
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chromo-pharmacophores: photochromic diarylmaleimide inhibitors for sirtuins†
C.
Falenczyk
a,
M.
Schiedel
b,
B.
Karaman
c,
T.
Rumpf
b,
N.
Kuzmanovic
a,
M.
Grøtli
d,
W.
Sippl
c,
M.
Jung
b and
B.
König
*a
aInstitute of Organic Chemistry, University of Regensburg, 93040 Regensburg, Germany. E-mail: burkhard.koenig@chemie.uni-regensburg.de
bInstitute of Pharmaceutical Sciences, University of Freiburg, 79104 Freiburg, Germany
cDepartment of Pharmaceutical Chemistry, Martin Luther University of Halle Wittenberg, 06120 Halle/Saale, Germany
dDepartment of Chemistry and Molecular Biology, University of Gothenburg, 41296 Göteborg, Sweden
First published on 11th August 2014
Abstract
Controlling the activity of sirtuins is of high biomedical relevance as the enzymes are involved in cancer, neurodegeneration and other diseases. Therefore structural elements of 3,4-bisindoylmaleimides (BIMs), which are known NAD+-dependent histone deacetylase (sirtuin) inhibitors, were merged with photochromic diarylmaleimides to yield photoswitchable enzyme inhibitors. The new inhibitors show excellent photophysical properties, are switchable even in polar solvents, and subtype selective against hSirt2. The inhibitory activity changes up to a factor of 22 for the two photoisomers and physiological properties can therefore be effectively toggled by irradiation with light of different wavelengths. Docking experiments using the enzyme crystal structure explain the observed activity changes based on the steric demand of the thiophene substitution and the rigidity of the molecular structure.
Introduction
Photochromism has recently received increasing attention due to a variety of potential applications in molecular optoelectronics, optical data storage and as molecular switches.1–3 The interest in photopharmacology is revealed by the increasing number of reported photochromic enzyme inhibitors,4–6 ion channel blockers7 and receptor ligands8,9 demonstrating the importance of such compounds as tools for the investigation of cellular processes and pathogeneses in life sciences.10 Two strategies are applied for controlling enzyme activity by light: the photochromic moiety can either be attached to the scaffold of a known substrate or is the linker between two ligands. The latter approach allows the light-induced change of the ligand spacing, which is useful for targets with two binding sites.2 Various types of photochromic compounds have been developed so far, for instance spiropyranes, azobenzenes, fulgides and diarylethenes (DTEs).2 Diarylmaleimide derivatives are well known for their photochromic properties and their ability to change conformational mobility reversibly upon irradiation (Fig. 1). Both photoisomers are thermally stable and show typically a high switching fatigue resistance. In comparison to other DTEs, the absorption of diarylmaleimides is shifted bathochromically and therefore the isomerization is possible at longer wavelengths, which reduces cell damage, otherwise caused by high energy light. The optical properties and the slightly better water solubility makes them interesting candidates for photochromic inhibitors.1 However, to be useful in biological applications, photochromic compounds should also undergo photochromic reactions in polar solvents. One handicap of diarylmaleimides is that these reactions are disfavoured in polar solvents due to a twisted intramolecular electron charge transfer (TICT) from the donor aryl part to the electron acceptor maleimide moiety.11–13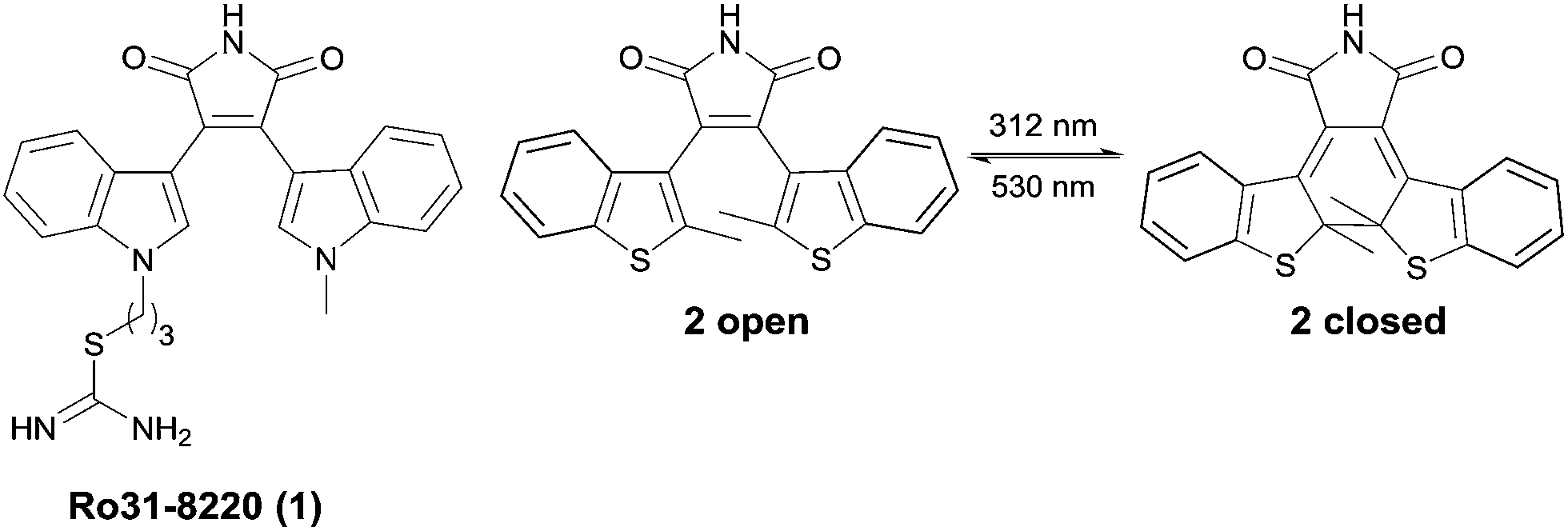 | ||
