
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In vivo anti-tumor activity of the organometallic ruthenium(II)-arene complex [Ru(η6-p-cymene)Cl2(pta)] (RAPTA-C) in human ovarian and colorectal carcinomas†
Andrea
Weiss
ab,
Robert H.
Berndsen
b,
Maxime
Dubois
c,
Cristina
Müller
c,
Roger
Schibli
cd,
Arjan W.
Griffioen
b,
Paul J.
Dyson
*a and
Patrycja
Nowak-Sliwinska
*a
aInstitute of Chemical Sciences and Engineering, Swiss Federal Institute of Technology (EPFL), Lausanne, Switzerland. E-mail: Patrycja.Nowak-Sliwinska@epfl.ch; Paul.Dyson@epfl.ch; Fax: +41 61 693 5110; Fax: +41 61 693 9780; Tel: +41 21 693 5169 Tel: +41 21 693 9853
bAngiogenesis Laboratory, Department of Medical Oncology, VU University Medical Center, Amsterdam, The Netherlands
cCenter for Radiopharmaceutical Sciences ETH-PSI-USZ, Villigen-PSI, Switzerland
dDepartment of Chemistry and Applied Biosciences, ETH Zurich, Zurich, Switzerland
First published on 5th August 2014
Abstract
Based on the clinical success of platinum-based anti-cancer drugs such as cisplatin, carboplatin and oxaliplatin, a variety of other metal-based anti-cancer compounds are being investigated. In particular, a number of ruthenium-based compounds have been identified which exhibit unique biochemical properties and reduced toxicity profiles compared to the clinically used platinum-based drugs. We have developed a series of organometallic ruthenium(II)-arene complexes that were shown to exert anti-metastatic activity with relatively minor activity on primary tumor growth. Here, we show that the prototype compound, [Ru(η6-p-cymene)Cl2(pta)], where pta = 1,3,5-triaza-7-phosphaadamantane (RAPTA-C), reduces the growth of primary tumors in preclinical models for ovarian and colorectal carcinomas. When administered daily at relatively low doses (0.2 mg kg−1), RAPTA-C was shown to significantly reduce the growth of the A2780 ovarian carcinoma transplanted onto the chicken chorioallantoic membrane model. Similar activity was observed in LS174T colorectal carcinoma in athymic mice, albeit at a higher dose. In both models, a clear inhibition of microvessel density was observed, confirming the previously discovered anti-angiogenic mechanism of RAPTA-C. Biodistribution studies with radiolabeled (103Ru) RAPTA-C indicate that the compound is rapidly cleared from the organs and the bloodstream through excretion by the kidneys. As such, RAPTA-C is a promising compound for translation to clinical evaluation.
Introduction
Cisplatin, carboplatin and oxaliplatin are widely used in the clinical treatment of a broad spectrum of cancers.1 Despite the success of these platinum-based drugs, their use is limited due to significant toxicity and both intrinsic and acquired drug resistance.2 In light of these limitations, a large focus in current research has been directed towards the development of compounds that are based on metals other than platinum.2,3 In recent years, the prominence of ruthenium-based complexes has significantly grown as they have been shown to possess anti-metastatic properties and overcome both of these main limitations of platinum-based drugs in a number of animal models.4,5 Two ruthenium(III)-based compounds (Chart 1), namely KP1019 (indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)]) and NAMI-A (imidazolium trans-[tetrachloro(dimethylsulfoxide)(1H-imidazole)ruthenate(III)]), have undergone phase I clinical evaluation. | ||
| Chart 1 Structures of KP1019 (A) and NAMI-A (B). | ||
Ruthenium(III) complexes, however, are prone to ligand exchange reactions in aqueous media/physiological buffer which hamper, to some extent, the rational design of such new compounds with relevant medicinal properties. For this reason, ruthenium(II)-arene compounds have attracted considerable attention in recent years following encouraging in vivo data on two prototypical compounds (Chart 2), [Ru(η6-p-cymene)Cl(en)], where en = ethylenediamine (termed RAED-C)6 and [Ru(η6-p-cymene)Cl2(pta)], where pta = 1,3,5-triaza-7-phosphaadamantane (termed RAPTA-C).7 RAED-C was shown to have a moderate effect on reducing the growth of primary tumors whereas RAPTA-C has shown moderate effects on solid tumor metastases. Recently, these differences have been tentatively attributed to differences in the preferential binding site of each molecule to chromatin, with RAED-C binding to DNA sites and RAPTA-C to the histone core.8
 | ||
| Chart 2 Structures of RAED-C (A) and RAPTA-C (B). | ||
The mechanisms by which ruthenium-based drugs exert their anticancer effects remain to be fully elucidated although, as mentioned above, chromatin binding appears to be an important mechanism. It has been proposed for ruthenium(III) complexes that their reduced toxicity to normal tissue may be attributed to the so-called “activation by reduction” mechanism, i.e. ruthenium(III) complexes act as prodrugs that can be reduced to active ruthenium(II) species in the hypoxic microenvironment of a tumor. Similar to KP1019 and NAMI-A, which are both based on a ruthenium(III) ion, the ruthenium(II) complex RAPTA-C also appears to be well tolerated in vivo showing considerably reduced side-effects compared to platinum-based drugs (as judged by the high doses that may be tolerated by animals).
