), NO2− (100 μM, 5 min), NO3− (100 μM, 5 min), ROO˙ (100 μM, 5 min), ˙O2− (100 μM, 5 min), ˙OH (100 μM, 5 min) and NO (100 μM, 5 min) in pH 7.30 buffer solution; (b) selectivity test of probe 1–D-fructose complex (probe 1, 2 μM; D-fructose, 100 mM) in the presence of various ROS/RNS in pH 7.3 buffer solution. The spectra were collected with excitation at 410 nm at 25 °C (Ex slit: 5.0, Em slit: 5.0). | ||
We also tested the fluorescence reaction of the 1–D-fructose complex towards ONOO− and H2O2 in pH 5.0 buffer solution in order to mimic the acidic conditions found in cancer cells (Fig. S9†). Under the same concentrations of H2O2 (500 μM) and ONOO− (500 μM), the fluorescence ratio only slightly decreased to (F − F0)/F = ca. 0.09 with H2O2, while the value decreased significantly to (F − F0)/F = ca. 0.65 for ONOO−. Therefore, our fluorescent probe can be employed for the selective and sensitive detection of peroxynitrite under physiological and pathological conditions where −OCl is not present or is at very low concentration. In addition, previous research reported that endogenous amines and thiols readily react with hypochlorite,37 which makes it unlikely that hypochlorite will compete with peroxynitrite for the boronate-based fluorescence probes in the cellular system.
Intracellular imaging for exogenous and endogenous ONOO−
We then evaluated the ability of the probe 1–D-fructose complex to visualize exogenous and endogenous ONOO− using cell imaging experiments. The HeLa cells were incubated with probe 1 (5 μM) and D-fructose (250 mM) for 30 min at 37 °C, after which we added 0 μM, 5 μM and 30 μM peroxynitrite–PBS buffer solutions for 10 min at 37 °C, and after washing with DPBS, the cells were observed using confocal laser scanning microscopy. The probe 1–D-fructose complex can