 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rules for priming and inhibition of glycosaminoglycan biosynthesis; probing the β4GalT7 active site†
Anna
Siegbahn
a,
Sophie
Manner‡
a,
Andrea
Persson‡
ab,
Emil
Tykesson‡
b,
Karin
Holmqvist
a,
Agata
Ochocinska
a,
Jerk
Rönnols
c,
Anders
Sundin
a,
Katrin
Mani
b,
Gunilla
Westergren-Thorsson
b,
Göran
Widmalm
c and
Ulf
Ellervik
*a
aCentre for Analysis and Synthesis, Centre for Chemistry and Chemical Engineering, Lund University, P.O. Box 124, SE-221 00 Lund, Sweden. E-mail: ulf.ellervik@chem.lu.se
bDepartment of Experimental Medical Science, Lund University, BMC A13, SE-221 00 Lund, Sweden
cDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden
First published on 12th June 2014
Abstract
β-1,4-Galactosyltransferase 7 (β4GalT7) is an essential enzyme in the biosynthesis of glycosaminoglycan (GAG) chains of proteoglycans (PGs). Mammalian cells produce PGs, which are involved in biological processes such as cell growth and differentiation. The PGs consist of a core protein, with one or several GAG chains attached. Both the structure of the PGs and the GAG chains, and the expression of the enzymes involved in their biosynthesis and degradation, vary between normal cells and tumor cells. The biosynthesis of GAG chains is initiated by xylosylation of a serine residue of the core protein, followed by galactosylation by β4GalT7. The biosynthesis can also be initiated by exogenously added β-D-xylopyranosides with hydrophobic aglycons, which thus can act as acceptor substrates for β4GalT7. To determine the structural requirements for β4GalT7 activity, we have cloned and expressed the enzyme and designed a focused library of 2-naphthyl β-D-xylopyranosides with modifications of the xylose moiety. Based on enzymatic studies, that is galactosylation and its inhibition, conformational analysis and molecular modeling using the crystal structure, we propose that the binding pocket of β4GalT7 is very narrow, with a precise set of important hydrogen bonds. Xylose appears to be the optimal acceptor substrate for galactosylation by β4GalT7. However, we show that modifications of the xylose moiety of the β-D-xylopyranosides can render inhibitors of galactosylation. Such compounds will be valuable tools for the exploration of GAG and PG biosynthesis and a starting point for development of anti-tumor agents.
Introduction
β-1,4-Galactosyltransferase 7 (β4GalT7) is a key enzyme in the biosynthesis of heparan sulfate (HS) and chondroitin sulfate/dermatan sulfate (CS/DS) glycosaminoglycan (GAG) chains. The GAG chains are linear polysaccharides of alternating uronic acids and aminosugars, which are most often linked to a core protein to form proteoglycans (PGs). PGs interact with growth factors and are important for a diverse set of biological processes, such as cell growth and differentiation. In addition, the PG/GAG expression is critical in the pathobiology of all stages of cancer progression, such as proliferation, tumor invasion, and metastasis.1–3 Both the structure of the PGs and the GAG chains, and the expression of enzymes involved in their biosynthesis and degradation vary between normal cells and tumor cells. For example, Sasisekharan and co-workers successfully used GAG fragments, released from tumor cell surfaces, to inhibit tumor growth and colonization in vivo.4 The enzymes involved in the biosynthesis of GAG chains are thus promising targets for drug development.The biosynthesis of HS and CS/DS is initiated by xylosylation of a serine residue of the core protein by xylosyltransferase (XT).5 The xylosylated protein is then galactosylated in a stepwise manner by two galactosyltransferases (β4GalT7, also known as GalTI,6 and β3GalT6, also known as GalTII) and subsequently glucuronated by glucuronyltransferase I (GlcAT-I) to form a linker tetrasaccharide that is the branching point for the biosynthesis of HS and CS/DS (Fig. 1a). During the polymerization of HS and CS/DS, posttranslational modifications, such as O- and N-sulfation, result in extensive structural diversity of the chains.
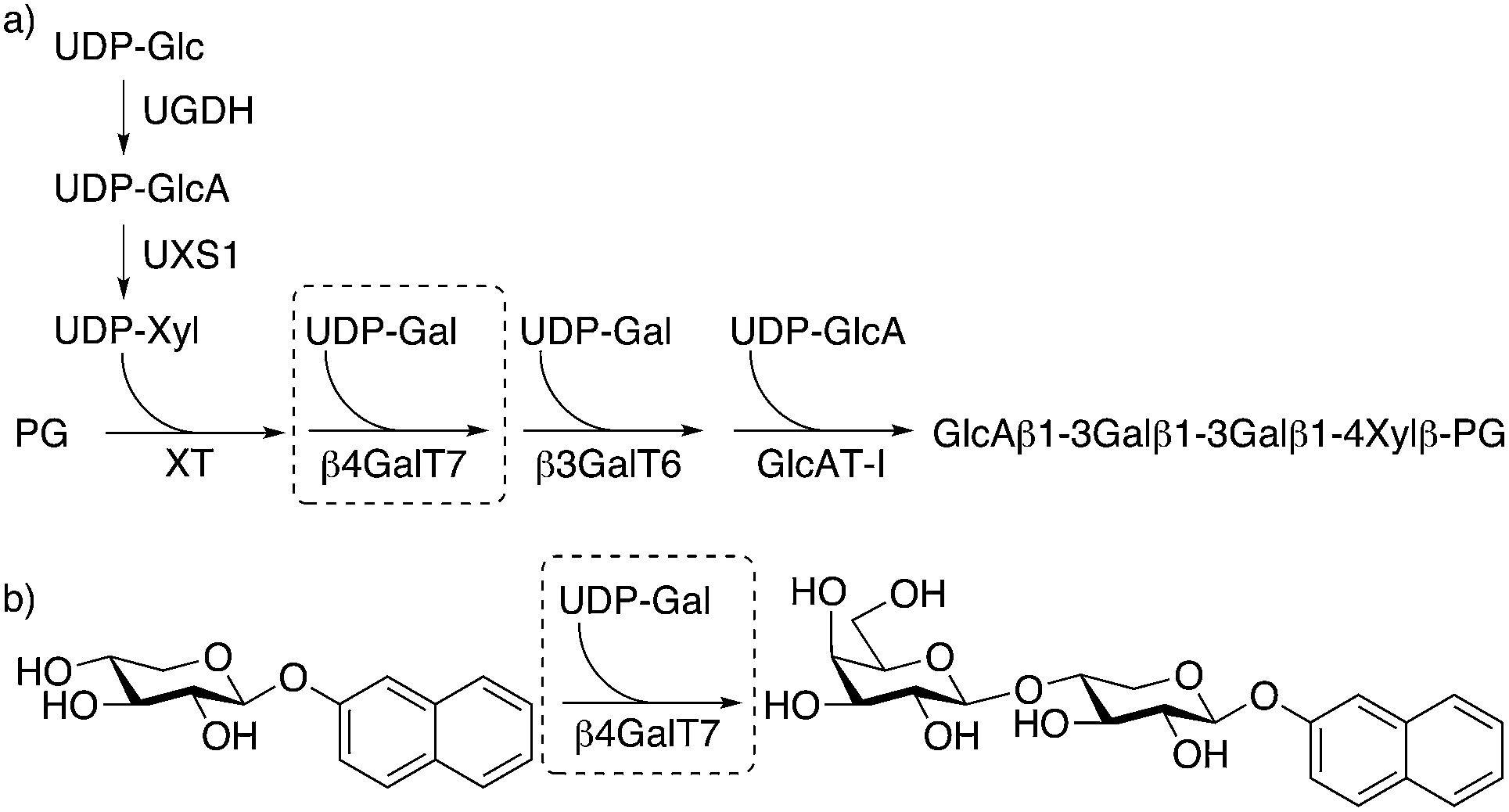 | ||
| Fig. 1 (a) Biosynthesis of the linker tetrasaccharide of HS and CS/DS proteoglycans. (b) Galactosylation of XylNap to form GalXylNap. | ||
The biosynthesis of GAG chains can also be initiated by xylosides carrying hydrophobic aglycons, such as 2-naphthyl β-D-xylopyranoside (XylNap, 1a, Fig. 1b and Chart 1).7,8 The xylosides can, when exogenously supplied to cells, act as acceptors in the first galactosylation step by β4GalT7, and thus provide a simplified model system for GAG biosynthesis, and serve as a starting point for development of anti-tumor drugs.