RAPTA-C was recently shown to also exert a strong anti-angiogenic effect,9 similar to that of sorafenib, a clinically used anti-angiogenic small molecule drug.10 Moreover, there are similarities shared by RAPTA-C and NAMI-A, both of which show limited direct cytotoxic effects on cancer cells in vitro while exhibiting anti-metastatic behaviour in vivo. NAMI-A also has an anti-angiogenic component to its mechanism as demonstrated by its ability to induce apoptosis in a spontaneously transformed human endothelial cell line (ECV304) through the inhibition of MEK/ERK signalling.11
RAPTA-C presents particular interest when viewed as an anti-angiogenic agent, as it has been shown to reduce the growth of lung metastases in CBA mice bearing the MCa breast carcinoma,12 while many clinically used VEGF-targeted therapies have been shown to induce pro-metastatic phenotypes in treated tumors, such as the VEGF targeted tyrosine kinase inhibitor sunitinib.13 Consequently, RAPTA-C is an attractive compound for further pre-clinical development. Therefore, we decided to evaluate the effects of RAPTA-C in more detail on primary tumors of human carcinomas. The results presented here demonstrate that RAPTA-C possesses anticancer activity, in conjunction with an intrinsic anti-angiogenic mechanism on primary tumors.
Results and discussion
The effect of RAPTA-C on tumor growth inhibition was investigated in a human A2780 ovarian carcinoma implanted on the chicken chorioallantoic membrane (CAM) model. A2780 cells were inoculated on embryo development day (EDD) 7 and monitored for 11 days. Established and vascularized tumors were detected 3 days post implantation (EDD 10). Treatment with RAPTA-C was performed by i.v. injections for five consecutive days (Fig. 1A). Tumors grew to an average size of approximately 160 mm3 by EDD 18 when left untreated (Fig. 1C). Tumor growth was efficiently inhibited following treatment with RAPTA-C and on the final experiment day, the growth of tumors was inhibited by about 75% (**p = 0.0007) at a dose of 0.2 mg kg−1 per day (Fig. 1B). An inhibition of ca. 45% was observed at a dose of 0.05 and 0.02 mg per kg per day, however, neither were statistically significant. It is noteworthy that READ-C (10 mg kg−1, i.p.) tested on tumor derived from the same cell line in athymic mice showed tumor growth inhibition of about 50% at 10-fold higher dose as compared to that used in our study, which resulted in the inhibition of tumor growth by 75%.6 The effect of RAPTA-C was, in fact, comparable to that of cisplatin in this study.6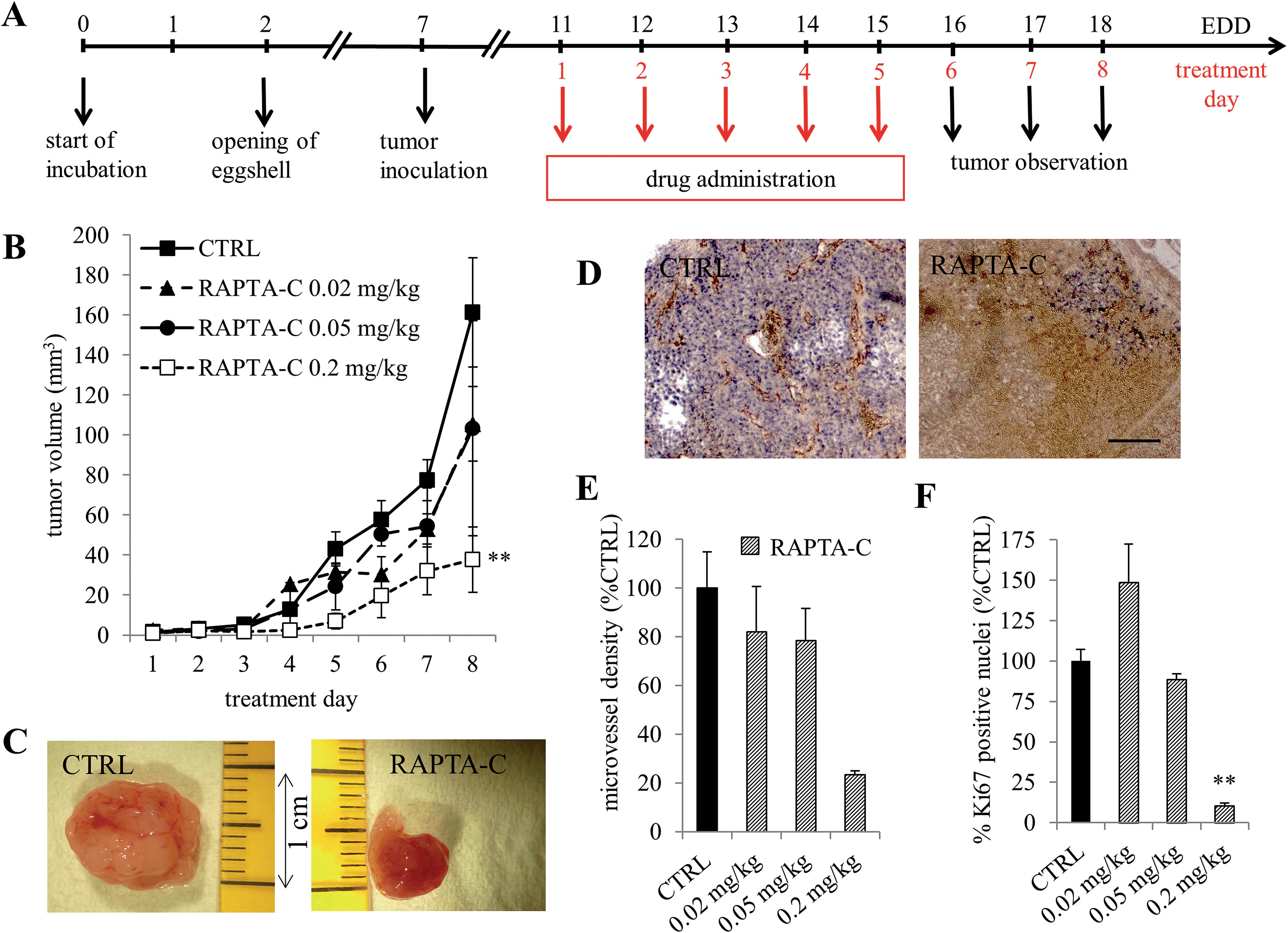 | ||