penetrate into the live cell membrane and displayed strong fluorescence throughout the cytoplasm region (Fig. 5A(b)). When external peroxynitrites were added to the cells, the fluorescence of the probe 1–D-fructose complex was quenched in a concentration-dependent manner (Fig. 5A(c) and A(d)).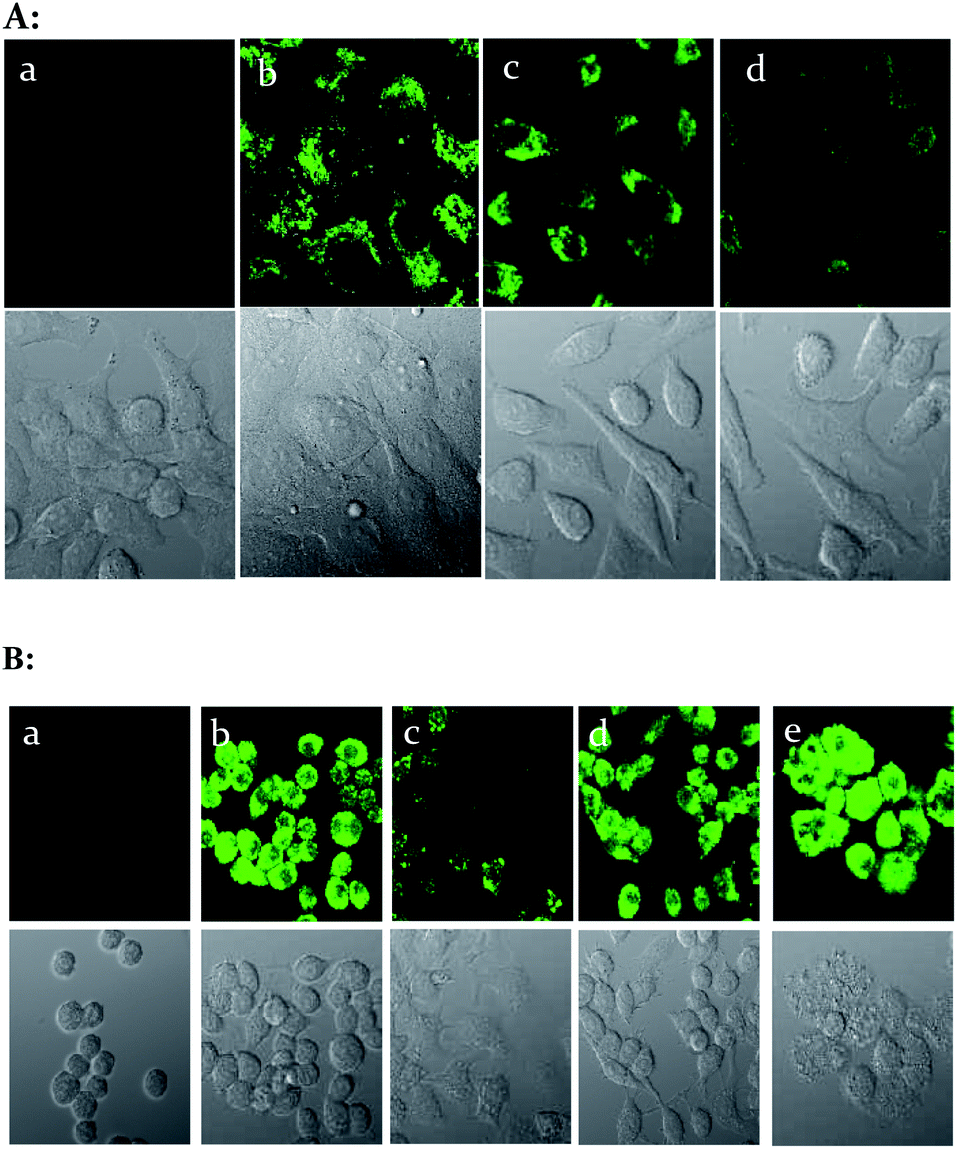 | ||
| Fig. 5 (A) Fluorescent imaging for exogenous ONOO− in HeLa cells. The probe 1–D-fructose was formed by mixing probe 1 (5 μM) and D-fructose (250 mM) in situ. (a) Blank without probe; (b) probe 1–D-fructose only; (c) probe 1–D-fructose and ONOO− (5 μM); (d) probe 1–D-fructose and ONOO− (30 μM). (B) Fluorescent imaging for endogenous ONOO− in RAW 264.7 cells. The probe 1–D-fructose was formed by mixing probe 1 (5 μM) and D-fructose (250 mM) in situ. (a) Blank without probe; (b) probe 1–D-fructose only; (c) probe 1–D-fructose and LPS, IFN-γ, PMA; (d) c + aminoguanidine; (e) c + TEMPO. | ||
Animal macrophage cell lines can produce peroxynitrite via immunogenic stimuli.41 Therefore, to detect peroxynitrite made naturally in the cell, we used RAW 264.7 cells (mouse macrophage). RAW 264.7 cells were stimulated with 1 μg ml−1 LPS (lipopolysaccharide, bacterial membrane component) for 16 h at 37 °C, 50 ng ml−1 IFN-γ for 4 h and 2.5 ng ml−1 PMA for 30 min, and then incubated with 5 μM sensor for 30 min. They showed weak fluorescence compared to control cells (Fig. 5B(b) and B(c)). For the inhibition test of peroxynitrite production, 100 μM TEMPO (superoxide scavenger) and 0.5 mM amino guanidine (nitric oxide synthase inhibitor) were pretreated with the media for 4 h and in this case strong fluorescence was observed (Fig. 5B(d) and B(e)). From these experiments, it is clear that the reaction of the sensor with peroxynitrite can be detected in live cells.