| Fig. 1 Sirtuin inhibitor 1 (Ro31-8220) and by way of example the structural related photochromic diarylmaleimide 2, which interconvert reversible by irradiation with light of different wavelength between the open and closed photoisomer. | ||
There is an ongoing interest in the regulation of enzyme activity in the field of epigenetics, particularly with regard to anticancer research. NAD+-dependent histone deacetylases also termed sirtuins are enzymes, which deacetylate histones and certain non-histone proteins. Sirtuins (Sirt1–7) represent the class III of four histone deacetylases (HDACs). Their activity is dependent on nicotinamide adenine dinucleotide (NAD+), whereas the other classes are zinc-dependent amidohydrolases.14–16 The biological activity of sirtuins has been linked to the pathogenesis of cancer17 as well as viral,18 metabolic19,20 and neurological diseases.21,22 As several disorders are linked to the perturbation of HDACs, they are a promising target for drug discovery. Various class I and II HDACs anticancer drugs are already in clinical trials or approved as therapeutics.23 To get more insight into the biological activity and molecular mechanism of all class III HDAC members there is a need to investigate potent and isotype selective small molecule modulators for human sirtuins. Several lead structures for small-molecule sirtuin inhibitors have been identified so far.16 Besides nicotinamide as the endogenous inhibitor, splitomicins, suramins, indoles as well as kinase inhibitors like bisindoylmaleimides (BIMs) are of particular interest.14–16 Diarylmaleimides are stable photochromic compounds and structurally related to BIMs like 1 (Ro31-8220), a potent sirtuin inhibitor (Fig. 1).14 We envisioned that merging the structure of the DTE chromophore and the BIM pharmacophore to new diarylmaleimides would lead to photochromic sirtuin inhibitors. Variation of the substitution pattern and molecular docking experiments provides insight into the binding mode of the diarylmaleimides and their photoisomers to the active site of the enzyme and thereby lead to potent and isotype selective inhibitors for sirtuins.
Results and discussion
Synthesis
A common strategy for the synthesis of 3,4-diarylmaleimides is the intramolecular Perkin-type condensation.24,25 The synthesis of compound 2 (Fig. 2) was previously described by Scandola et al.3 Adapted from this synthesis, the non-symmetric maleimide 3 was prepared in a similar manner with further functionalization by Suzuki coupling (see ESI†).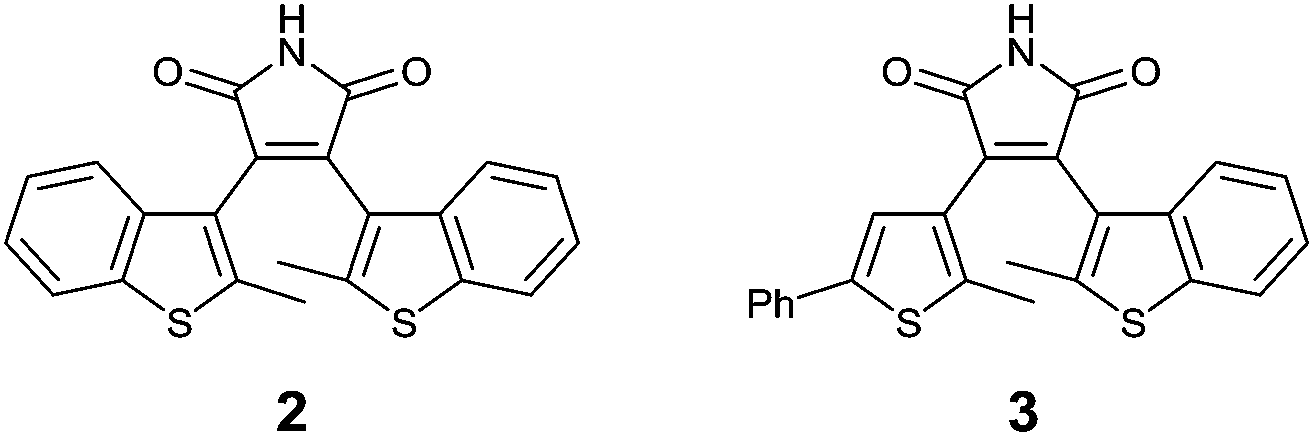 | ||
| Fig. 2 Structures of diarylmaleimides 2 and 3. | ||
Unfortunately, compound 11 could not be obtained by this strategy. Hence, we developed an alternative route for the synthesis of 11, which is outlined in Scheme 1. Starting from 5-chloro-2-methyl-thiophene (4) standard Friedel–Crafts conditions yielded ketone 5 and glyoxylate ester 8. The ester 6 was obtained via an oxidative rearrangement from ketone 5. Suzuki coupling yielded the functionalized esters 7 and 9. Compound 10 was prepared by aminolysis of glyoxylate ester 9, which was subsequently used in a Perkin condensation with ester 7 to obtain maleimide 11. Scheme 1 summarizes the synthesis of the symmetric diarylmaleimide 11.