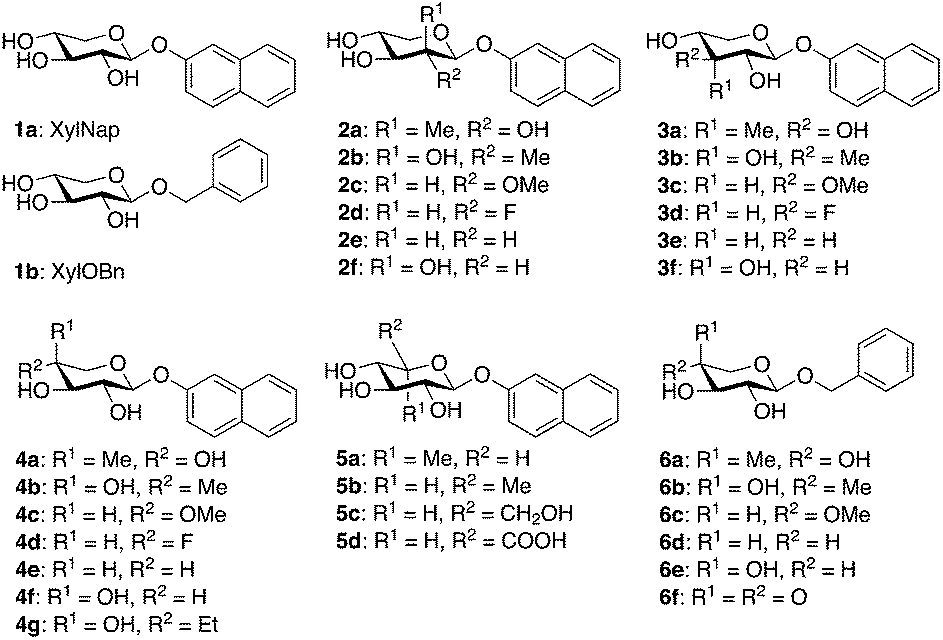 | ||
| Chart 1 Structures of xylosides and xyloside analogs. | ||
Up until the cloning of β4GalT7, the exploration of the biosynthesis of GAG chains has been limited to synthesis and testing of xylosides and xyloside analogs in cell assays. These studies have established the structural importance of the aglycon for the amount of GAG priming as well as for the type of chain primed (i.e., HS or CS/DS).9–18 Much less is known regarding modifications of the xylose moiety. In 2011, we reported the synthesis of a series of modified 2-naphthyl β-D-xylopyranosides, where each hydroxyl group was epimerized or replaced by a hydrogen atom, a fluorine atom, or a methoxy group (Chart 1, compounds 2c–f, 3c–f, and 4c–f).19 The amount of GAG priming induced on these compounds was investigated and showed that treatment with XylNap initiated the expected priming of GAG chains, whereas none of the tested analogs showed priming of GAG chains. However, we observed a small inhibitory effect on the endogenous glycosylation of PG for the 4-fluoro and the 4-deoxy analogs (4d, 33%, 4e, 24%). These results were in accordance with findings from Kuberan and co-workers, who presented GAG inhibitors based on 4-deoxy-4-fluoro-xylopyranosides with up to 90% inhibition.20,21 However, some of our results were contradictory to earlier investigations by Esko and co-workers who reported GAG priming by xyloside analogs with modifications in positions 2, 3, and 4 of the corresponding benzyl β-D-xylopyranosides.22 Interestingly, the authors also reported GAG priming by the 4-keto compound 6f (Chart 1), at similar concentrations as XylOBn (1b), and by benzyl 4-C-methyl-β-D-xylopyranoside (6a) at approximately 100-fold higher concentrations of 6a compared to 1b.
In 2013, Qasba and co-workers published the crystal structure of a substrate-enzyme complex of human β4GalT7, as well as that of the similar enzyme from Drosophila melanogaster.23,24 The two enzymes show a significant overlap in the catalytic pocket, and the Drosophila variety was used in co-crystallization with UDP-galactose (UDP-Gal) and xylobiose (i.e., Xylβ1-4Xyl). To suppress decomposition of the UDP-Gal by the enzyme, the authors used a mutated D211N-β4GalT7. They proposed that the catalytic reactions are initiated by binding of UDP-Gal and a manganese ion to β4GalT7. This binding initiates a conformational reorganization from an open form to a closed conformation, with simultaneous formation of a xylose-binding pocket. The xylose moiety is then perfectly positioned for formation of the glycosidic bond and the authors proposed an SN2-like mechanism, similar to the catalytic mechanism shown by β4GalT1.25,26 Furthermore, aromatic residues at the entrance of the catalytic pocket could explain the strong GAG priming shown by some xylosides with aromatic aglycons.
We hypothesize that β4GalT7 does not tolerate any modifications of the xylose moiety, and that it is possible to design effective inhibitors of GAG biosynthesis by modifying xylose. To pinpoint the requirements and limitations for priming and inhibition of GAG chains we synthesized, and unequivocally characterized, a collection of xyloside analogs (Chart 1). To rule out any effects due to cellular uptake of these compounds, we cloned and expressed β4GalT7, the enzyme that catalyzes the second step of the formation of the linker tetrasaccharide, and tested xyloside analogs in a cell-free assay.
Results
Synthesis
Benzylation of XylNap (1a) under phase transfer conditions generated a separable mixture of dibenzylated 7, 8, and 9 (Scheme 1).19 Next, the methyl group was introduced using a Dess–Martin oxidation–Grignard addition sequence, which resulted in stereoisomeric mixtures, separable by column chromatography. Finally, to avoid saturation of the naphthyl moiety, debenzylation was performed using catalytic hydrogenation in HCl/DMF,27 which gave the expected methylated 2-naphthyl β-D-xylosides 2a, 2b, 3a, 3b, 4a, and 4b. A similar procedure, using isopropylidene protection, gave the corresponding compound 4g, carrying an ethyl group in position 4. | ||
| Scheme 1 Synthesis of compounds modified in positions 2, 3, and 4. Reagents and conditions: (i) BnBr, TBAI (cat.), KOH (10% aq), CH2Cl2, r.t., 16 h, 7 (6%), 8 (66%), 9 (14%); (ii) (a) 15% Dess–Martin periodinane in CH2Cl2, CH2Cl2, r.t., N2, 2 h; (b) 3 M MeMgBr in Et2O, r.t., N2, 1.5 h, 10 (57%), 11 (10%), 12 (19%), 13 (68%); (iii) (a) 15% Dess–Martin periodinane in CH2Cl2, CH2Cl2, r.t., N2, 2 h; (b) 3 M MeMgBr in Et2O, 0 °C to r.t., N2, 1.5 h, 14 (42%), 15 (42%); (iv) 10% Pd/C (14%), HCl (22 eq.), DMF, H2, r.t., 3 h, 2a (67%), 2b (44%), 3a (76%), 3b (49%), 4a (70%), 4b (54%); (v) 2-methoxy propene, CSA (cat.), DMF, r.t., 1.5 h, (51%); (vi) (a) 2 M oxalyl chloride in CH2Cl2, DMSO, CH2Cl2, −78 °C, 2 h; (b) 3 M EtMgBr in Et2O, THF, r.t., 12 h, (38%); (vii) 70% AcOH, 1 h, (56%). | ||
Compound 5a was synthesized from per-O-acetylated L-idose (18)28 in a standard aromatic glycosylation, while compound 5b was synthesized from the commercially available 2-naphthyl β-D-glucopyranoside (5c) by a selective tosylation of position 6 followed by reduction using LiAlH4 (Scheme 2, top). Compound 5d is commercially available.