Conclusions
In conclusion, we have developed a new approach for the intracellular detection of ONOO− through the use of a boronic acid fluorescent probe in the presence of monosaccharide (e.g.D-fructose). The reaction between peroxynitrite and the boronic ester triggered a significant fluorescence on–off response which is selective over a variety of reactive oxygen and nitrogen species. Notably, from the perspective of a mechanistic explanation, the enhanced interaction between the amine and the boron provides a novel strategy to design new fluorescent probes for the detection of peroxynitrite which can distinguish ONOO− from H2O2 and other ROS/RNS. More importantly, the simple, sensitive fluorescent probe was successfully manipulated to visualize exogenous and endogenous ONOO− in living cells. We believe that our novel system will enable the investigation of diseases states (such as inflammation) in biological systems involving the production of peroxynitrite.2,42,43Methods
Experimental methods
In the corresponding experiments, in vitro fluorescence titrations with peroxynitrite and other ROS/RNS were carried out at 25 °C in pH 7.30 PBS buffer (KH2PO4, 1/15 M; Na2HPO4, 1/15 M) and pH 5.00 buffer (NaOAc–HOAc, 50 mM). The saccharide–boronic acid complexes were formed by mixing free boronic acid (2 μM) with D-fructose (100 mM) for 10 min in situ. The peroxynitrite alkaline stock was prepared following a literature procedure44 and assayed using a UV-Vis spectrophotometer via ε302 nm = 1670 cm−1 M−1. The preparation of other ROS and RNS solutions is described in the ESI† (General methods).Confocal microscopy imaging
Cells were seeded in 35 mm glass bottomed dishes at a density of 3 × 105 cells per dish in culture media. After 24 h, 5 μM sensor (final 0.25% DMF, N,N-dimethylformamide) and 250 mM D-fructose were added to the cells and the cells were incubated for 30 min at 37 °C. For HeLa cells, the 5 or 30 μM peroxynitrite–PBS solutions were added to the cells for 10 min at 37 °C. For RAW 264.7 cells, 1 μg ml−1 LPS (lipopolysaccharide) was added to the media for 16 h, 50 ng ml−1 IFN-γ (interferon-gamma) for 4 h and 2.5 ng ml−1 PMA (phorbol 12-myristate-13-acetate) for 30 min, and the cells were then incubated with 5 μM sensor for 30 min. For the inhibition test, 100 μM TEMPO and 0.5 mM amino guanidine were added to the media for 4 h. After washing with the DPBS twice to remove the residual probe, the cells were imaged by confocal laser scanning microscopy (FV1200, Olympus, Japan). Cells were excited by a 405 nm diode laser and detected at BA 490–590 nm.Acknowledgements
TDJ and XS are grateful for financial support from China Scholarship Council (CSC) and University of Bath Full Fees Scholarship. The Catalysis And Sensing for our Environment (CASE) network is thanked for research exchange opportunities. JSF, SDB and TDJ thank ECUST for guest professorships. JSF thanks the University of Birmingham for support. JY thanks the support from the National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIP) (no. 2012R1A3A2048814).Notes and references
- J. S. Beckman, T. W. Beckman, J. Chen, P. A. Marshall and B. A. Freeman, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 1620 CrossRef CAS.
- P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315 CrossRef CAS PubMed.
- K.-T. Lin, J.-Y. Xue, M. C. Lin, E. G. Spokas, F. F. Sun and P. Y.-K. Wong, Am. J. Physiol.: Cell Physiol., 1998, 274, C855 CAS.
- A. G. Estévez, R. Radi, L. Barbeito, J. T. Shin, J. A. Thompson and J. S. Beckman, J. Neurochem., 1995, 65, 1543 CrossRef.
- C. Szabo, H. Ischiropoulos and R. Radi, Nat. Rev. Drug Discovery, 2007, 6, 662 CrossRef CAS PubMed.
- H. Ischiropoulos and J. S. Beckman, J. Clin. Invest., 2003, 111, 163 CrossRef CAS.
- P. Sarchielli, F. Galli, A. Floridi, A. Floridi and V. Gallai, Amino Acids, 2003, 25, 427 CrossRef CAS PubMed.
- F. Torreilles, S. d. Salman-Tabcheh, M.-C. Guérin and J. Torreilles, Brain Res. Rev., 1999, 30, 153 CrossRef CAS.
- N. Ito, U. T. Ruegg, A. Kudo, Y. Miyagoe-Suzuki and S. i. Takeda, Nat. Med., 2013, 19, 101 CrossRef CAS PubMed.
- X. Chen, X. Tian, I. Shin and J. Yoon, Chem. Soc. Rev., 2011, 40, 4783 RSC.
- D. Yang, H.-L. Wang, Z.-N. Sun, N.-W. Chung and J.-G. Shen, J. Am. Chem. Soc., 2006, 128, 6004 CrossRef CAS PubMed.
- Z.-N. Sun, H.-L. Wang, F.-Q. Liu, Y. Chen, P. K. H. Tam and D. Yang, Org. Lett., 2009, 11, 1887 CrossRef CAS PubMed.
- T. Peng and D. Yang, Org. Lett., 2010, 12, 4932 CrossRef CAS PubMed.
- Q. Zhang, Z. Zhu, Y. Zheng, J. Cheng, N. Zhang, Y. T. Long, J. Zheng, X. Qian and Y. Yang, J. Am. Chem. Soc., 2012, 134, 18479 CrossRef CAS PubMed.