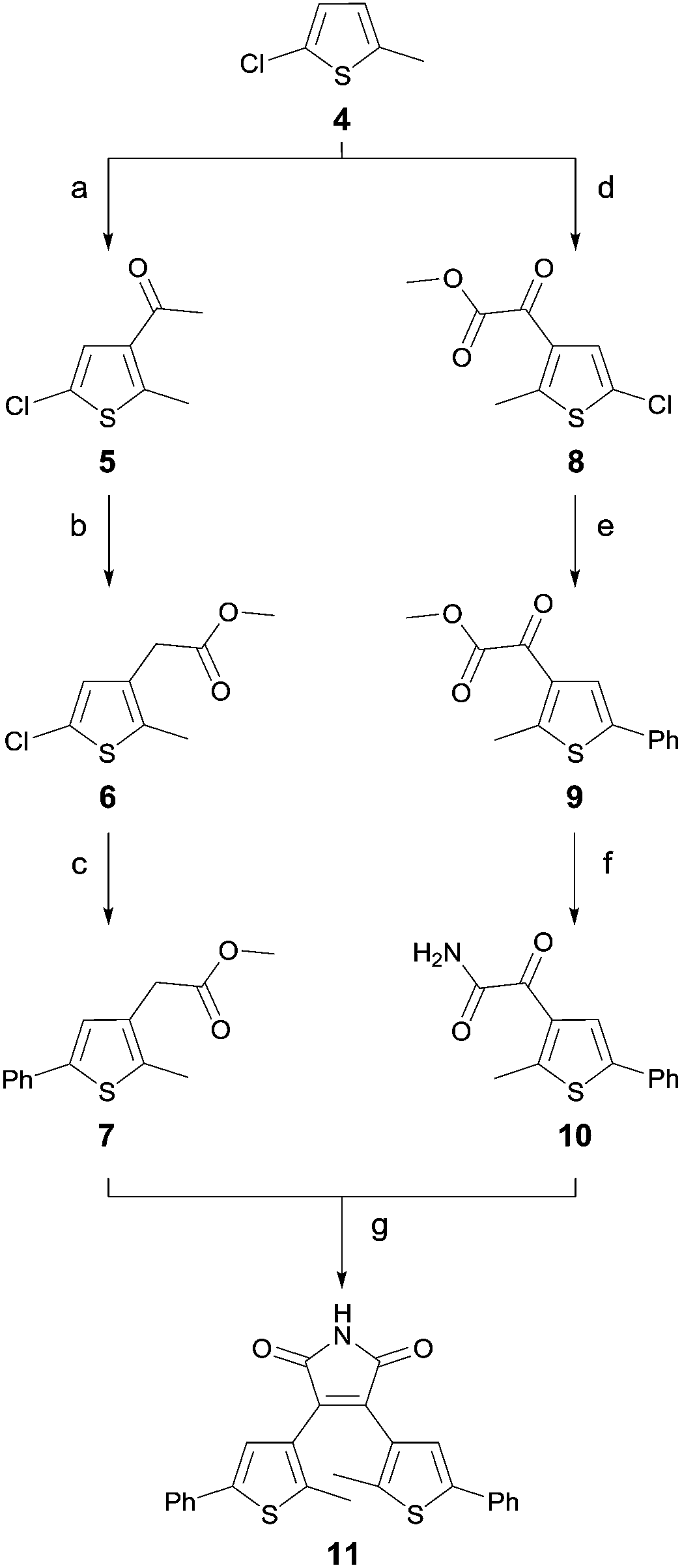 | ||
| Scheme 1 Synthetic strategy with reagents and conditions for 11: (a) acetyl chloride, AlCl3, CH2Cl2, 0 °C, 2 h, (84%); (b) TTN, HClO4, MeOH, RT, overnight, (83%); (c) Pd2(dba)3, XPhos, phenylboronic acid, K3PO4, 1,4-dioxane, 100 °C, overnight, (52%); (d) methyl chlorooxoacetate, AlCl3, CH2Cl2, 0 °C, 2 h, (73%); (e) Pd2(dba)3, XPhos, phenylboronic acid, K3PO4, 1,4-dioxane, 100 °C, overnight, (63%); (f) aq. NH3, THF, RT, overnight, (80%); and (g) KOtBu (1 M in THF), THF, 0 °C → RT, overnight, (17%). | ||
Photochromic properties
The photoisomerization of photochromic compounds in polar solvents is essential for applications in biological assays. As dimethyl sulfoxide is a standard solvent for inhibitor screening, this solvent was used to investigate the switching ability of the diarylmaleimides. The ring closure is suppressed in polar solvents due to a twisted intramolecular electron transfer. This phenomenon is well-investigated for maleimides bearing two benzothiophenes.12 To enhance the switching ability of DTEs the introduction of sterical hindrance either by bulky substituents on the aryl moieties or on the bridge is a widely applied concept.26–28 As seen from the perspective of the pharmacophore only the introduction of sterically demanding substituents on the aryl moieties are possible. Otherwise the ability to form relevant hydrogen bonds between the maleimide and the backbone of the enzyme is lost. We decided to replace the benzothiophene moieties by phenyl substituted thiophenes. Despite this change the structure of the chromophore is still similar to the pharmacophore, but the photochromic properties improve significant. The exchange of the methyl-benzothiophenes in 2 by one or two methyl-phenyl substituted thiophenes in 3 or 11, resp., stabilizes the antiparallel conformation, which favors the photochemical ring closure reaction.The photoisomerization was monitored by absorption spectroscopy (Fig. 3) and the photostationary states (PSS) were determined by HPLC measurements (see ESI†).