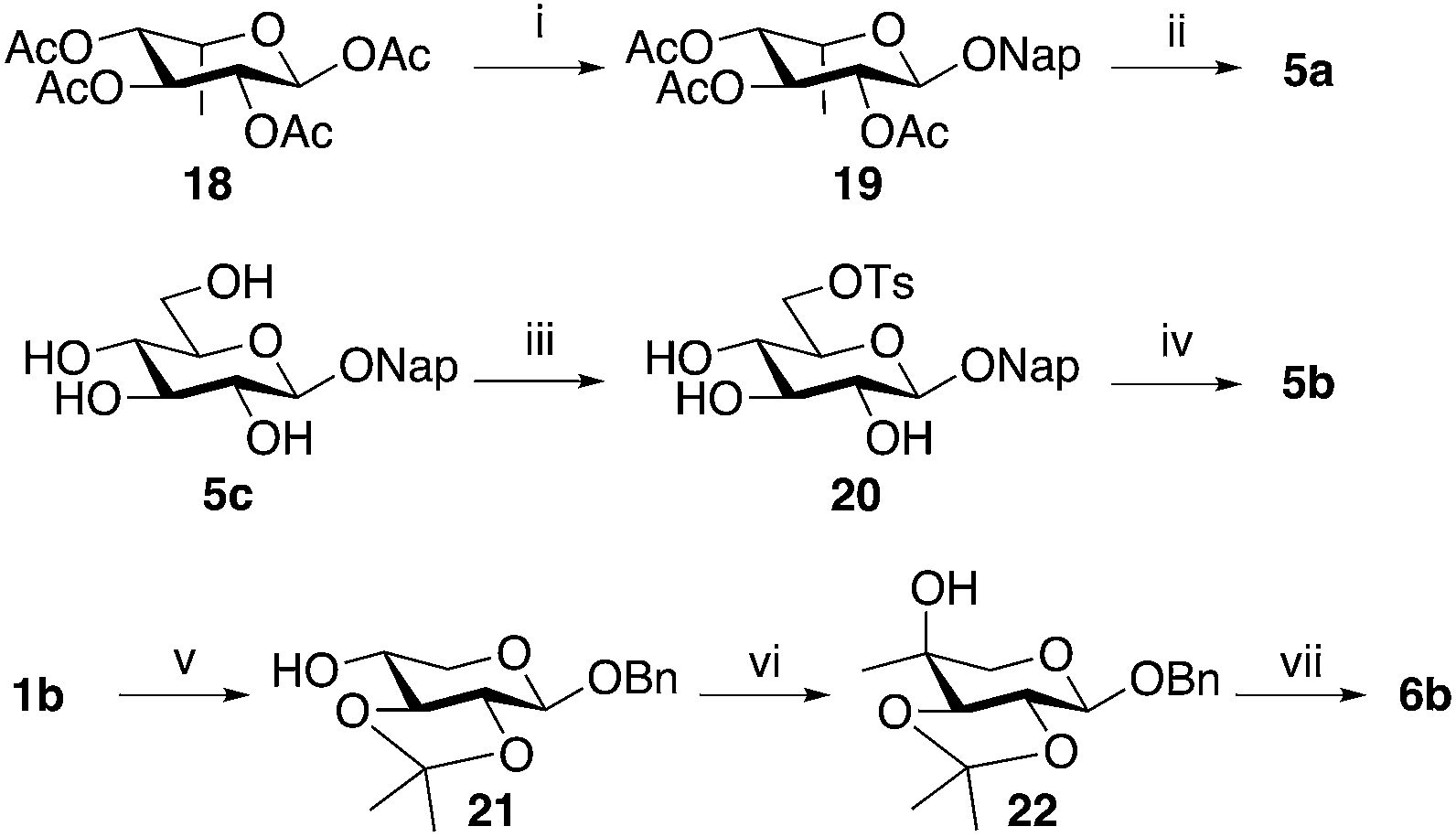 | ||
| Scheme 2 Synthesis of compounds modified in position 5 and synthesis of compound 6b. Reagents and conditions: (i) BF3·OEt2 (2.5 eq.), Et3N (0.5 eq.), 2-naphthol (1.5 eq.), CH2Cl2, 0 °C, 2 h, (78%); (ii) 1 M NaOMe (0.1 eq.), MeOH, r.t., 2 h, (81%); (iii) TsCl (1.5 eq.), ZnBr2 (1.1 eq.), pyridine, −25 °C, 1 h, (86%); (iv) LiAlH4 (6 eq.), THF, 30 °C, 22 h, (61%). (v) 2-Methoxy propene (5.8 eq.), TFA (cat.), DMF, r.t., 6 h, (47%); (vi) (a) 2 M oxalyl chloride in CH2Cl2 (3 eq.), DMSO (6 eq.), CH2Cl2, −78 °C, 2 h (b) 3 M MeMgBr in Et2O (3 eq.), THF, r.t., 12 h, (55%); (vii) Dowex 50Wx4, MeOH, r.t., 4 h, (56%). | ||
In addition, we intended to synthesize 6a, previously synthesized by Esko,22 due to its structural similarities with 4a, following the original procedure (Scheme 2, bottom). Starting from benzyl β-D-xylopyranoside (1b),29 selective protection of 2-OH and 3-OH, using 2-methoxy propene and CSA in DMF, gave 21 in 47% yield with an additional 12% of the corresponding 3,4-protected analog. Next, the methyl group was introduced in 21, using a Swern oxidation–Grignard addition sequence. To our surprise, compound 22 was the only isolated product. Finally, deprotection of 22 with acidic ion exchange resin in methanol, gave benzyl 4-methyl-β-D-arabinopyranoside (6b) in 56% yield. Conformational analysis, vide infra, unequivocally confirmed an equatorial orientation of the 4-C-methyl group. It is reasonable to believe that the structure of the compound synthesized and tested by Esko and co-workers was 6b rather than 6a.
Configurational and conformational analysis
The stereochemical assignments of the endocyclic tertiary carbon atoms are based on 1H,1H nuclear Overhauser effects (NOEs), 3JC,H and 3JH,H coupling constants, 13C and 1H chemical shifts. With these at hand, the extent of the major conformation of each compound was assessed. The 1H and 13C chemical shifts of the xylose residues were assigned by 1D 1H and 13C experiments in conjunction with a few 2D 1H, 1H- and 1H, 13C-correlated experiments (ESI†).Axially oriented methyl groups are in close proximity to axial hydrogen atoms on γ-carbons as well as vicinal hydrogen atoms, whereas equatorial methyl groups are proximate only to vicinal hydrogen atoms (in the 4C1 conformation). Employing 1D 1H,1H-DPFGSE-NOESY experiments,30 selective excitation of the methyl groups in compounds 2a, 2b, 3a, 3b, 4a, and 4b resulted in the NMR spectra displayed in Fig. 2. The 2-(R)-configuration (i.e., axial orientation of methyl group) of compound 2a was assigned via a prominent NOE-correlation from the methyl protons to H4 (Fig. 2). For compound 2b, NOE-correlations were only observed to H1 and H3, which supports an equatorially oriented methyl group and hence a 2-(S)-configuration. The 3-(S)-configuration of compound 3a was assigned via NOE-correlations from the methyl protons to H1 and H5pro-S, whereas these were essentially absent in compound 3b. The stereochemical assignment at C4 of compound 4a was partly obstructed due to overlap of the H2 and H3 resonances in the 1H NMR spectrum. However, a stronger correlation between the methyl group and H5pro-R than between the methyl group and H5pro-S was observed, which indicates an axial orientation and consequently the 4-(R)-configuration. For compound 4b, NOEs of similar magnitudes were observed for the signals of H3, H5pro-R, and H5pro-S, when irradiating the methyl resonance, which clearly indicates an equatorial orientation of the methyl group and thus the 4-(S)-configuration. Selective excitation of the methyl group in compound 6b gave a nearly identical NOE result to that observed for compound 4b.