- Z. J. Chen, W. Ren, Q. E. Wright and H. W. Ai, J. Am. Chem. Soc., 2013, 135, 14940 CrossRef CAS PubMed.
- E. Galbraith and T. D. James, Chem. Soc. Rev., 2010, 39, 3831 RSC.
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1106 RSC.
- S. D. Bull, M. G. Davidson, J. M. van den Elsen, J. S. Fossey, A. T. Jenkins, Y. B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312 CrossRef CAS PubMed.
- X. Sun, S.-Y. Xu, S. E. Flower, J. S. Fossey, X. Qian and T. D. James, Chem. Commun., 2013, 49, 8311 RSC.
- M. C. Y. Chang, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2004, 126, 15392 CrossRef CAS PubMed.
- A. E. Albers, V. S. Okreglak and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 9640 CrossRef CAS PubMed.
- E. W. Miller, O. Tulyathan, E. Y. Isacoff and C. J. Chang, Nat. Chem. Biol., 2007, 3, 263 CrossRef CAS PubMed.
- D. Srikun, E. W. Miller, D. W. Domaille and C. J. Chang, J. Am. Chem. Soc., 2008, 130, 4596 CrossRef CAS PubMed.
- G. C. Van de Bittner, E. A. Dubikovskaya, C. R. Bertozzi and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21316 CrossRef CAS PubMed.
- D. Srikun, A. E. Albers, C. I. Nam, A. T. Iavarone and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 4455 CrossRef CAS PubMed.
- B. C. Dickinson, C. Huynh and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 5906 CrossRef CAS PubMed.
- E. W. Miller, A. E. Albers, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2005, 127, 16652 CrossRef CAS PubMed.
- G. C. Van de Bittner, C. R. Bertozzi and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 1783 CrossRef CAS PubMed.
- B. C. Dickinson, Y. Tang, Z. Chang and C. J. Chang, Chem. Biol., 2011, 18, 943 CrossRef CAS PubMed.
- A. R. Lippert, G. C. Van de Bittner and C. J. Chang, Acc. Chem. Res., 2011, 44, 793 CrossRef CAS PubMed.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601 RSC.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936 RSC.
- S. Banerjee, J. A. Kitchen, S. A. Bright, J. E. O'Brien, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Commun., 2013, 49, 8522 RSC.
- R. B. P. Elmes, M. Erby, S. A. Bright, D. C. Williams and T. Gunnlaugsson, Chem. Commun., 2012, 48, 2588 RSC.
- L. Zhu, S. H. Shabbir, M. Gray, V. M. Lynch, S. Sorey and E. V. Anslyn, J. Am. Chem. Soc., 2006, 128, 1222 CrossRef CAS PubMed.
- J. Zielonka, A. Sikora, M. Hardy, J. Joseph, B. P. Dranka and B. Kalyanaraman, Chem. Res. Toxicol., 2012, 25, 1793 CrossRef CAS PubMed.
- A. Sikora, J. Zielonka, M. Lopez, J. Joseph and B. Kalyanaraman, Free Radical Biol. Med., 2009, 47, 1401 CrossRef CAS PubMed.
- Q. Xu, K.-A. Lee, S. Lee, K. M. Lee, W.-J. Lee and J. Yoon, J. Am. Chem. Soc., 2013, 135, 9944 CrossRef CAS PubMed.
- S. Trupp, A. Schweitzer and G. J. Mohr, Org. Biomol. Chem., 2006, 4, 2965 CAS.
- T. D. James, K. R. A. S. Sandanayake, R. Iguchi and S. Shinkai, J. Am. Chem. Soc., 1995, 117, 8982 CrossRef CAS.
- M. Pekarova, L. Kubala, H. Martiskova, L. Bino, M. Twarogova, A. Klinke, T. K. Rudolph, Z. Kuchtova, H. Kolarova, G. Ambrozova, R. Kuchta, J. Kadlec and A. Lojek, Eur. J. Pharmacol., 2013, 713, 68 CrossRef CAS PubMed.
- S. Pfeiffer, A. Lass, K. Schmidt and B. Mayer, FASEB J., 2001, 15, 2355 CrossRef CAS PubMed.
- J. C. Sullivan and J. S. Pollock, Circ. Res., 2006, 98, 717 CrossRef CAS PubMed.
- J. W. Reed, H. H. Ho and W. L. Jolly, J. Am. Chem. Soc., 1974, 96, 1248 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc01417k |
| This journal is © The Royal Society of Chemistry 2014 |