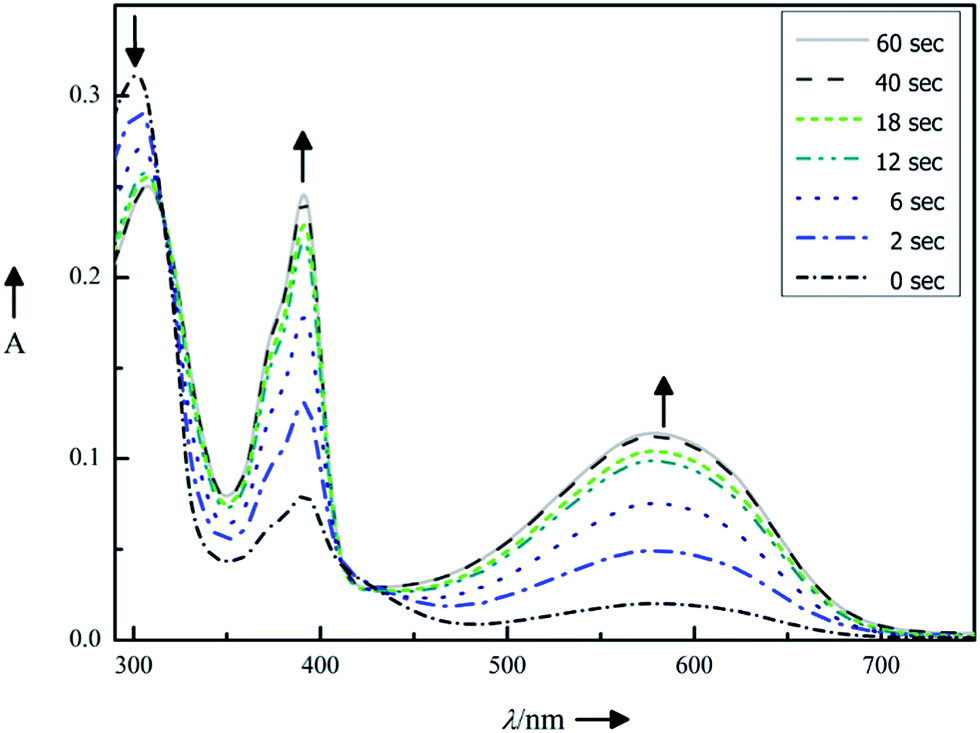 | ||
| Fig. 3 Photochromic properties of compound 11 (10 μm in DMSO): absorption spectra changes upon continuous irradiation for 0, 2, 6, 12, 18, 40 and 60 s with light of λmax = 312 nm. | ||
Upon irradiating (λ = 312 nm) the dissolved compounds 2, 3 and 11, a rapid color change was observed, resulting in new absorption bands characteristic for each compound. The photochromic properties are summarized in Table 1. Replacing the benzothiophene moieties by phenyl-substituted thiophenes has two consequences: (1) the absorbance maxima for the closed isomers shift bathochromically from compound 2 to compound 11. Therefore light of longer wavelength is sufficient for the isomerization. (2) The photoconversion becomes more efficient as indicated by PSS increasing from 62% to 94%. Such high PSS is advantageous for binding investigations as the interaction with the enzyme can be restricted to mainly one photoisomer.
By way of example, Fig. 3 illustrates the photoisomerization of compound 11. Upon irradiating (λ = 312 nm) of compound 11 in dimethyl sulfoxide solution, a rapid color change from light green to deep purple was observed, resulting in new absorption bands at 390 nm and 580 nm (marked with black arrows in Fig. 3). The isosbestic points (Table 1) indicate a clean two-component switching. Repetition of the photoisomerization of compound 11 shows a good fatigue resistance over five cycles (see ESI†).