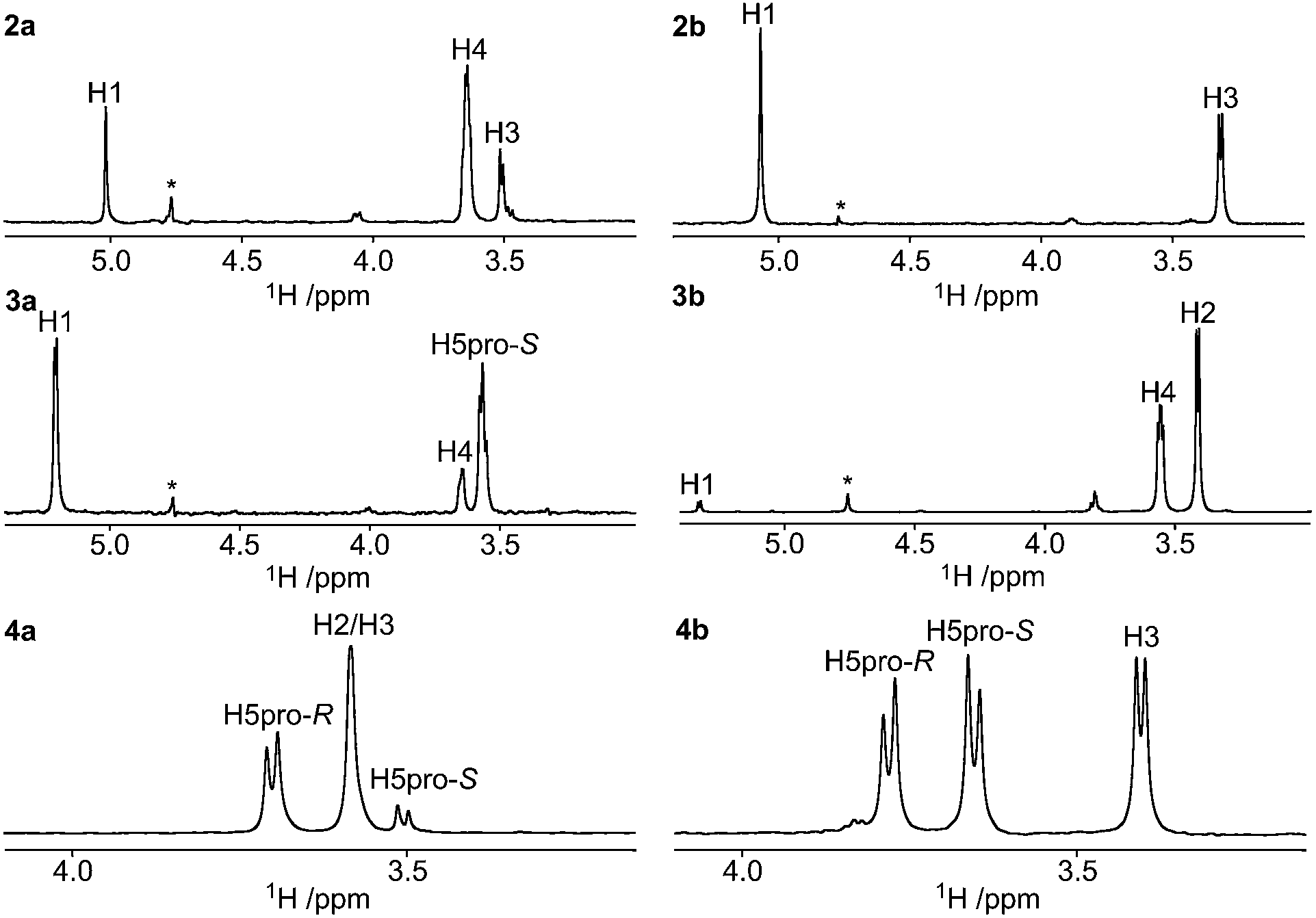 | ||
| Fig. 2 Selected regions of 1D 1H,1H-NOESY spectra of compounds 2a–b, 3a–b, and 4a–b (top to bottom) in methanol-d4 as solvent. Selective excitation was carried out on the proton resonance from the C-methyl group using a τmix of 0.41 s in the experiment. An asterisk marks residual CD3OH solvent peak. | ||
According to Karplus-type equations,31–33 the 3JCH coupling constants from the 13C nuclei of axially oriented methyl groups to axial vicinal protons should be larger compared to those from equatorially oriented methyl groups. Selected 3JCH coupling constants were measured via the 1DLR experiment,34,35 that relies on selective excitation of 13C resonances followed by evolution of the heteronuclear coupling and detection on the 1H nuclei. Heteronuclear coupling constants were determined by modified J doubling in the frequency domain.36 In all cases of the pairwise comparisons for the diastereomeric pairs, 2a and 2b, 3a and 3b, as well as 4a and 4b, we observed larger 3JCH coupling constants for 2a, 3a, and 4a, compared to 2b, 3b, and 4b. This is consistent with an antiperiplanar arrangement in the former set of molecules. The observed 3JCH coupling constants for compound 6b were similar to those observed for compound 4b, verifying the 4-(S)-configuration of 6b.22
In all diastereomeric pairs, the 13C chemical shifts of axial methyl groups were shifted upfield compared to those of equatorial methyl groups,37 with inter-pair differences ranging from 2–6 ppm. The 1H chemical shifts of axial protons in γ-position to the methyl groups were shifted downfield when the methyl groups were oriented equatorially, hydroxyl groups thus being axial, in compounds 2b, 3b, and 4b. This is in accordance with what is seen for δH5 of hexapyranosides with respect to a change in anomeric configuration.38 Typically, a downfield chemical shift displacement ΔδH of 0.2 ppm was observed upon a 1,3-syn-axial interaction with an oxygen atom.
Deviations from the 4C1 conformation can be observed and quantified to some extent by 3JHH coupling constant analysis. The compounds bearing axial methyl groups, 2a, 3a, and 4a, have smaller 3JHH coupling constants than their respective diastereoisomers. The maximum values of 3JH1,H2, 3JH2,H3, 3JH3,H4, and 3JH4,H5pro-S, correspond to the 4C1 conformation; thus, the observed coupling differences arise due to population contributions of non-4C1 conformers for these compounds. This is in agreement with the expected larger 1,3-diaxial steric repulsion of a methyl group compared to a hydroxyl group. The larger coupling constant deviations observed for compound 3a, compared to all other compounds reflect, with respect to steric repulsion, the significance of the number of 1,3-diaxial hydrogen atoms in relation to the axial methyl substituent, two for 3a and one for 2a and 4a.
Plausibly populated non-4C1 conformers are the skew conformers 1S5 and 2SO, and the 1C4 conformation. The 2SO conformation has previously been shown to be present in xylose derivatives.19 In compound 4a, the methyl group would be oriented pseudo-equatorially in the 2SO conformation. In compound 2a, however, the 2SO conformation would increase steric interactions between the methyl group and H5pro-R. Rather, the 1S5 conformation seems beneficial for compound 2a, by placing the methyl group in pseudo-equatorial orientation and the anomeric substituent in pseudo-axial orientation. For compound 3a, the 1S5 and/or 2SO conformations would be feasible non-4C1 states. The 1C4 conformation would relieve steric strain in compounds 2a, 3a, and 4a by orienting the respective methyl groups equatorially, and benefit from the anomeric effect by placing the exo-anomeric oxygen in an axial orientation. Estimates based on the available 3JHH data suggest that up to about 20% of the conformational space for compounds 2a and 3a are occupied by non-4C1 conformations, whereas up to 10% is the suggested figure for compound 4a and <10% for compounds 2b, 3b, 4b, and 6b.
Employing the above described methodology to additional xyloside compounds resulted in >90% of the 4C1 conformation for 4f, solely the 4C1 conformation for 4g, an equilibrium between the 4C1, 2SO, and 1C4 conformations being ∼1/4, ∼1/4, and ∼1/2, respectively, for 5a where the 2SO conformation is supported by a conspicuous NOE between the H5 and H2 protons,39 and solely the 4C1 conformation for 5b.
To summarize, we have unequivocally determined the configurations of each stereocenter in the synthesized target compounds described above (Schemes 1 and 2). Furthermore, we observed that the majority of our xyloside analogs preferentially adopts the 4C1 conformation. Compounds bearing axial methyl groups are exceptions, and for compound 5a, the 1C4 conformation seems to be most populated.
β4GalT7 assay
A truncated version of β4GalT7 fused with glutathione S-transferase (GST) was expressed in E. coli, yielding 12 mg of purified (>95% by SDS PAGE) protein per liter of growth medium. As previously reported,40 the truncated E. coli expression product of β4GalT7 fused to GST does not substantially affect substrate binding and catalytic properties of β4GalT7, compared to the membrane-bound mammalian product. This enabled us to use the bacterial product as a model for the mammalian enzyme.XylNap (1a) is known to be a primer of GAG chains, and is thereby a substrate for β4GalT7 (Fig. 1b). The activity of β4GalT7 was thus determined using XylNap as acceptor substrate and UDP-Gal as donor substrate. The reaction time and the amount of enzyme were chosen so that the production of disaccharide, that is GalXylNap, showed first-order dependence of these two parameters. The reaction progress was analyzed by HPLC using fluorescence detection. We thus observed a new peak with a shorter retention time than XylNap and with a mass spectra corresponding to GalXylNap (Fig. 3a). The recombinant β4GalT7 showed Michaelis–Menten kinetics for XylNap with a KM value of 0.78 mM (Fig. 3b). In accordance with the literature,41 the enzyme showed excess-substrate inhibition from UDP-Gal, and the KM value for UDP-Gal (0.35 mM) was comparable to previously reported data.40–42 The solubilities of the xylosides in DMSO limited the maximum concentration of substrate that could be added in the assay to approximately 2.5 mM.