| Fig. 1 Inhibition of A2780 tumor growth in the CAM model by RAPTA-C. (A) Treatment protocol: RAPTA-C was administered i.v. on treatment days 1–5 (corresponding to EDD 11–15) and tumors were excised on the last day of the experiment. (B) Growth curve of A2780 tumors grafted on the CAM showing tumor volume with respect to treatment day. (C) Images show representative tumors from the vehicle treated (CTRL) and RAPTA-C (0.2 mg kg−1) treated CAMs. (D) Representative images of the immunohistochemical staining of the endothelial cell marker CD31 (in brown) showing reduced microvessel density per mm2 in tumors treated with RAPTA-C normalized to the tumor surface area and provided as a % of the control (E) and Ki-67 positive nuclei (in blue) (D) and quantification of the percentage of the tumor surface area staining positive for Ki-67 (as a % of CTRL) (F). Black bar in the right image of (D) represents 500 μm and is valid for both images. Error bars represent standard error of the mean, SEM; N = 5 in all treatment groups; N > 5 CTRL group; **p < 0.01. | ||
The effect of RAPTA-C treatment on the process of angiogenesis was assessed by measuring the vessel density using immunohistochemical staining of the CAM tissue sections with the endothelial cell marker CD31.14 Staining of tumor sections for the nuclear proliferation marker Ki-67 was performed to determine intra-tumor cell viability.15 Tumors were resected on the last day of the experiment (EDD 18). The control samples contained viable tumor tissue with many blood vessels whereas the treated tissues contained large areas of non-proliferating tumor cells (Fig. 1D and F, **p = 0.0099). Additionally, quantification of the microvessel density in the sparse viable tissue remaining revealed significantly reduced microvessel density (0.2 mg kg−1 per day), as compared to the control tumors (Fig. 1E). These results confirm the in vivo antitumor activity of RAPTA-C was achieved in parallel to an anti-angiogenic effect.
RAPTA-C was also evaluated in Swiss athymic mice inoculated on the right flank with human LS174T colorectal adenocarcinoma cells. When tumor size reached approximately 5–6 mm in diameter (day 5 after inoculation), the mice were randomized into groups and treated once daily with RAPTA-C (i.p. 10, 40 or 100 mg kg−1) for 11 days or with PBS only (containing 0.9% DMSO) as a control. Spectroscopic characterization of RAPTA-C in DMSO solutions with or without albumin is presented in Fig. S1 (ESI†), showing that the compound is stable under the treatment conditions, as expected from previous studies.16 Tumor growth was monitored during treatment and, after this period, mice were sacrificed and tumors were resected. Tumor growth of RAPTA-C treated tumors (i.p. 100 mg kg−1 per day) was inhibited (**p = 0.008) by approximately 50% compared to the control group (Fig. 2A) on the last experiment day. Lower doses (10 and 40 mg kg−1 per day) did not lead to significant tumor growth inhibition. We did not observe toxicity during the course of treatment. The average body weight of animals in the treatment groups did not change significantly over the course of treatment (Fig. 2B).