Enzyme inhibition
Next, the inhibitory effect of the photoresponsive compounds 2, 3 and 11 against sirtuins in a homogenous fluorescent assay, previously established by Jung et al. was determined. The enzymatic conversion was quantified using (S)-[5-acetylamino-1-(4-methyl-2-oxo-2H-chromen-7-ylcarbamoyl)-pentyl]-carbamic acid benzyl ester also termed ZMAL29 as fluorescent substrate for deacetylation followed by a tryptic digestion step.301 (Ro31-8220) has an IC50 value of 0.8 μm and is the most potent BIM inhibitor reported so far for human Sirt2 (hSirt2).14,16 In comparison to 1 the photoresponsive diarylmaleimides 3 and 11 showed similar inhibition strength against hSirt2. The respective IC50 values are given in Table 2.| Entry | Compound | hSirt1 IC50 [μM] | hSirt2 IC50 [μM] | hSirt3 IC50 [μM] | hSirt2 IC50 closed/IC50 open |
|---|---|---|---|---|---|
| a n.i.: no inhibition [<10% @ 20 μm]. b —: not tested. | |||||
| 1 | 1 (ref. 14 and 15) | 3.5 ± 0.4 | 0.8 ± 0.2 | 3.7 ± 0.2 | |
| 2 | 11 open | n.i.a | 4.2 ± 0.8 | n.i. | 22 |
| 3 | 11 closed | n.i. | 92.3 ± 8.0 | n.i. | |
| 4 | 3 open | —b | 2.3 ± 0.4 | — | 1 |
| 5 | 3 closed | — | 2.1 ± 0.3 | — | |
| 6 | 2 open | — | 12.9 ± 5.4 | — | 2 |
| 7 | 2 closed | — | 23.1 ± 5.6 | — | |
The scaffold of compound 2 is structurally closest related to 1. Its open isomer exhibits a lower IC50 value against hSirt2 (Table 2, entry 6) than its closed photoisomer. By the exchange of one of the benzothiophenes in 2 by a phenyl substituted thiophene as in compound 3 the inhibition activity increases. However, the open and the closed photoisomer of compound 3 show the same inhibition (Table 2, entries 4 and 5). The replacement of the second benzothiophene from the maleimide moiety by a phenyl substituted thiophene in compound 11 yields an inhibition corresponding to an IC50 value of 4.2 μm of the open photoisomer. The ring closed photoisomer of 11 shows a twentyfold lower enzyme inhibition with 92.3 μm (Table 2, entries 2 and 3). Diarylmaleimide 11 gives the most significant change in hSirt2 inhibition activity for both photoisomers and therefore its isotype selectivity against human Sirt1 (hSirt1) and human Sirt3 (hSirt3) under the same assay conditions30 as used for hSirt2 were determined. The isotype selectivity for BIM 1 (Ro31-8220) is only small (Table 2, entry 1). Therefore inhibitor concentrations at 20 μm and 200 μm were used to determine the enzyme activity using nicotinamide as a reference. Neither the open isomer nor the closed isomer of 11 inhibited hSirt1 or hSirt3 significantly (Table 2, entries 2 and 3).
Next, the enzyme activity of hSirt2 was controlled by light under assay conditions. Therefore, the photochemical conversion from the closed isomer to the open isomer of 11 and the corresponding inhibitory activity were measured as a function of time (Fig. 4). Hence the less potent closed isomer of 11 was incubated with the enzyme under assay conditions and irradiated over a period of 30 min with a green LED (2.5 W, 530 nm emission maximum). Then the cofactor NAD+ and ZMAL were added and the assay was performed under the usual conditions. Fig. 4 illustrates the time course of the isomerization (dotted line) and inhibitory activity (straight line) of 11. The irradiated samples switched from the less potent closed form of 11 to the significantly more potent open form.
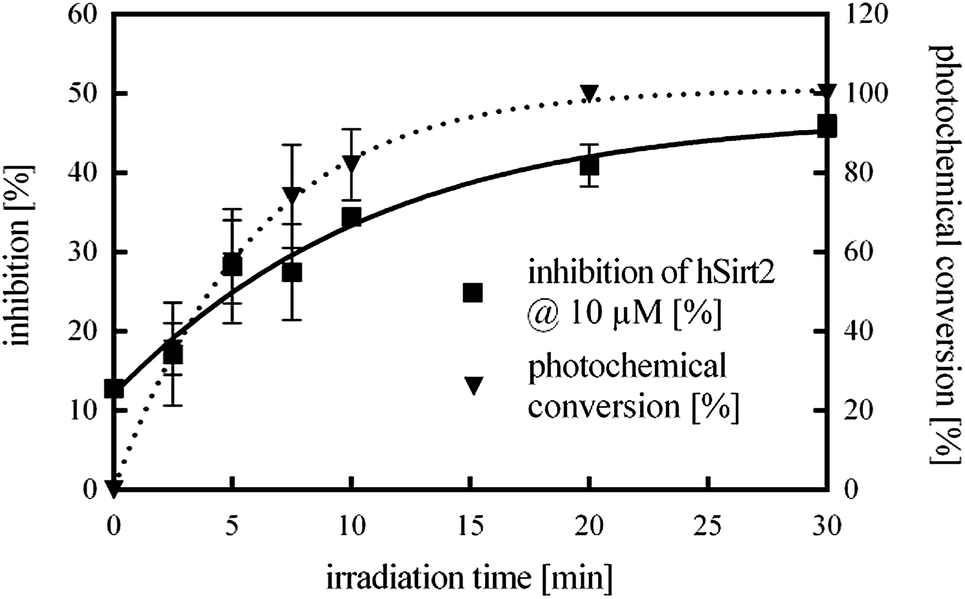 | ||
| Fig. 4 Time course of the photochemical conversion from the closed to the open form of 11 (dotted line) and the corresponding inhibitory activity against hSirt2 (straight line) upon continuous irradiation with light of λmax = 530 nm for 0, 2.5, 5, 7.5, 10, 20 and 30 min at an assay concentration of 10 μM. | ||
After 20 min complete photochemical conversion of 11 and a constant inhibition value are observed. The determined values correspond to the inhibition activity of the open isomer of 11 (Table 2) proving that the photoisomerization is possible in the presence of the enzyme under assay conditions.