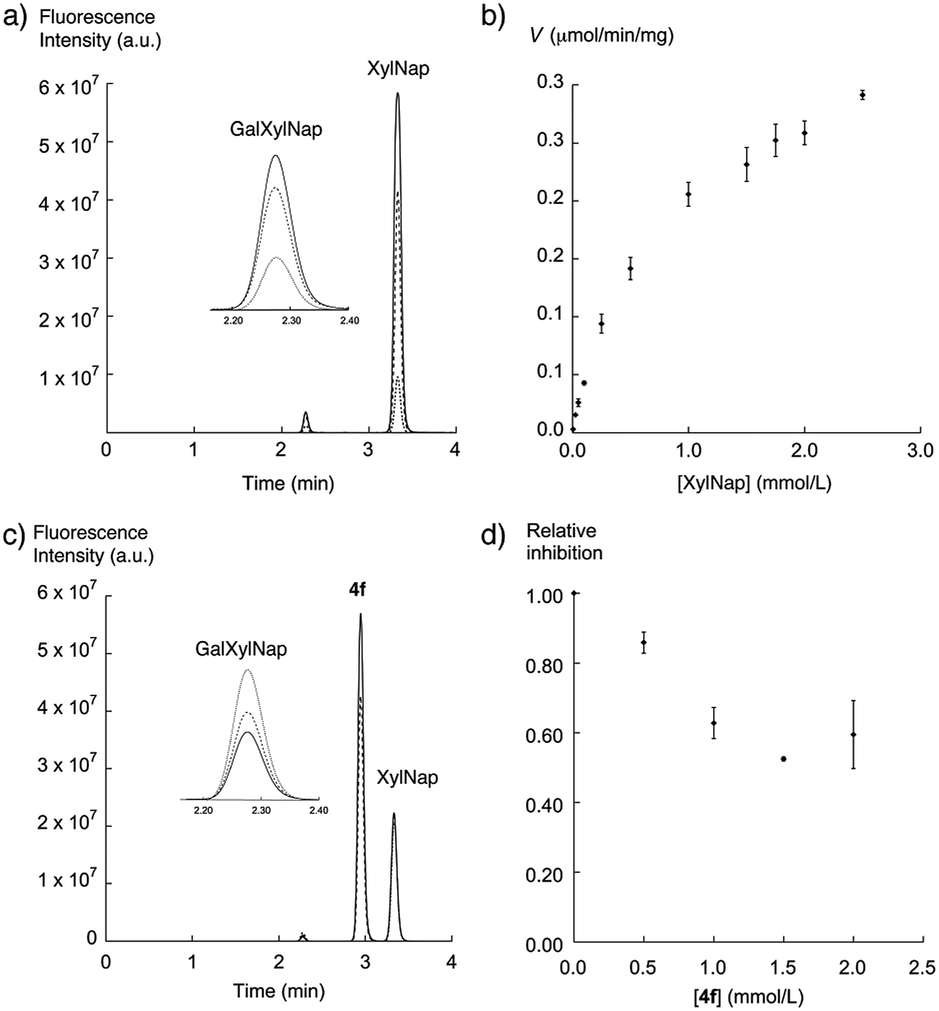 | ||
| Fig. 3 Galactosylation and inhibition of galactosylation by β4GalT7. (a) HPLC chromatograms for galactosylation experiments with XylNap. The GalXylNap peak is increasing with increasing concentrations of XylNap; 0.25 mM (dotted line), 1.0 mM (dashed line), and 2.0 mM (solid line). (b) Michelis–Menten representation of the activity of β4GalT7 (V) as a function of the XylNap concentration. (c) HPLC chromatograms for inhibition experiments with 4f. The GalXylNap peak is decreasing with increasing concentrations of 4f; 0 mM (dotted line), 0.5 mM (dashed line), and 1.5 mM (solid line). (d) Relative inhibition of GalXylNap plotted versus the concentration of 4f. The areas of the GalXylNap peaks, upon addition of 4f, are compared to the areas of the GalXylNap peaks without inhibitor. | ||
To determine if our synthesized xyloside analogs could serve as substrates for β4GalT7 (i.e., if the analogs are galactosylated), each compound was added as the only acceptor substrate in the β4GalT7 assay (Fig. 3c and d). As expected, most of these xyloside analogs failed to act as substrates for β4GalT7, with exceptions for compounds 2a, 2d, 2e, and 5a that showed weak galactosylation, compared to XylNap (Fig. 4, white bars). In each case of galactosylation, the mass spectra for the corresponding galactosylated products were determined, and were in all cases found to be accurate.
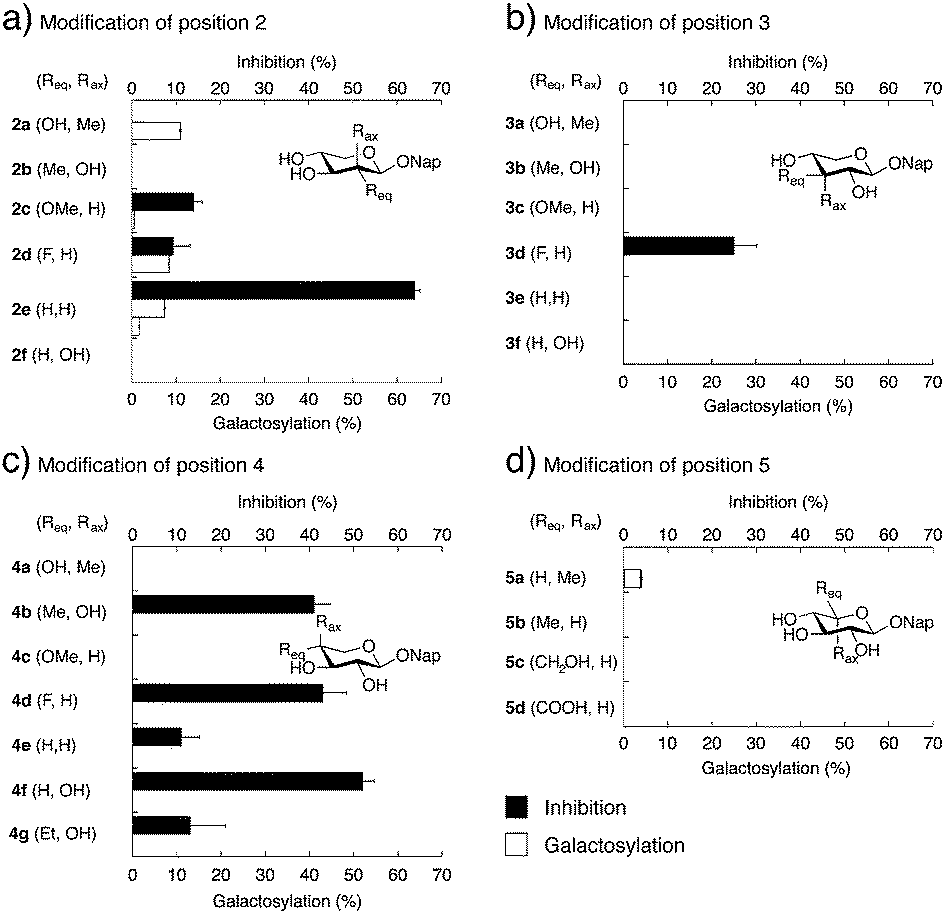 | ||
| Fig. 4 Galactosylation and inhibition of galactosylation by β4GalT7. White bars indicate amount of galactosylation (% of the effect shown by XylNap). Black filled bars show inhibition of galactosylation of XylNap (% of control without added xyloside analog; 0.5 mM of XylNap and 2.0 mM of xyloside analog). The y-axis displays compound number in bold face (the substituent in equatorial orientation, axial orientation). Compound 2e is displayed by two white bars. This compound is probably galactosylated at two different positions. | ||
Previous studies have shown GAG inhibitory effects of specific xyloside analogs.19–21 To determine the inhibitory activity of our compounds, XylNap was used as mimic of the natural substrate (i.e., the xylosylated PG core protein). A fixed amount of XylNap was added, together with different concentrations of the xyloside analogs, and the amount of formed GalXylNap was measured. A decrease in GalXylNap formation, compared to the control, indicates that the compound is an inhibitor of the enzyme (Fig. 3c and d). Interestingly, we observed a weak decrease in GalXylNap formation in the presence of compounds 2c, 2d, 3d, 4e, and 4g, and significant reduction of GalXylNap formation for compounds 2e, 4b, 4d, and 4f. The results are shown in Fig. 4 (black bars) and discussed below (detailed data in ESI†). To conclude, any modification of the xylose moiety rendered analogs unable to function as efficient substrates for β4GalT7, and several compounds showed effective inhibition of β4GalT7.
Effects on GAG priming in cells
As previously mentioned, the GAG priming abilities of XylNap (1a), as well as of compounds 2c–f, 3c–f, and 4c–f have been investigated earlier in HCC70 cells and CCD-1095Sk cells.19 Here, we investigated the GAG priming abilities of XylNap and compounds 2a, 2b, 3a, 3b, 4a, 4b, 4g, 5a, 5b, and 6b using XT deficient pgsA-745 cells. Since these cells are incapable of synthesizing complete PGs, they produce very small amounts of endogenous GAG chains. This make them suitable for investigations of xyloside priming. The cells were incubated with the xyloside analogs in low sulfate medium supplemented with [35S]sulfate. After 5 h of incubation, the GAG chains were isolated from the culture media and the amount of radioactivity in each sample was counted. Except for XylNap, no xyloside analog induced significant priming of GAG chains (data not shown).To investigate if the inhibitory effects observed in the β4GalT7 assay were transferable to cell culture studies, HCC70 cells were treated with either XylNap alone or with equimolar amounts (0.1 mM) of compounds 4b or 4d. After 24 h of incubation in low-sulfate medium, supplemented with [35S]sulfate and xyloside analogs, the GAG chains were isolated from the culture media. The samples were run on a Superose 6 column and the radioactivity was measured (Fig. 5). The addition of either 4b or 4d resulted in a decrease of GAG chains primed from XylNap, corresponding to 50% and 25%, respectively. To conclude, this indicates that the results from the enzymatic investigations of β4GalT7 (that is galactosylation and its inhibition) also can be observed in cellular systems.