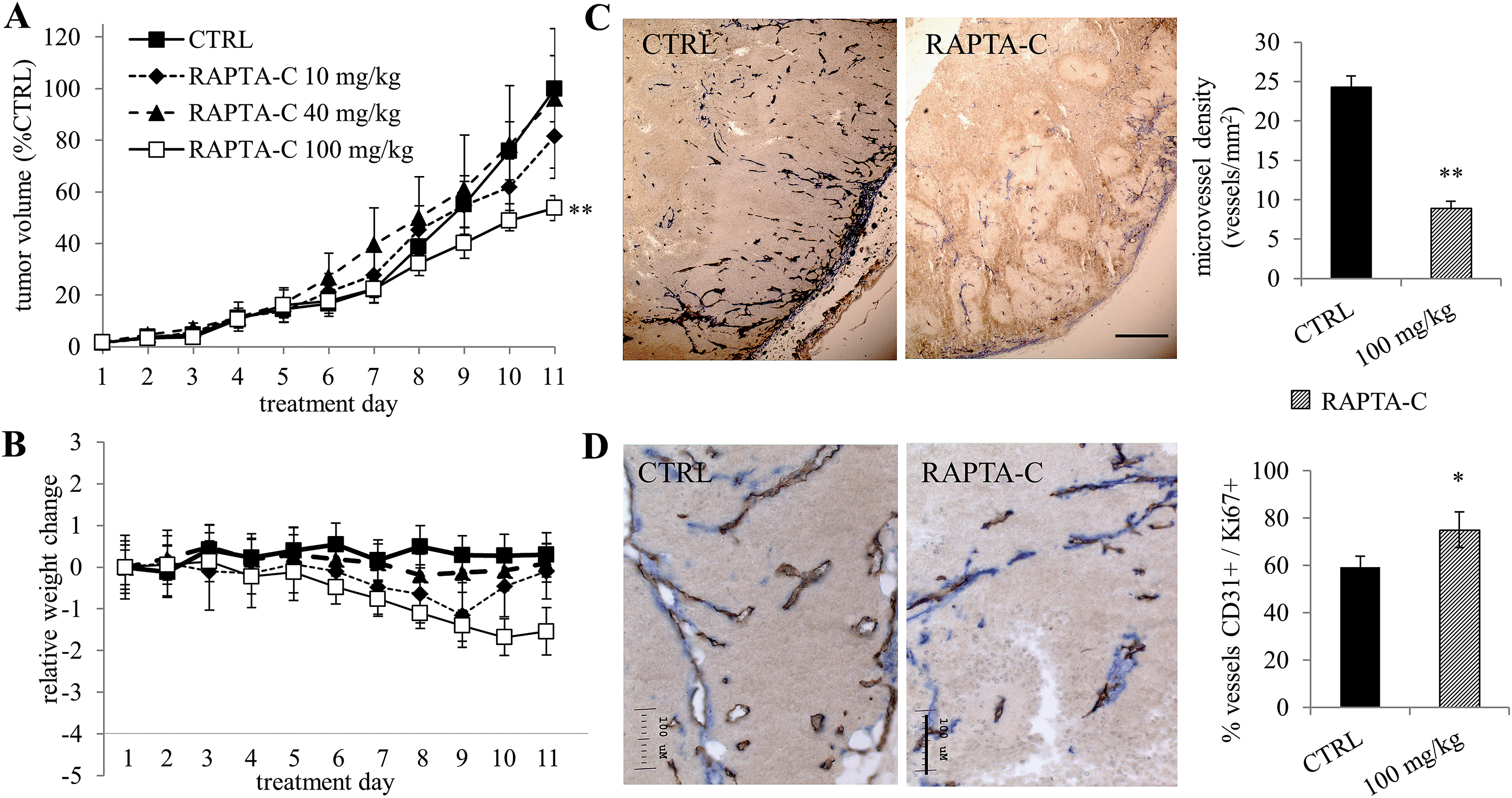 | ||
| Fig. 2 RAPTA-C induced inhibition of human LS174T colorectal carcinoma tumor growth in athymic mice. (A) RAPTA-C dose dependent growth curves of LS174T tumors. (B) Relative weight change of treated mice during the experiment. The average weight of mice in the RAPTA-C 100 mg kg−1 treatment group was not significantly different between treatment days 1 and 11 (p = 0.06). (C) Representative images of CD31 positive immunohistochemical sections of CTRL and RAPTA-C (100 mg kg−1) treated tumors on the last day of experiment and microvessel density quantification, expressed as the number of vessels per mm2 (**p = 9.6 × 10−7). Magnification 4×, bar in the right image represents 500 μm and is valid for both images. (D) Representative images of SMA and CD31 positive immunohistochemical sections of CTRL and RAPTA-C (100 mg kg−1) treated tumors on the last day of experiment and their quantification (*p = 0.0166). Magnification 20×, bar in the right image represent 100 μm and is valid for both images. Error bars represent SEM; N = 3–9 mice per group. *p < 0.05, **p < 0.01. | ||
KP1019 has been evaluated in various colorectal tumor models and was found to have a strong activity in a chemo-resistant colon carcinoma model (MAC15A) and was subsequently tested in an autochthonous colorectal tumor model in rats, which more accurately reflects both the histological appearance and malignant progression of human disease.17 In this model, KP1019 was highly effective, resulting in approximately 90% tumor growth inhibition. It was more effective that cisplatin, which was completely ineffective, and 5-fluorouracil treatment. 5-Fluorouracil therapy, which is a standard treatment for colorectal carcinoma and is the most effective single agent therapy, inhibited tumor growth by 60% as compared to the control tumors.18,19 Treatment of CBA mice with MCa mammary carcinoma with KP1019 administered at 40 mg per kg per day from day 6 to day 11 post tumor implantation (6 daily administrations) resulted in a reduction of primary tumor weight by 67% relative to the tumor mass in the control animals.20
Treated tumor tissue sections were stained for endothelial cells (with CD31 staining in brown) and smooth muscle actin (with anti-SMA in blue), indicating the presence of mature blood vessels (Fig. 2C and D). Tumor size measurements appeared to be an underestimation of the RAPTA-C activity, as treated tumors showed large areas of necrosis. In addition, around the remaining blood vessels small patches of Ki-67 negative tumor cells were observed, indicating that that tumor cells are in a quiescent state (Fig. S2, ESI†). Quiescence describes reversible growth or proliferation arrest and is considered to represent a state induced by diverse anti-mitogenic signals leading to cell division arrest.21 Interestingly, assessment of microvessel density revealed a significant decrease in the number of blood vessels in treated tumors (Fig. 2C, for 100 mg kg−1 per day), indicating a strong anti-angiogenic effect. SMA staining revealed that RAPTA-C treatment resulted in normalized, more mature blood vessels that were covered by pericytes. While control tumors were characterized by numerous, newly generated and immature blood vessels lacking pericytes (CD31+/SMA−), we noticed that treatment with RAPTA-C resulted in the disappearance of this newly formed angiogenic vasculature and led to an increased number of mature vessels covered by pericytes (CD31+/SMA+, Fig. 2D). This observation further confirms the angiostatic activity of RAPTA-C.
The low toxicity of RAPTA-C when applied at quite high doses is remarkable and therefore the accumulation of RAPTA-C was studied in order to rationalize, at least in part, the absence of toxicity. The biodistribution of radioactively labelled 103Ru-RAPTA-C in healthy Balb/c mice was determined (Fig. 3). The procedure used to prepare 103Ru-RAPTA-C is provided in the Experimental section. Two hours after injection, it was found that the level of ruthenium in the urine was ca. 28-fold higher than in liver, spleen, lungs and heart, indicative of fast and efficient renal excretion. Ruthenium was not found to specifically accumulate in any of the tested organs. The slightly higher levels in the kidneys (approx. 2-fold) and intestines (approx. 3-fold) may be expected only 2 hours after injection. These data parallel a previous pharmacokinetic study in which RAPTA-C was injected i.p. in healthy Swiss CD-1 mice.7 In this previous study, the content of ruthenium in the blood, plasma, and in selected organs (liver, kidney, spleen, and lung) was quantified by atomic absorption spectroscopy. A half-life (T1/2) of about 12 hours was estimated that varied slightly depending on the dose and administration schedule. Two hours after RAPTA-C administration, the concentration of RAPTA-C in kidneys was 2-fold higher than in the spleen, while the majority of ruthenium was eliminated in urine within 1 hour of administration. The previous and current data show that the rate of clearance of RAPTA-C is faster than that of NAMI-A.22 A 106Ru analogue of RAED-C revealed that the highest levels of the compound were associated with the liver and kidney.23
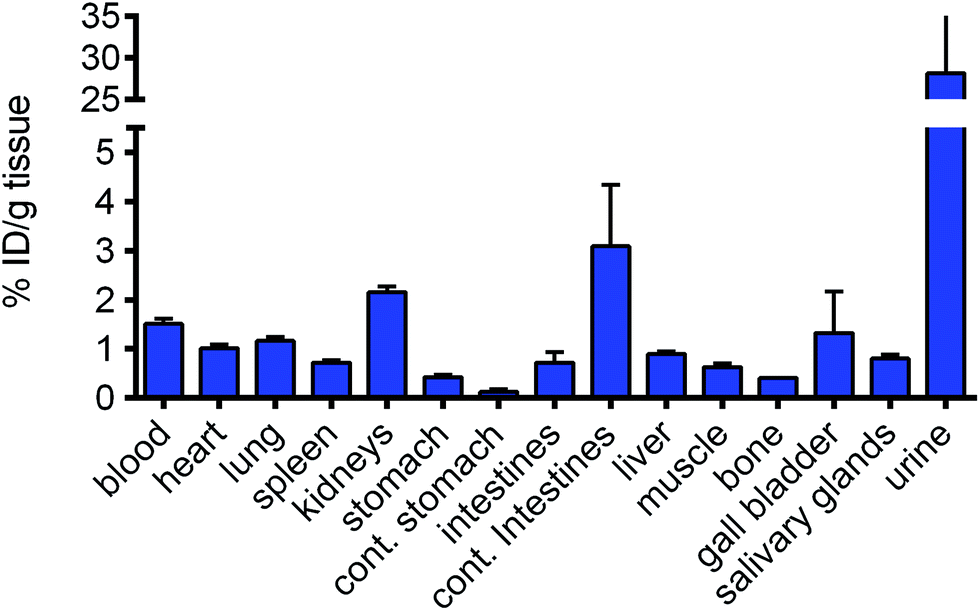 | ||
| Fig. 3 RAPTA-C biodistribution after injection in Balb/c mice. Mice were injected i.p. with 103Ru-RAPTA-C (1.9 MBq per mouse) and were sacrificed 2 hours post injection and their organs were resected. The data are presented as % injected dose per gram of tissue (% ID per g). Indicated values are means, error bars represent SEM. | ||
Experimental
General chemicals
Ruthenium(III) chloride hydrate (35–40% Ru) was obtained from Merck and 1,3,5-triaza-7-phosphaadamantane (pta) 97% and (R)-(−)-α-phellandrene ≥95% (sum of enantiomers) were obtained from Sigma-Aldrich. Purification of the 103Ru-RAPTA-C was performed using Silica gel 60 from Fluka.103RuCl3·3H2O production
Neutron activation of natural RuCl3·3H2O gave the 103RuCl3·3H2O. Irradiations were performed with the spallation neutron source SINQ at the PSI. Irradiation of natural ruthenium with a neutron flux of 1014 n cm−2 s−1 gave the following radionucleides: 97Ru (T1/2 = 2.9 days), 103Ru (T1/2 = 39.3 days) and 105Ru (T1/2 = 4.44 h). The thermal cross section of 102Ru (σ = 1.27 barn) and the relative long half-life of 103Ru compared to the other radionucleides produced allowed for the production of 103Ru with high radionuclidic purity with 97Ru or 105Ru activities undetectable after 1 week of decay (Fig. S3, ESI†). Neutron irradiation of the chlorides gave negligible 36Cl and 38Cl activity.Example: 15 mg natural ruthenium (39.9 mg RuCl3·3H2O) were irradiated for 16.2 days with the 1014 n cm−2 s−1 flux giving a 103Ru activity of 109.8 MBq from the 124.7 MBq expected. 103Ru activity was calculated according to the equation given in the ESI.† The radiosynthesis of 103Ru-RAPTA-C was monitored with a Cyclone phosphor imager (Perkin-Elmer).
103Ru-RAPTA-C radiosynthesis
103RuCI3·3H2O (13.6 mg, 0.05 mmol, 36.21 MBq) was suspended in ethanol (6 ml) and underwent reflux for 3 h to obtain a dark green solution. (R)-(−)-α-Phellandrene (0.31 mmol) was added to the reaction mixture and the reaction was stirred under reflux for 6 h. The reaction was cooled to room temperature and diethyl ether (3 ml) was added to precipitate the [(p-cymene)103RuCl2]2 dimer. The dimer was washed repeatedly with diethyl ether to remove unreacted (R)-(−)-α-phellandrene. The supernatant was removed with a flow of nitrogen and replaced with dichloromethane (6 ml) and pta (8 mg, 0.05 mmol) was added to the solution. The reaction was stirred for 4 h at room temperature. Purification using column chromatography (Silica gel 60) with a methanol–dichloromethane 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 eluent gave the pure 103Ru-RAPTA-C (60.5%, 21.9 MBq). HPLC: 8.27 min at 342 nm; ESI-MS: 486 m/z [Ru(η6-p-cymene)Cl2(pta) + Na]+ (calc. 486.358), 464 m/z [Ru(η6-p-cymene)Cl2(pta) + H]+ (calc. 464.376), 428 m/z [Ru(η6-p-cymene)Cl(pta)]+ (calc. 427.918), see Chart 3.
:30 eluent gave the pure 103Ru-RAPTA-C (60.5%, 21.9 MBq). HPLC: 8.27 min at 342 nm; ESI-MS: 486 m/z [Ru(η6-p-cymene)Cl2(pta) + Na]+ (calc. 486.358), 464 m/z [Ru(η6-p-cymene)Cl2(pta) + H]+ (calc. 464.376), 428 m/z [Ru(η6-p-cymene)Cl(pta)]+ (calc. 427.918), see Chart 3.
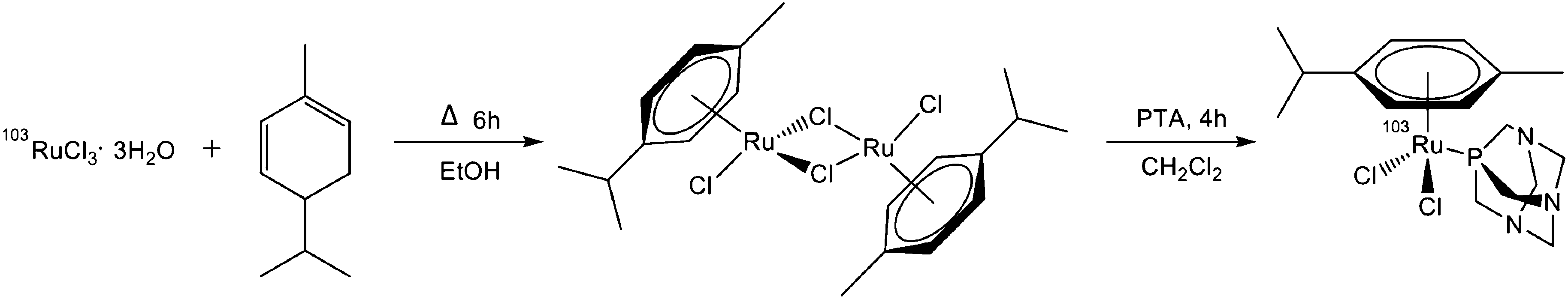 | ||
| Chart 3 Radio synthesis of 103Ru-RAPTA-C. | ||
Cell culture, preparation and implantation on the CAM model
A2780 human ovarian carcinoma cells (ECACC, Salisbury, UK) were maintained in RPMI-1640 cell culture medium supplemented with GlutaMAX™ (Gibco, Carlsbad, USA), 10% heat-inactivated bovine calf serum (Sigma-Aldrich, St. Louis, USA) and 1% antibiotics (Sigma-Aldrich). Fertilized chicken eggs were incubated in a hatching incubator (relative humidity 65%, 37 °C), as previously described.24 A2780 cells (106) were prepared in serum-free RPMI-1640 as a spheroid in a 25 μl hanging drop and 3 h later were transplanted on the surface of the in ovo CAM on EDD 825 (Fig. 1A). Vascularized three-dimensional tumors were visible 3 days after tumor cell implantation, on EDD 11, when treatment was initiated. Tumors were treated daily by i.v. injection of freshly prepared RAPTA-C into a main blood vessel of the CAM. After preliminary experiments, we observed that the most efficient tumor growth inhibition was obtained with RAPTA-C after 5 intravenous injections, in the tested dose range, on 5 consecutive days. Therefore, we used this treatment schedule in the current study. RAPTA-C was dissolved in DMSO to afford a stock solution, which was subsequently diluted to the desired concentrations in 0.9% NaCl. Controls were treated with vehicle (DMSO in 0.9% NaCl). The concentration of drug was administered each day for 5 consecutive days and doses were calculated assuming an average embryo weight of 8.23 grams (i.e. the average embryo weight during the course of the 5 days of treatment). Tumors were monitors for a total of 11 days after cell implantation, at which time embryos were sacrificed and tumors were resected and fixed in zinc-fixative for additional analysis. N = 5 in all treatment groups; N > 5 in the control group.Cell implantation in athymic mice
Experiments in female 7-week old Swiss female athymic mice purchased from Charles River (Orleans, France) were carried out according to a protocol approved by the Committee for Animal Experiments for the Canton Vaud, Switzerland (license 2736). Mice were injected subcutaneously in the right flank with 0.5 × 106 LS174T cells (purchased from Cell Line Service GmbH Eppelheim, Germany), previously cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum and 1% antibiotics). When tumors reached a size of 5–6 mm in diameter, mice received 10, 40 or 100 mg kg−1 of freshly prepared RAPTA-C in 100 μl solution via i.p. injections. This treatment was repeated daily for 11 consecutive days. Tumors dimensions and body weight were measured daily. Tumor volumes (mm3) were calculated as follows: volume = (largest diameter in mm) × (perpendicular diameter in mm)2 × 0.5. Mice were euthanized when the CTRL tumor size reached 1000 mm3. N = 9, 4, 6 and 3 mice for CTRL, 10 mg kg−1, 40 mg kg−1 and 100 mg kg−1 treatment groups, respectively.Immunohistochemistry
On the last day of the experiment tumors were resected and fixed overnight in zinc fixative solution. CAM and mice tumors were stained as previously described.10,14 Briefly, 5 μm sections were blocked with 5% BSA in PBS followed by incubation with primary antibodies against CD31 (rat anti-CD31; 1:200, clone SZ31, Dianova, Hamburg, Germany). Secondary donkey anti-rat biotinylated antibodies (1:200; Jackson, Suffolk, UK) were then incubated, followed by Streptavidin-HRP (1:50; Dako, Glostrup, Denmark), and visualized by 3,3′-diaminobenzidine (DAB), resulting in a brown-colored precipitate at the antigen site. Subsequently, incubation with antibodies against smooth muscle actin (SMA) (mouse anti-human SMA; 1:200, clone MIB-1, Dako, Glostrup, Denmark) or Ki-67 (mouse anti-human Ki-67; 1:200, clone 1A4, Dako, Gostrup, Denmark) was followed by alkaline phosphatase conjugated polymer Mach 2 (Biocare Medical, Concord, USA) and visualized by Fast Blue BB/Naphthol-AS-MX-Phosphate, resulting in a blue/purple-coloured precipitate.
Biodistribution studies
The in vivo experiments were approved by the local veterinarian department and conducted in accordance with the Swiss law of animal protection. Three 6–8-week-old female, Balb/c mice were purchased from Charles River Laboratories (Sulzfeld, Germany). 103Ru-RAPTA-C (1.9 MBq, 2.6 μmol, 100 μL per mouse) was injected into a lateral tail vein. The animals were sacrificed 2 h after drug administration. Selected tissues and organs were collected, weighed, and counted for radioactivity using a γ-spectrometer running with InterWinner software. The results were listed as a percentage of the injected dose per gram of tissue mass (% ID per g). Counts of a defined volume of the original injection solution counted at the same time were used as a reference.Statistical analysis
Values are given as mean values ± standard error of the mean (SEM). Data are represented as averages of independent experiments. Statistical analysis was done using the two-way ANOVA test and t-test using GraphPad Prism software® (GraphPad Software Inc., CA). *p indicating p-values lower than 0.05 and **p indicating p-values lower than 0.01 were considered statistically significant.Conclusions
Certain ruthenium-based compounds show promise as anticancer agents with different characteristics compared to clinically applied platinum-based drugs.12,26–29 Our studies were focused on an organometallic ruthenium(II)-arene complex, i.e. RAPTA-C, that was shown to exhibit both anti-metastatic and anti-angiogenic effects.9,30 Herein, we describe the effect of RAPTA-C on the growth inhibition of primary tumors. In this respect, RAPTA-C treatment was found to efficiently reduce the growth of tumors by approx. 75% in one model, with the inhibition of angiogenesis (determined by vessel density assessment) being at least partly responsible for this action. An efficient and general induction of tumor cell death was observed in the tumor tissue, with remaining viable tumor islands completely negative for the proliferation marker Ki-67. These results support the translational development of RAPTA-C into clinical studies.Acknowledgements
We thank the Swiss National Science Foundation, the National Competence Center for Biomedical Imaging, EPFL and Dr J. Jacobi for financial support. We thank Dr Konstantin Zhernosekov (PSI) for production of 103RuCI3·3H2O and Prof. Hubert van den Bergh (EPFL) for helpful discussions.References
- F. Muggia, Gynecol. Oncol., 2009, 112, 275–281 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- A. Levina, A. Mitra and P. A. Lay, Metallomics, 2009, 1, 458–470 RSC.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- Z. Adhireksan, G. E. Davey, P. Campomanes, M. Groessl, C. M. Clavel, H. Yu, A. A. Nazarov, H. F. Yeo, W. H. Ang, P. Droge, U. Rothlisberger, P. J. Dyson and C. A. Davey, Nat. Commun., 2014, 5, 3462 Search PubMed.
- P. Nowak-Sliwinska, J. R. van Beijnum, A. Casini, A. A. Nazarov, G. Wagnières, H. van den Bergh, P. J. Dyson and A. W. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS PubMed.
- A. Weiss, J. R. van Beijnum, D. Bonvin, P. Jichlinski, P. J. Dyson, A. W. Griffioen and P. Nowak-Sliwinska, J. Cell. Mol. Med., 2014, 18, 480–491 CrossRef CAS PubMed.
- B. Sanna, M. Debidda, G. Pintus, B. Tadolini, A. M. Posadino, F. Bennardini, G. Sava and C. Ventura, Arch. Biochem. Biophys., 2002, 403, 209–218 CrossRef CAS.
- A. Bergamo, A. Masi, A. F. Peacock, A. Habtemariam, P. J. Sadler and G. Sava, J. Inorg. Biochem., 2010, 104, 79–86 CrossRef CAS PubMed.
- J. M. Ebos, C. R. Lee, W. Cruz-Munoz, G. A. Bjarnason, J. G. Christensen and R. S. Kerbel, Cancer Cell, 2009, 15, 232–239 CrossRef CAS PubMed.
- A. W. Griffioen, L. A. Mans, A. M. de Graaf, P. Nowak-Sliwinska, C. L. de Hoog, T. A. de Jong, F. A. Vyth-Dreese, J. R. van Beijnum, A. Bex and E. Jonasch, Clin. Cancer Res., 2012, 18, 3961–3971 CrossRef CAS PubMed.
- M. Ramires, L. David, D. Leitao, M. Seixas, F. Sansonetty and M. Sobrinho-Simoes, J. Pathol., 1997, 182, 62–67 CrossRef CAS.
- C. Scolaro, C. G. Hartinger, C. S. Allardyce, B. K. Keppler and P. J. Dyson, J. Inorg. Biochem., 2008, 102, 1743–1748 CrossRef CAS PubMed.
- B. K. Keppler, Metal Complexes in Cancer Chemotherapy, Wiley-VCH, 1993 Search PubMed.
- M. R. Berger, F. T. Garzon, B. K. Keppler and D. Schmahl, Anticancer Res., 1989, 9, 761–765 CAS.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS PubMed.
- A. Bergamo, A. Masi, M. A. Jakupec, B. K. Keppler and G. Sava, Met.-Based Drugs, 2009, 2009, 681270 CAS.
- H. A. Coller, L. Sang and J. M. Roberts, PLoS Biol., 2006, 4, e83 Search PubMed.
- L. Weiss, Cancer Metastasis Rev., 2000, 19, 193–383 CrossRef.
- J. D. Hoeschele, A. Habtemariam, J. Muir and P. J. Sadler, Dalton Trans., 2007, 4974–4979 RSC.
- S. H. Lim, P. Nowak-Sliwinska, F. A. Kamarulzaman, H. van den Bergh, G. Wagnieres and H. B. Lee, Photochem. Photobiol., 2010, 86, 397–402 CrossRef CAS PubMed.
- Y. Adar, M. Stark, E. E. Bram, P. Nowak-Sliwinska, H. van den Bergh, G. Szewczyk, T. Sarna, A. Skladanowski, A. W. Griffioen and Y. G. Assaraf, Cell Death Dis., 2012, 3, 1–10 Search PubMed.
- A. Bergamo, R. Gagliardi, V. Scarcia, A. Furlani, E. Alessio, G. Mestroni and G. Sava, J. Pharmacol. Exp. Ther., 1999, 289, 559–564 CAS.
- G. Sava, R. Gagliardi, A. Bergamo, E. Alessio and G. Mestroni, Anticancer Res., 1999, 19, 969–972 CAS.
- E. S. Antonarakis and A. Emadi, Cancer Chemother. Pharmacol., 2010, 66, 1–9 CrossRef CAS PubMed.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- A. Bergamo, A. Masi, P. J. Dyson and G. Sava, Int. J. Oncol., 2008, 33, 1281–1289 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc01255k |
| This journal is © The Royal Society of Chemistry 2014 |