Additionally, the open photoisomer of 11 was tested for cellular activity using hyperacetylation of tubulin31 as a marker. 11 showed tubulin hyperacetylation and hence target engagement similar to the control inhibitor 1 (Ro31-8220)14 (see ESI†). But further studies are required as we cannot rule out indirect effects as the compounds have not been optimized for selectivity, e.g. against kinases.
Molecular docking
In order to rationalize the observed in vitro activities docking of the open and closed form of the diarylmaleimides to hSirt2 (PDB-ID 3ZGO) was carried out. Docking of the open form of the isomers 2, 3 and 11 to hSirt2 resulted in a similar interaction at the acetyl-lysine channel of hSirt2 (see ESI†) favoured by a hydrogen bond with the backbone of Val233 and van-der-Waals interactions with hydrophobic and aromatic residues (Phe96, Phe119, Ile169, His187, Phe234, and Phe235). Val233 represents a conserved amino acid in sirtuins that binds the amide group of the acetyl-lysine substrate. The open photoisomers fit nicely to the acetyl-lysine pocket of hSirt2. The angle between the aromatic rings and the maleimide is for all open photoisomers around −50 deg., which allows the accommodation of both aromatic substituents in the acetyl-lysine pocket. The closed photoisomer of 11 might be too big (distance from both ends of the phenyl ring 14.7 Å compared to 12.9 Å for 3 and 10.9 Å for 2) and rigid to interact in a similar way within the binding pocket as the open and flexible isomer of 11 (Fig. 5). The docking results suggest that the terminal aromatic rings and methyl groups clash with His187 and Phe235 resulting in a different orientation at the binding pocket compared to the closed isomers of 2 and 3 (Fig. 6). As a consequence, the distance between the maleimide and Val233 backbone carbonyl group is increased (3.85 Å) disabling the hydrogen bond formation (see ESI†). Only the closed photoisomers of 2 and 3 fit well to the binding pocket and make favourable van-der-Waals interactions with the aromatic and hydrophobic residues (see ESI†).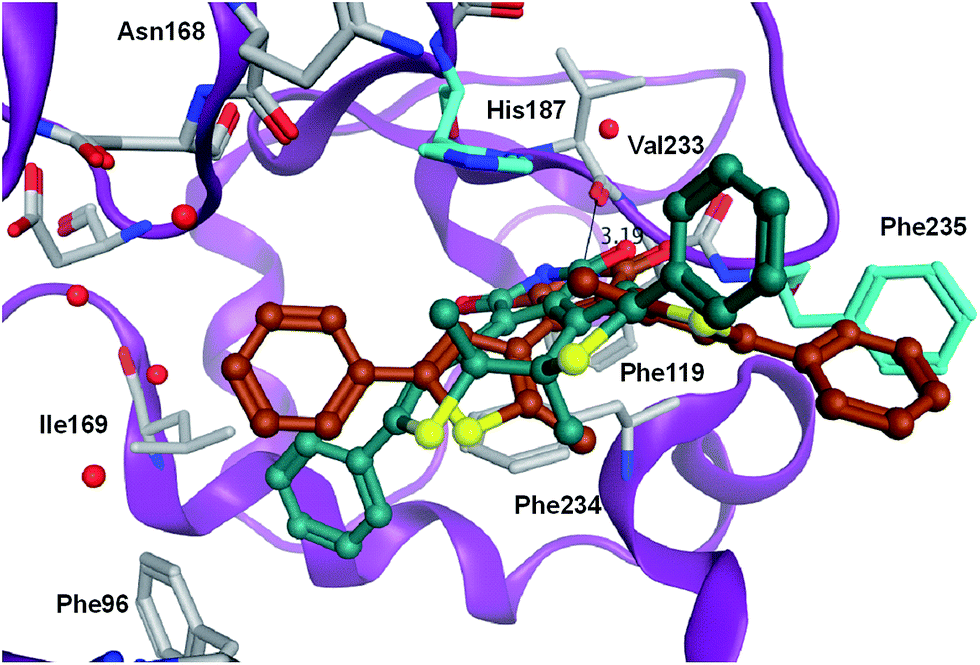 | ||
| Fig. 5 Comparison of the binding mode of photoisomer 11 at hSirt2 in the open (brown) and the closed form (dark green). The two residues (Phe235 and His187) that hinder the proper orientation of the closed photoisomer 11 are colored cyan. Water molecules are shown as red spheres. The hydrogen bond to the backbone of Val233 is shown as black line (distance between the NH group of closed 11 and the backbone carbonyl of Val233 is 3.19 Å). | ||
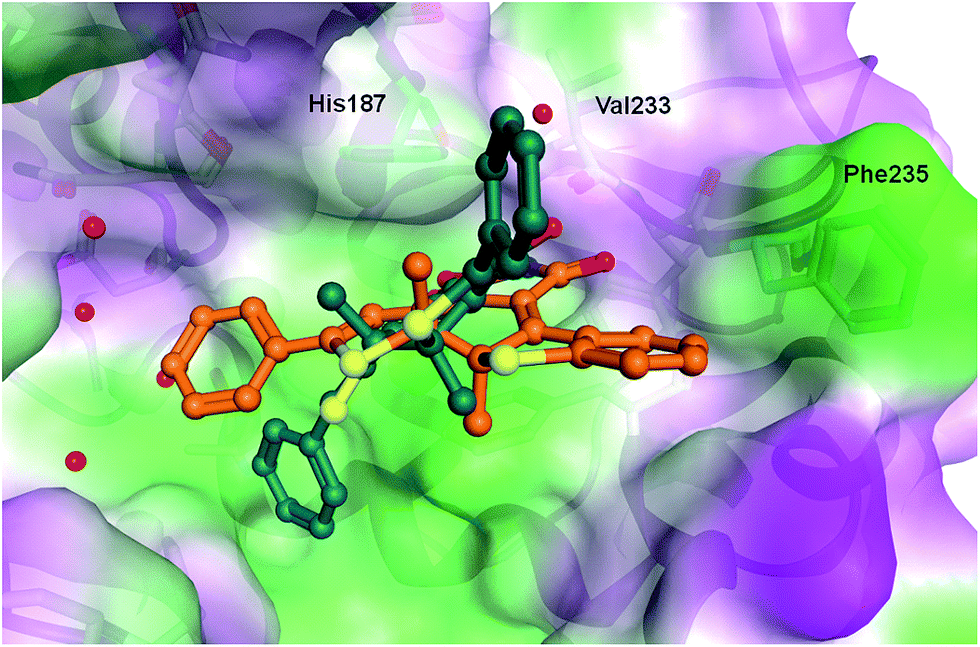 | ||
| Fig. 6 Comparison of the binding mode of closed photoisomer 3 (orange) and closed photoisomer 11 (dark green) at hSirt2. Water molecules are shown as red spheres. | ||
Docking to the apo Sirt3 (PDB-ID 3GLS) substrate binding pocket was not possible due to a different conformation of the flexible loop flanking the acetyl-lysine channel. Especially, the residues Phe157, Arg158 and Phe294 block part of the substrate binding pocket observed in hSirt2 (see ESI†). Thus, the docking suggests that the observed selectivity of the photochromic inhibitor 11 arises from a different orientation of amino acid residues located at the entrance of the acetyl-lysine channel of hSirt3.