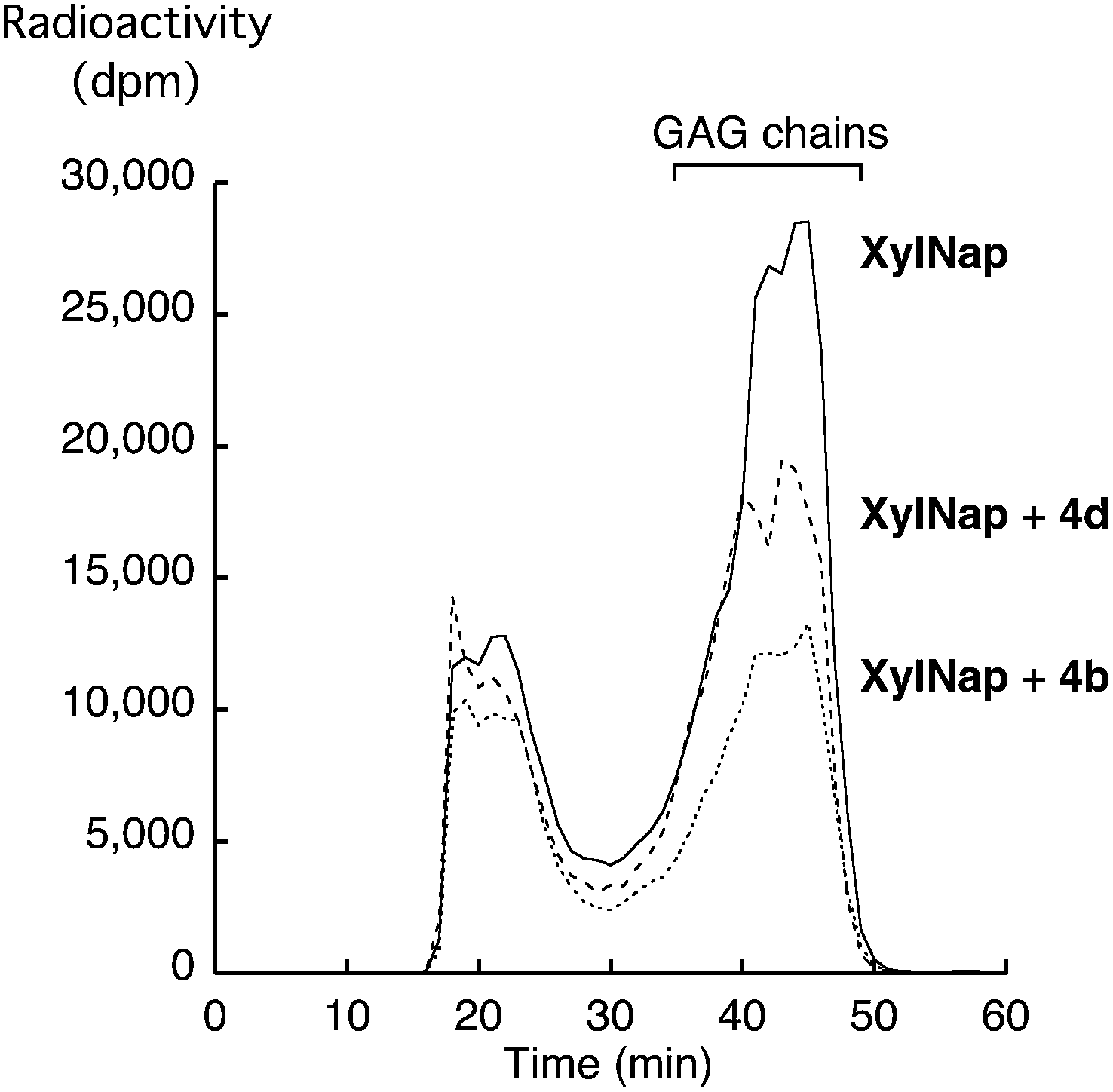 | ||
| Fig. 5 Superose 6-profiles for [35S]sulfate-labeled material isolated from HCC70 cells after treatment with XylNap (solid line), XylNap and 4d (dashed line), and XylNap and 4b (dotted line). | ||
Discussion
By combining our structure-activity relationship studies with the known crystal structure of β4GalT7, we can now provide a detailed model for binding of xylosides and xyloside analogs to β4GalT7.Most modifications of position 2 in xylose, rendered analogs less prone to galactosylation, compared to XylNap (1a). However, we observed weak galactosylation of compound 2a, carrying an axial methyl group, in contrast to 2b. The 2-fluoro analog 2d showed galactosylation and inhibition, whereas the 2-methoxy analog 2c showed inhibition similar to 2d and very weak galactosylation. This indicates that the hydroxyl group in position 2 might acts as a hydrogen bond acceptor. Interestingly, the 2-deoxy analog 2e showed formation of two different compounds. Compound 2e lacks substituents in both C2 and C5 of the xylose moiety, and thus shows pseudo-rotational symmetry. It is reasonable that it fits the active site in the enzyme in two different orientations and can be galactosylated at both position 3 and 4. Compound 2e, also showed very efficient inhibition of galactosylation (64%), possibly explained by the flexibility in binding orientation.
Modifications of position 3 resulted in inactive compounds, with the exception of the 3-deoxy-3-fluoro analog 3d. The fluoro substituent is similar in size as a hydroxyl group and can probably accept a hydrogen bond from Asp211, but with a reversed hydrogen bond pattern (Fig. 6). From inspection of the crystal structure,23 it is obvious that the binding pocket is narrow and compound 3c, carrying a sterically more demanding methoxy group, does not fit, and is thus inactive. The importance of the 3-OH is further validated by the deoxy compound 3e, which showed no galactosylation or inhibition.
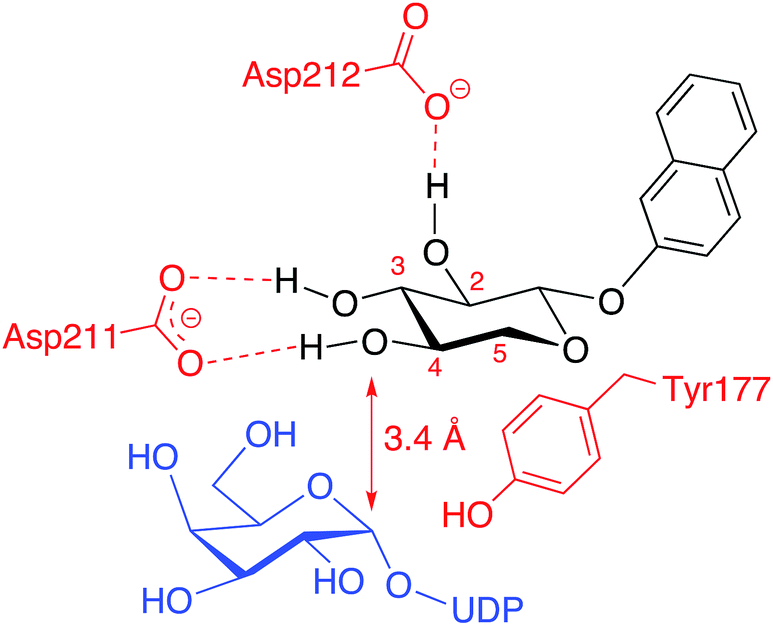 | ||
| Fig. 6 Proposed binding pattern of xylosides and UDP-Gal in the binding pocket of β4GalT7. | ||
From the perspective of inhibitor design, our results indicate that position 4 is the most interesting. The addition of an axial methyl group, compound 4a, rendered an inactive compound while the epimeric analog 4b, carrying an equatorial methyl group and an axial hydroxyl group, showed strong inhibition. In order to understand these results, we performed molecular modeling based on X-ray diffraction data of the Drosophila β4GalT7 mutant D211N (PDB ID 4M4K), using Schrödinger 2013 software suite, with the OPLS-2005 force field and GB/SA solvation for water. The xylose moiety of XylNap adopts a very similar orientation and binding mode as the xylobiose in the crystal structure (Fig. 7a).
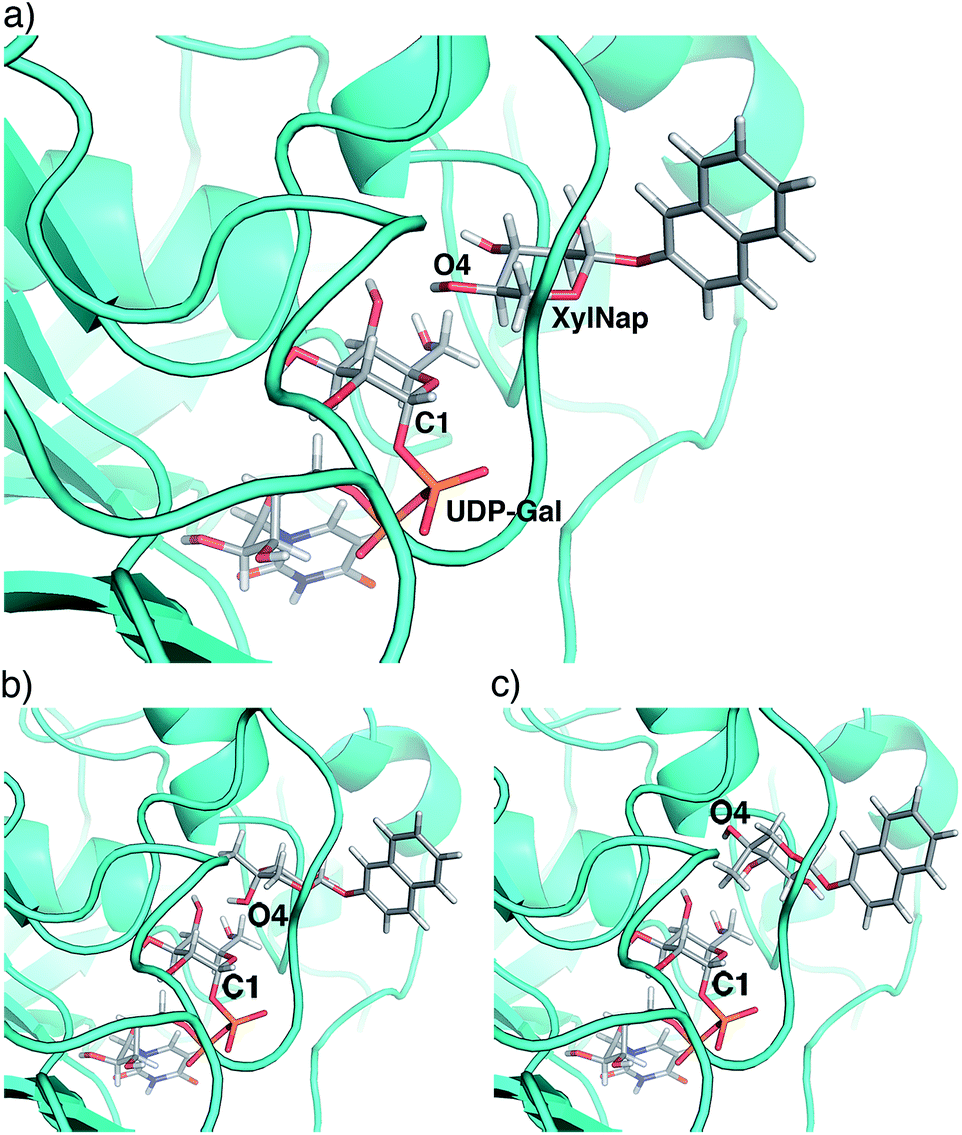 | ||
| Fig. 7 Molecular modeling of xylosides in β4GalT7. (a) O4 of the xylose moiety in XylNap (1a) is oriented for nucleophilic substitution on C1 of UDP-Gal. The xylose moiety adopts a very similar orientation and binding mode as the original xylobiose in the crystal structure. (b) In compound 4b, the axial 4-OH is situated in an orientation suitable for nucleophilic substitution, similar to XylNap. However, the loss of one hydrogen bond results in diminished nucleophilicity. (c) In 4a, the possibility for nucleophilic substitution is eliminated due to steric hindrance from the axial methyl group. | ||
From our conformational analysis vide supra, we conclude that compound 4b is locked to a 4C1 conformation. Interestingly, in this conformation, the axial 4-OH group is positioned in an orientation suitable for nucleophilic attack with ample space for the equatorial methyl group (Fig. 7b). However, in the axial orientation it is not possible to form the important hydrogen bond from 4-OH to Asp211, which results in diminished nucleophilicity of O4 of xylose moiety (Fig. 6). Consequently, this compound acts as an inhibitor of galactosylation rather than a substrate for galactosylation. On the contrary, the axial methyl group in compound 4a clearly eliminates the possibility for nucleophilic substitution due to steric hindrance, and this compound showed no galactosylation or inhibition (Fig. 7c).
As previously mentioned, Esko and co-workers reported a study of substituted benzyl xylosides and the amount of GAG priming on these compounds in pgsA-745 cells.22 As expected, most modifications of the 4-hydroxyl group, such as methylation, deoxygenation, or epimerization (Chart 1, compounds 6c–e), rendered inactive compounds. On the contrary, methylation of positions 2 or 3 gave analogs that initiated GAG priming, although at higher concentrations compared to XylOBn (1b). The 3-deoxy-3-fluoro analog induced priming at approximately 10-fold higher concentrations compared to the parent compound. Interestingly, the authors reported priming of GAG chains by benzyl 4-C-methyl-β-D-xylopyranoside (6a) at approximately 100-fold higher concentrations compared to XylOBn. Our attempts to synthesize compound 6a, using the same method as Esko and co-workers, resulted in the epimeric 6b, a compound very difficult to distinguish from 6a. As mentioned, when investigating the structurally similar compounds 4a and 4b, no significant galactosylation could be observed. Instead, compounds 4b and 6b acted as inhibitors of β4GalT7 and compound 6b showed 43% inhibition (ESI†).
It is worth to notice that the galactosylation of XylNap is very efficient and in our hands, using pgsA-745 cells, we observed maximum GAG priming on XylNap at concentrations of 0.1 mM. In our initial studies, we did observe significant priming for compounds 2b, 4b, and 6b at 1 mM in pgsA-745 cells, very similar to the results of Esko and co-workers. However, upon close inspection using fluorescence detection, we realized that these samples were contaminated with trace amounts of XylNap. This was enough to give false dose response curves in our studies. After careful HPLC purification, we observed no significant GAG priming from these compounds (see ESI† for an elaborate discussion).
The absence of an equatorial methyl group, as in compound 4f, resulted in an even stronger inhibition (52%) whereas the addition of a sterically demanding ethyl group resulted in a diminished inhibitory effect (4g, 13%), compared to 4b (41%). We propose that xyloside analogs with axial hydroxyl groups in position 4 can act as strong inhibitors of galactosylation and that the corresponding equatorial groups induce steric hindrance and diminished inhibition.
As expected, in the absence of a hydroxyl group, the deoxy compound 4e and the 4-fluoro analog 4d did not show any galactosylation. However, 4d showed strong inhibition of galactosylation. These results were transferable to cell studies and we observed 50% inhibition of XylNap induced GAG synthesis in HCC70 cells by compound 4b and 25% by compound 4d. As previously mentioned, Kuberan and co-workers reported a 4-fluoro analog with up to 90% inhibition in CHO cells.20,21
The methyl analog 5a showed very weak galactosylation and no inhibition. Interestingly, this analog adopts the unusual 2SO conformation. It is reasonable to assume that the addition of an axial methyl group results in a compound that is tolerated by the enzyme, due to a conformational change, but that this analog is a less preferred substrate for β4GalT7. Any equatorial groups in position 5, compounds 5b–d gave complete loss of activity. These analogs are conformationally rigid and locked to the 4C1 conformation, in which the group in position 5 induces strong steric hindrance. From the crystal structure, it is apparent that both O3 and O4 of the xylose moiety form hydrogen bonds to the side chain of Asp211, whereas the hydrophobic moiety of Tyr177 interacts with C5 of xylose, and thus determines the specificity. Consequently, it was shown that a mutation of Tyr177 to Gly changes the specificity from xylose to glucose.23,24
Conclusions
We have synthesized a number of xyloside analogs for exploration of the β4GalT7 active site. The identity and conformation of these compounds have been determined by several NMR techniques in combination with molecular modeling. The abilities of these compounds to act as substrates or inhibitors of β4GalT7 have been investigated in a cell-free assay and further validated by cell studies.We conclude that the binding pocket of β4GalT7 is very narrow and decorated with a precise set of hydrogen bond acceptors. From our data, we propose that xylose is the optimal substrate for galactosylation. Every single hydrogen bond is necessary for efficient galactosylation of xylosides, and any modification renders less active or inactive compounds. Consequently, we anticipate that it is difficult to synthesize better substrates by modification of the xylose moiety. However, modifications of the aglycon could provide more efficient substrates.