In case of apo hSirt1 (PDB-ID 4IG9), which shows also an open conformation of the substrate binding site, the docking of the photoisomers of 11 resulted in less favourable complexes. Due to the orientation of Phe414 the pocket shows a different shape compared to hSirt2 which hinders the formation of a hydrogen bond between Val233 and the maleimide group of the docked inhibitors (see ESI†). Compound 11 is therefore isotype selective for hSirt2 and although 11 shows no higher potency than 1, it is the first example for a potent photochromic maleimide sirtuin inhibitor with such selectivity and the first example for a photochromic inhibitor combining elements of a chromophore and a pharmacophore within one structure.
Conclusions
In summary, we have merged structural elements of the well investigated photochromic diarylmaleimides with biological relevant bisindoylmaleimides, which are known for their sirtuin inhibitory activity, yielding potent and isotype selective photochromic inhibitors. The new compounds are the first photochromic epigenetic inhibitors described so far. The photochromic and enzyme inhibition properties of the new diarylmaleimides were optimized by variation of the maleimide scaffold. Exchange of the benzothiophene moieties by phenyl substituted thiophenes led to nearly quantitative photostationary states of the diarylmaleimides in dimethyl sulfoxide and subtype selective inhibition of hSirt2 in the low micromolar range. The photoisomers of diarylmaleimide 11 differ more than 20-fold in their inhibition ability. Irradiation of the closed photoisomer of 11 in the presence of the enzyme isomerizes the compound into the more potent open photoisomer allowing the remote control of the enzyme activity by green light. The inhibitory activity of the open photoisomer of 11 was also tested in cell culture. 11 induced hyperacetylation of tubulin, but further studies are necessary to eliminate indirect effects.The new diarylmaleimides combine in their molecular structure photochromic and selective enzyme inhibiting properties. They are valuable tools for the investigation of molecular binding mechanisms of sirtuins and the application of the concept to other privileged structures of the bisindoylmaleimide family may be readily envisaged.
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG) within the GRK 1910 and within the CRC992 (MEDEP, Medical Epigenetics) is gratefully acknowledged. We thank Karin Schmidtkunz for western blot experiments.Notes and references
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef PubMed.
- M. T. Indelli, S. Carli, M. Ghirotti, C. Chiorboli, M. Ravaglia, M. Garavelli and F. Scandola, J. Am. Chem. Soc., 2008, 130, 7286–7299 CrossRef CAS PubMed.
- D. Vomasta, A. Innocenti, B. König and C. T. Supuran, Bioorg. Med. Chem. Lett., 2009, 19, 1283–1286 CrossRef CAS PubMed.
- D. Vomasta, C. Högner, N. R. Branda and B. König, Angew. Chem., Int. Ed., 2008, 47, 7644–7647 CrossRef CAS PubMed.
- B. Reisinger, N. Kuzmanovic, P. Löffler, R. Merkl, B. König and R. Sterner, Angew. Chem., Int. Ed., 2014, 53, 595–598 CrossRef CAS PubMed.
- M. R. Banghart, A. Mourot, D. L. Fortin, J. Z. Yao, R. H. Kramer and D. Trauner, Angew. Chem., Int. Ed., 2009, 48, 9097–9101 CrossRef CAS PubMed.
- M. Volgraf, P. Gorostiza, S. Szobota, M. R. Helix, E. Y. Isacoff and D. Trauner, J. Am. Chem. Soc., 2006, 129, 260–261 CrossRef PubMed.
- M. Schönberger and D. Trauner, Angew. Chem. Int. Ed., 2014, 53, 3264–3267 CrossRef PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- T. Yamaguchi and M. Irie, Chem. Lett., 2004, 33, 1398–1399 CrossRef CAS.
- T. Yamaguchi, K. Uchida and M. Irie, J. Am. Chem. Soc., 1997, 119, 6066–6071 CrossRef CAS.
- M. Irie and K. Sayo, J. Phys. Chem., 1992, 96, 7671–7674 CrossRef CAS.
- J. Trapp, A. Jochum, R. Meier, L. Saunders, B. Marshall, C. Kunick, E. Verdin, P. Goekjian, W. Sippl and M. Jung, J. Med. Chem., 2006, 49, 7307–7316 CrossRef CAS PubMed.
- M. Freitag, J. Schemies, T. Larsen, K. El Gaghlab, F. Schulz, T. Rumpf, M. Jung and A. Link, Bioorg. Med. Chem., 2011, 19, 3669–3677 CrossRef CAS PubMed.
- M. Lawson, U. Uciechowska, J. Schemies, T. Rumpf, M. Jung and W. Sippl, Biochim. Biophys. Acta, Gene Regul. Mech., 2010, 1799, 726–739 CrossRef CAS PubMed.
- B. Peck, C.-Y. Chen, K.-K. Ho, P. Di Fruscia, S. S. Myatt, R. C. Coombes, M. J. Fuchter, C.-D. Hsiao and E. W.-F. Lam, Mol. Cancer Ther., 2010, 9, 844–855 CrossRef CAS PubMed.
- M. R. Pinzone, B. Cacopardo, F. Condorelli, M. Di Rosa and G. Nunnari, Curr. Drug Targets, 2013, 14, 648–652 CrossRef CAS.
- G. Leonard, Nature, 2006, 444, 868–874 CrossRef PubMed.
- H. H. Riekelt, P. Eija and A. Johan, Nat. Rev. Mol. Cell Biol., 2012, 13, 225–238 Search PubMed.
- T. F. Outeiro, E. Kontopoulos, S. M. Altmann, I. Kufareva, K. E. Strathearn, A. M. Amore, C. B. Volk, M. M. Maxwell, J.-C. Rochet, P. J. McLean, A. B. Young, R. Abagyan, M. B. Feany, B. T. Hyman and A. G. Kazantsev, Science, 2007, 317, 516–519 CrossRef CAS PubMed.
- G. Jun, W. Wen-Yuan, M. Ying-Wei, G. Johannes, G. Ji-Song, P. Ling, M. Gloria, K. Dohoon, C. S. Susan and T. Li-Huei, Nature, 2010, 466, 1105–1109 CrossRef PubMed.
- M. Slingerland, H.-J. Guchelaar and H. Gelderblom, Anti-Cancer Drugs, 2014, 25, 140–149 CrossRef CAS PubMed.
- M. M. Faul, L. L. Winneroski and C. A. Krumrich, J. Org. Chem., 1998, 63, 6053–6058 CrossRef CAS.
- M. M. Faul, L. L. Winneroski and C. A. Krumrich, Tetrahedron Lett., 1999, 40, 1109–1112 CrossRef CAS.
- R. Göstl, B. Kobin, L. Grubert, M. Pätzel and S. Hecht, Chem. –Eur. J., 2012, 18, 14282–14285 CrossRef PubMed.
- W. Li, C. Jiao, X. Li, Y. Xie, K. Nakatani, H. Tian and W. Zhu, Angew. Chem., Int. Ed., 2014, 53, 4603–4607 CrossRef CAS PubMed.
- C. Elsner, T. Cordes, P. Dietrich, M. Zastrow, T. T. Herzog, K. Rück-Braun and W. Zinth, J. Phys. Chem. A, 2009, 113, 1033–1039 CrossRef CAS PubMed.
- B. Heltweg, F. Dequiedt, E. Verdin and M. Jung, Anal. Biochem., 2003, 319, 42–48 CrossRef CAS.
- B. Heltweg, J. Trapp and M. Jung, Methods, 2005, 36, 332–337 CrossRef CAS PubMed.
- B. J. North, B. L. Marshall, M. T. Borra, J. M. Denu and E. Verdin, Mol. Cell, 2003, 11, 437–444 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures of the synthesis of 3 and 11, additional spectral data, photochemical measurements, biological data and docking studies of 2, 3 and 11. See DOI: 10.1039/c4sc01346h |
| This journal is © The Royal Society of Chemistry 2014 |