In contrast to the limited options for design of GAG primers, we open up for development of very tight binders that can function as inhibitors of galactosylation, and presumably, depending on cell permeability, act as strong inhibitors of the GAG biosynthesis. Such compounds would be valuable tools for the exploration of GAG and PG biosynthesis in normal cells and tumor cells, and we speculate in that such inhibitors may be a starting point for development of pharmaceuticals. The main target for modifications of the xylose moiety is position 4, where a number of structural analogs show strong inhibition. Of special interest is epimerization of the 4-OH group, which results in potent inhibitors and consequently absence of galactosylation.
Acknowledgements
This work was supported by grants from the Alfred Österlund Foundation, the Crafoord Foundation, the Evy and Gunnar Sandberg Foundation, Greta and John Kock, the Gunnar Nilsson Cancer Foundation, the Heart & Lung Foundation, the Knut and Alice Wallenberg Foundation, the Koch Foundation, Lars Hiertas Memorial Foundation, the Magnus Bergwall Foundation, the Medical Faculty of Lund University, Lund University, the Royal Physiographic Society in Lund, Syskonen Svenssons Foundation for Medical Research, the Swedish Cancer Society, the Swedish Medical Research Council (11550), and the Swedish Research Council.Notes and references
- G. W. Yip, M. Smollich and M. Gotte, Mol. Cancer Ther., 2006, 5, 2139 CrossRef CAS PubMed.
- M. M. Fuster and J. D. Esko, Nat. Rev. Cancer, 2005, 5, 526 CrossRef CAS PubMed.
- R. Sasisekharan, Z. Shriver, G. Venkataraman and U. Narayanasami, Nat. Rev. Cancer, 2002, 2, 521 CrossRef CAS PubMed.
- D. Liu, Z. Shriver, G. Venkataraman, Y. El Shabrawi and R. Sasisekharan, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 568 CrossRef CAS PubMed.
- C. Götting, J. Kuhn, R. Zahn, T. Brinkmann and K. Kleesiek, J. Mol. Biol., 2000, 304, 517 CrossRef PubMed.
- R. Almeida, S. B. Levery, U. Mandel, H. Kresse, T. Schwientek, E. P. Bennett and H. Clausen, J. Biol. Chem., 1999, 274, 26165 CrossRef CAS PubMed.
- H. C. Robinson, M. J. Brett, P. J. Tralaggan, D. A. Lowther and M. Okayama, Biochem. J., 1975, 148, 25 CAS.
- N. B. Schwartz, L. Galligani, P. L. Ho and A. Dorfman, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 4047 CrossRef CAS.
- M. Jacobsson, U. Ellervik, M. Belting and K. Mani, J. Med. Chem., 2006, 49, 1932 CrossRef CAS PubMed.
- C. O. Abrahamsson, U. Ellervik, J. Eriksson-Bajtner, M. Jacobsson and K. Mani, Carbohydr. Res., 2008, 343, 1473 CrossRef CAS PubMed.
- T. A. Fritz, F. N. Lugemwa, A. K. Sarkar and J. D. Esko, J. Biol. Chem., 1994, 269, 300 CAS.
- M. Jacobsson, K. Mani and U. Ellervik, Bioorg. Med. Chem., 2007, 15, 5283 CrossRef CAS PubMed.
- M. Jacobsson, C. Winander, K. Mani and U. Ellervik, Bioorg. Med. Chem. Lett., 2008, 18, 2451 CrossRef CAS PubMed.
- R. Johnsson, K. Mani and U. Ellervik, Bioorg. Med. Chem., 2007, 15, 2868 CrossRef CAS PubMed.
- R. Johnsson, K. Mani and U. Ellervik, Bioorg. Med. Chem. Lett., 2007, 17, 2338 CrossRef CAS PubMed.
- S. O. Kolset, K. Sakurai, I. Ivhed, A. Overvatn and S. Suzuki, Biochem. J., 1990, 265, 637 CAS.
- F. N. Lugemwa and J. D. Esko, J. Biol. Chem., 1991, 266, 6674 CAS.
- X. V. Victor, T. K. Nguyen, M. Ethirajan, V. M. Tran, K. V. Nguyen and B. Kuberan, J. Biol. Chem., 2009, 284, 25842 CrossRef CAS PubMed.
- A. Siegbahn, U. Aili, A. Ochocinska, M. Olofsson, J. Rönnols, K. Mani, G. Widmalm and U. Ellervik, Bioorg. Med. Chem., 2011, 19, 4114 CrossRef CAS PubMed.
- D. R. Garud, V. M. Tran, X. V. Victor, M. Koketsu and B. Kuberan, J. Biol. Chem., 2008, 283, 28881 CrossRef CAS PubMed.
- Y. Tsuzuki, T. K. Nguyen, D. R. Garud, B. Kuberan and M. Koketsu, Bioorg. Med. Chem. Lett., 2010, 20, 7269 CrossRef CAS PubMed.
- F. N. Lugemwa, A. K. Sarkar and J. D. Esko, J. Biol. Chem., 1996, 271, 19159 CrossRef CAS PubMed.
- Y. Tsutsui, B. Ramakrishnan and P. K. Qasba, J. Biol. Chem., 2013, 288, 31963 CrossRef CAS PubMed.
- B. Ramakrishnan and P. K. Qasba, J. Biol. Chem., 2010, 285, 15619 CrossRef CAS PubMed.
- M. Krupicka and I. Tvaroska, J. Phys. Chem. B., 2009, 113, 11314 CrossRef CAS PubMed.
- B. Ramakrishnan, V. Ramasamy and P. K. Qasba, J. Mol. Biol., 2006, 357, 1619 CrossRef CAS PubMed.
- A. Ochocinska, A. Siegbahn and U. Ellervik, Tetrahedron Lett., 2010, 51, 5200 CrossRef CAS PubMed.
- S. C. Hung, S. R. Thopate and R. Puranik, Carbohydr. Res., 2001, 331, 369 CrossRef CAS.
- A. Debruyn, M. J. Anteunis, G. Aerts and E. Saman, Carbohydr. Res., 1975, 41, 290 CrossRef CAS.
- K. Stott, J. Keeler, Q. N. Van and A. J. Shaka, J. Magn. Reson., 1997, 125, 302 CrossRef CAS.
- R. Wasylishen and T. Schaefer, Can. J. Chem., 1973, 51, 961 CrossRef CAS.
- R. Aydin and H. Gunther, Magn. Reson. Chem., 1990, 28, 448 CrossRef CAS.
- J. E. Anderson and A. I. Ijeh, J. Chem. Soc., Perkin Trans. 2, 1994, 1965 RSC.
- T. Nishida, G. Widmalm and P. Sandor, Magn. Reson. Chem., 1995, 33, 596 CrossRef CAS.
- T. Nishida, G. Widmalm and P. Sandor, Magn. Reson. Chem., 1996, 34, 377 CrossRef CAS.
- F. del Río-Portilla, E. Sánchez-Mendoza, V. U. Constantino-Castillo and J. A. del Río, ARKIVOC, 2003,(xi), 213 Search PubMed.
- P.-E. Jansson, J. Lindberg and G. Widmalm, Acta Chem. Scand., 1993, 47, 711 CrossRef CAS PubMed.
- J. F. Stoddard, Stereochemistry of Carbohydrates, John Wiley and Sons, New York, 1971 Search PubMed.
- A. Canales, J. Angulo, R. Ojeda, M. Bruix, R. Fayos, R. Lozano, G. Gimenez-Gallego, M. Martin-Lomas, P. M. Nieto and J. Jimenez-Barbero, J. Am. Chem. Soc., 2005, 127, 5778 CrossRef CAS PubMed.
- I. Talhaoui, C. Bui, R. Oriol, G. Mulliert, S. Gulberti, P. Netter, M. W. H. Coughtrie, M. Ouzzine and S. Fournel-Gigleux, J. Biol. Chem., 2010, 285, 37342 CrossRef CAS PubMed.
- J. F. Garcia-Garcia, G. Corrales, J. Casas, A. Fernandez-Mayoralas and E. Garcia-Junceda, Mol. Biosyst., 2011, 7, 1312 RSC.
- F. Daligault, S. Rahuel-Clermont, S. Gulberti, M. T. Cung, G. Branlant, P. Netter, J. Magdalou and V. Lattard, Biochem. J., 2009, 418, 605 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental parts for configurational and conformational analysis, biological testing, and synthesis. See DOI: 10.1039/c4sc01244e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |