 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sunlight photocatalyzed regioselective β-alkylation and acylation of cyclopentanones†
Megumi
Okada
a,
Takahide
Fukuyama
a,
Keiichi
Yamada
a,
Ilhyong
Ryu
*a,
Davide
Ravelli
b and
Maurizio
Fagnoni
*b
aDepartment of Chemistry, Graduate School of Science Osaka Prefecture University, Sakai, Osaka 599-8531, Japan. E-mail: ryu@c.s.osakafu-u.ac.jp; Fax: +81 72 254 9695; Tel: +81 72 254 9695
bPhotoGreen Lab, Department of Chemistry, University of Pavia, viale Taramelli 12, 27100 Pavia, Italy. E-mail: fagnoni@unipv.it; Fax: +39 0382 987323; Tel: +39 0382 987198
First published on 8th May 2014
Abstract
Sunlight-induced direct regioselective β-alkylation of cyclopentanones with electron-deficient alkenes was accomplished by using tetrabutylammonium decatungstate (TBADT) as the catalyst. The regiochemistry can be rationalized by a polar transition state for an SH2 reaction. In the presence of CO, the reaction gave the three-component β-acylation product in good yield.
Introduction
The site-selective conversion of sp3 C–H bonds into C–C bonds remains a challenge in synthetic organic chemistry1 and in recent years, much attention has been directed to both transition metal-catalyzed2 and radical approaches to this goal.3 Of special note is a unique method to selectively convert β-C–H bonds (rather than α-C–H bonds) in ketones, reported by MacMillan and coworkers. This method employed radical generation from enamines formed via an organocatalytic reaction between the ketone and an amine.4 Alternatively, the ketone can be converted in situ to the corresponding α,β-unsaturated derivative and then functionalized.5We hypothesized that a photocatalyzed radical approach6 based on the use of tetrabutylammonium decatungstate (TBADT) as the catalyst,7–9 would be promising to promote the β-regioselective alkylation of cyclopentanones. In cyclopentanone (1a), the α-C–H bond is weaker than the β-C–H bond.10 However, we reasoned that β-selective C–H bond cleavage would be promoted by polar effects11 in a system requiring a highly polar SH2 (bimolecular homolytic substitution) transition state, as in the case of hydrogen abstraction by TBADT. When electronegative oxygen-centered radicals, such as those present in the excited decatungstate anion (cat*), abstract hydrogen from C–H bonds, the transition state should be polar in order to balance the positively charged carbon atom. Therefore, TS-a (Scheme 1), leading to Avia α-C–H cleavage from cyclopentanone, has to create an unstabilized electron-deficient α-carbon that can be regarded as an Umpolung type,12 rendering β-selective C–H bond cleavage to give BviaTS-b more feasible. Thus, we assumed that regioselective C–H cleavage would be followed by C–C bond formation which would be suitable for the straightforward β-C–H functionalization of cyclopentanone (1a). In this paper, we report that using TBADT as the photocatalyst, the reaction of cyclopentanones with electron-deficient alkenes proceeds with complete regioselectivity to give β-alkylated cyclopentanones. Interestingly, in most cases replacing artificial xenon light with sunlight irradiation led to similar or even better results. In addition, we also achieved TBADT-catalyzed β-acylation of cyclopentanone under CO pressurized conditions.
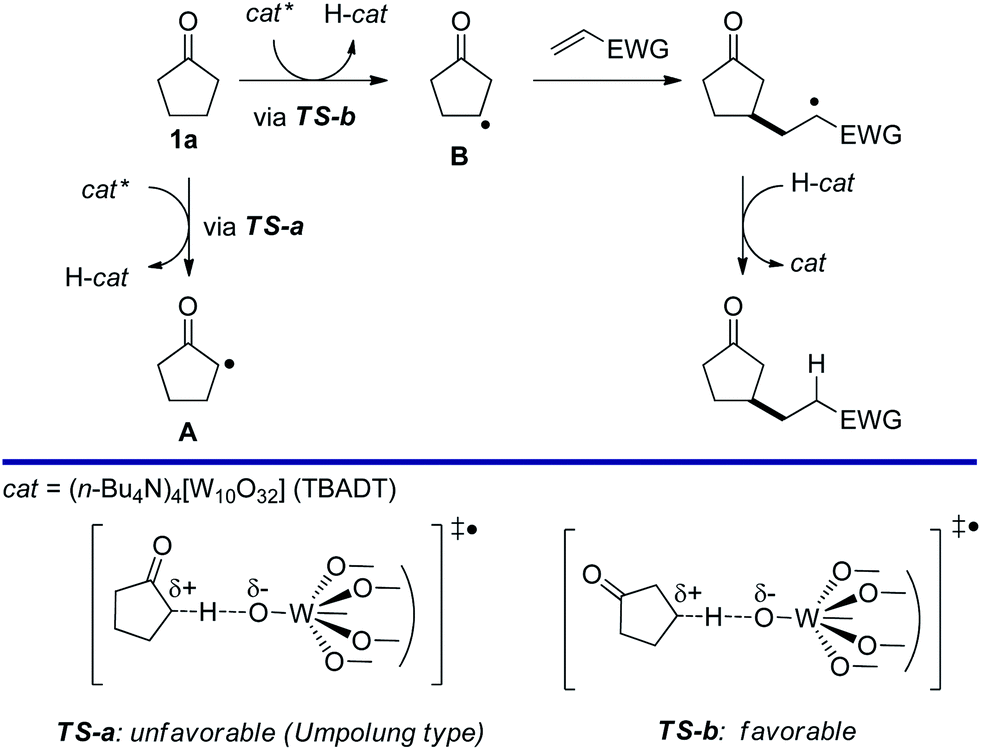 | ||
| Scheme 1 Concept: photocatalyzed β-selective alkylation of cyclopentanone based on the radical polar effect. | ||
Results and discussion
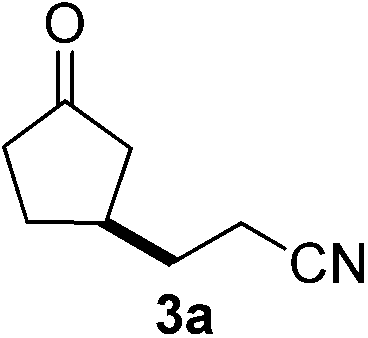
We screened the reaction between cyclopentanone (1a) and several electron-deficient alkenes as radical acceptors, including acrylonitrile (2a), methyl vinyl ketone (2b) and ethyl acrylate (2c). All these reactions resulted in an exclusive selectivity for β-C–H/C–C conversion (Scheme 2). As an example, when an acetonitrile solution of 1a (5 mmol), 2a (1 mmol) and TBADT (2 mol%) was irradiated for 24 h using a SolarBox equipped with a 1.5 kW xenon lamp (500 W m−2), β-alkylated ketone 3a was obtained in 61% yield. Not even trace amounts of 3a′ were detected in the 1H NMR spectrum of the crude product.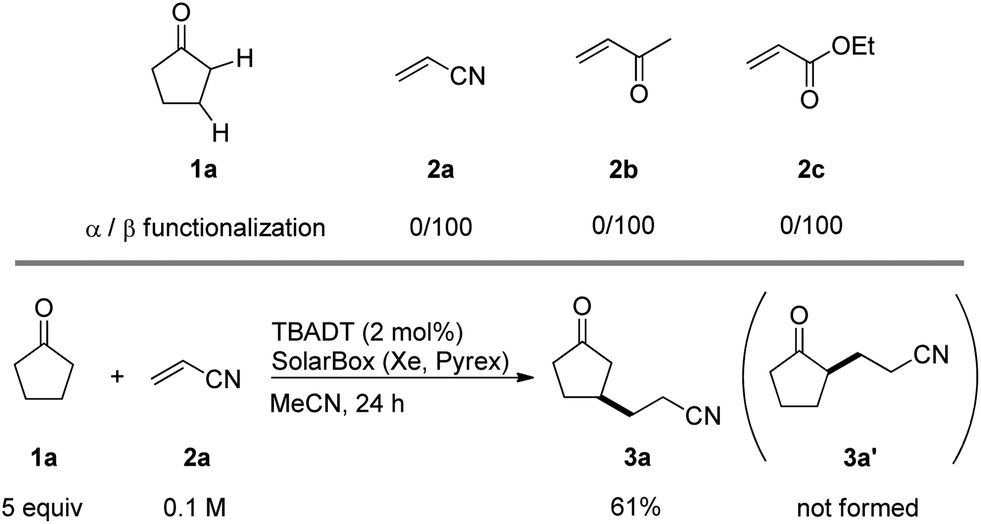 | ||
| Scheme 2 Regioselectivity in the TBADT-photocatalyzed C–H/C–C conversion in cyclopentanone (1a). | ||
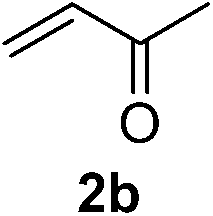
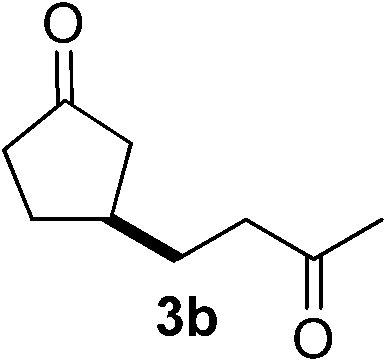
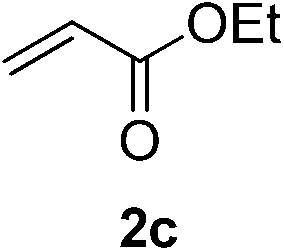
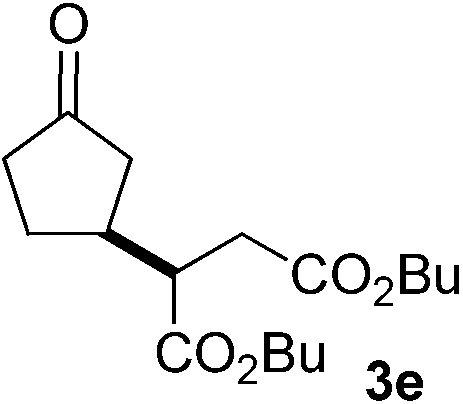
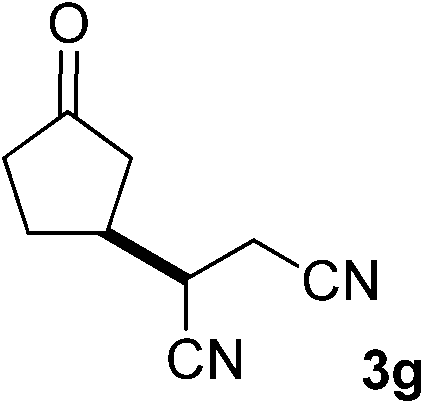
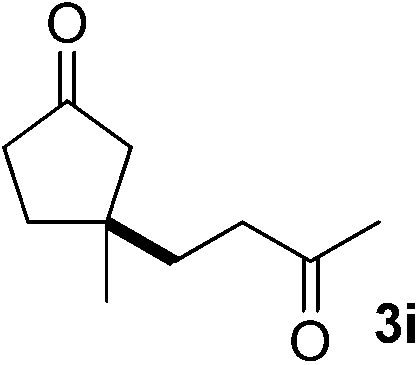
With these favorable results in hand, we then examined the β-regioselective C–H/C–C reaction of cyclopentanones 1 with a variety of electron-deficient olefins 2, and the results obtained are summarized in Table 1. The reaction of cyclopentanone (1a) with methyl vinyl ketone (2b) gave the expected 1,6-diketone 3b in 41% yield after isolation by silica gel chromatography (entry 2). Interestingly, direct sunlight irradiation of the reaction mixture (3 days, ca. 24 h) gave 3b in a higher yield (58%, entry 3). The reaction of 1a with ethyl acrylate (2c) or tert-butyl acrylate (2d) gave moderate yields of keto esters 3c and 3d, respectively (entries 4 and 5). The reaction between 1a and dibutyl maleate (2e) gave the corresponding ketone 3e in 61% yield as a mixture of 1.1/1 diastereomers (entry 6). The direct sunlight-promoted reaction of 1a with vinyl sulfone 2f and fumaronitrile (2g) gave β-alkylated products 3f and 3g in 70 and 73% yield, respectively (entries 7 and 8). Reaction of 1a with methylene norbornanone (2h) gave the corresponding 1,6-diketone 3h exclusively as the endo-isomer in a 1.7/1 diastereomer ratio (entry 9). Gratifyingly, the reaction of 3-methylcyclopentanone (1b), which has one methine, one methyl, and three different methylene carbons, with 2b proceeded with high selectivity for the abstraction of the β-methine C–H hydrogen, to afford 1,6-diketone 3i having a quaternary carbon (entry 10). Similarly, 1b reacted with vinyl sulfone 2f to give 3j in 62% yield (entry 11). We also examined the reaction of 2-methylcyclopentanone with 2b; however, it yielded an inseparable 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 4- and 3-alkylated products (see ESI†).
:1 mixture of 4- and 3-alkylated products (see ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | Alkene 2 | Methoda | Product 3 | Yieldb (%) |
| a Method A: 1 (5 mmol), 2 (1 mmol), TBADT (2 mol%), MeCN (10 mL), irradiation by a SolarBox equipped with a 1.5 kW Xe lamp (500 W m−2) for 24 h. Method B: 1 (5 mmol), 2 (1 mmol), TBADT (2 mol%), MeCN (10 mL), irradiation by sunlight for 3–5 days (8 h per day; see ESI1 for details). b Isolated yields by silica gel chromatography. If necessary, further purification was made by preparative HPLC. c TBADT (5 mol%), MeCN (5 mL), 12 h. d 12 h. e Determined by GC analysis. f Determined by 13C NMR and GC analysis. g Containing less than 5% of 4-alkylated product. | |||||
| 1 |
|
|
A |
|
61 |
| 2c | 1a |
|
A |
|
41 |
| 3 | B | 58 | |||
| 4 | 1a |
|
A |
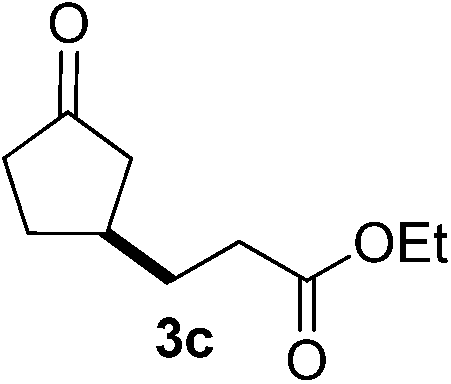
|
46 |
| 5d | 1a |
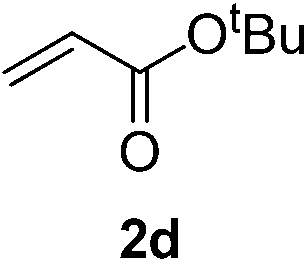
|
A |
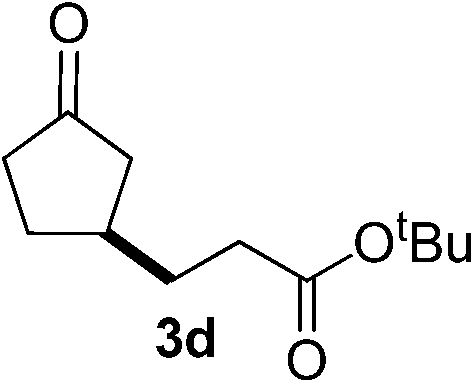
|
51 |
| 6 | 1a |
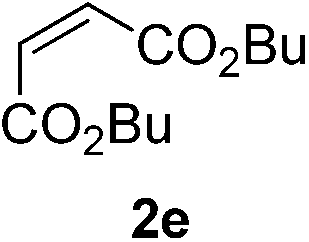
|
A |
|
61 (d.r. 1.1/1)e |
| 7 | 1a |
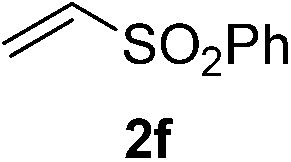
|
B |
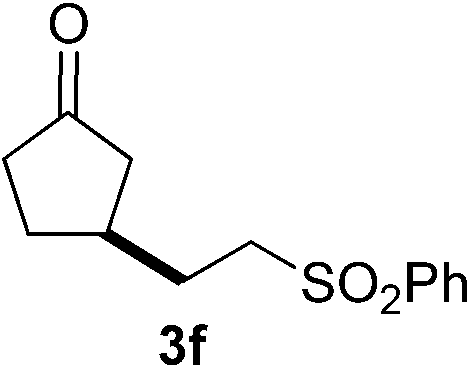
|
70 |
| 8 | 1a |
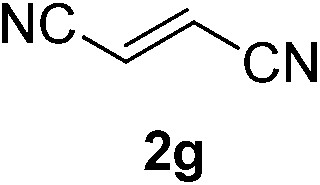
|
B |
|
73 (d.r. 1/1)f |
| 9 | 1a |
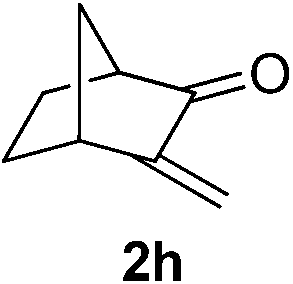
|
B |
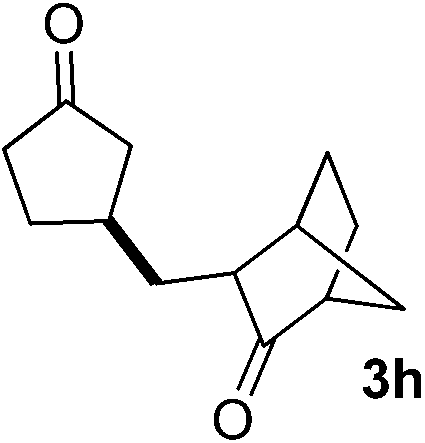
|
55 (only endo-, d.r. 1.7/1)f |
| 10 |
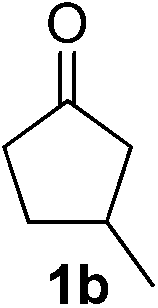
|
2b | A |
|
44g |
| 11 | 1b | 2f | A |
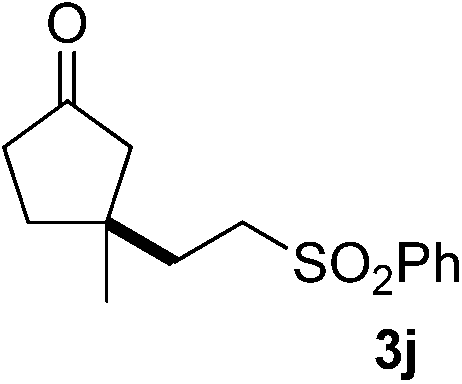
|
62g |
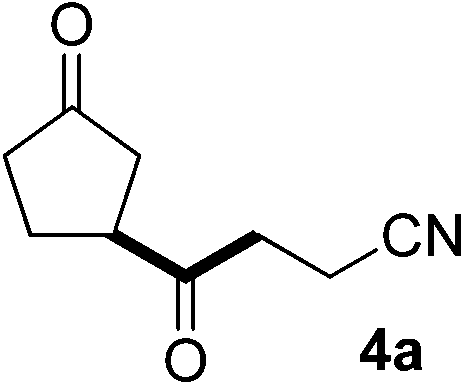
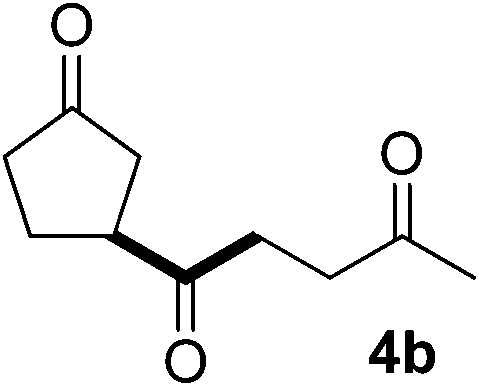
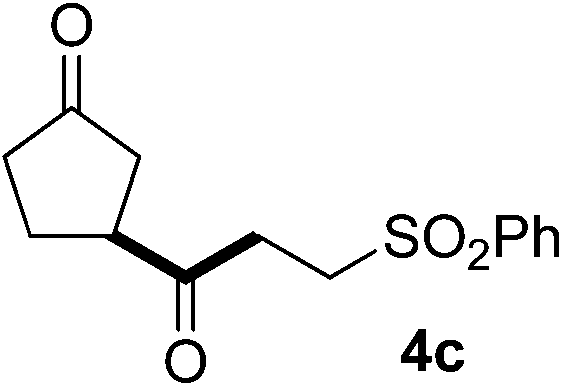
In our previous work, we have shown that radical carbonylations were feasible under TBADT-photocatalyzed conditions.9 Indeed, β-acylation of cyclopentanone (1a) under pressurized CO proceeded well. In this reaction the β-keto radical formed was trapped consecutively by CO and electron-deficient alkenes (Table 2).13 Thus, when an acetonitrile solution of 1a, 2a and TBADT (2 mol%) was irradiated for 20 hours under CO (200 atm) using a 300 W xenon lamp, the anticipated β-acylated product, diketo nitrile 4a, was obtained in 58% yield (entry 1). Similarly, 1,4,7-triketone 4b was obtained in 50% yield by a three-component reaction comprising 1a, CO and 2b (entry 2). The reaction with vinyl sulfone 2f gave the corresponding diketo sulfone 4c in 61% yield (entry 3).
For comparison, we examined the regioselectivities for cyclohexanone (1c), 4-methylcyclohexanone (1d) and cycloheptanone (1e) in the reaction with 2b (Scheme 3). As a result, when both β- and γ-hydrogens were present, H-abstraction by TBADT showed a slight preference for γ-C–H cleavage (statistically corrected γ:β ratio ca. 1:0.8) with cyclohexanone (1c) and cycloheptanone (1e) and, as expected, no α-C–H/C–C conversion took place. On the other hand, the presence of a methine group in compound 1d led to preferential selectivity (16:1, again statistically corrected) for the more hindered/labile γ-C–H bond, despite the bulkiness of the photocatalyst. This behavior is quite similar to that observed above in the case of 1b. In the reaction of 2-pentanone (1f) with methyl acrylate (2i), a selectivity for β-C–H over γ-methyl C–H was found (statistically corrected γ:β ratio ca. 1:8.5).
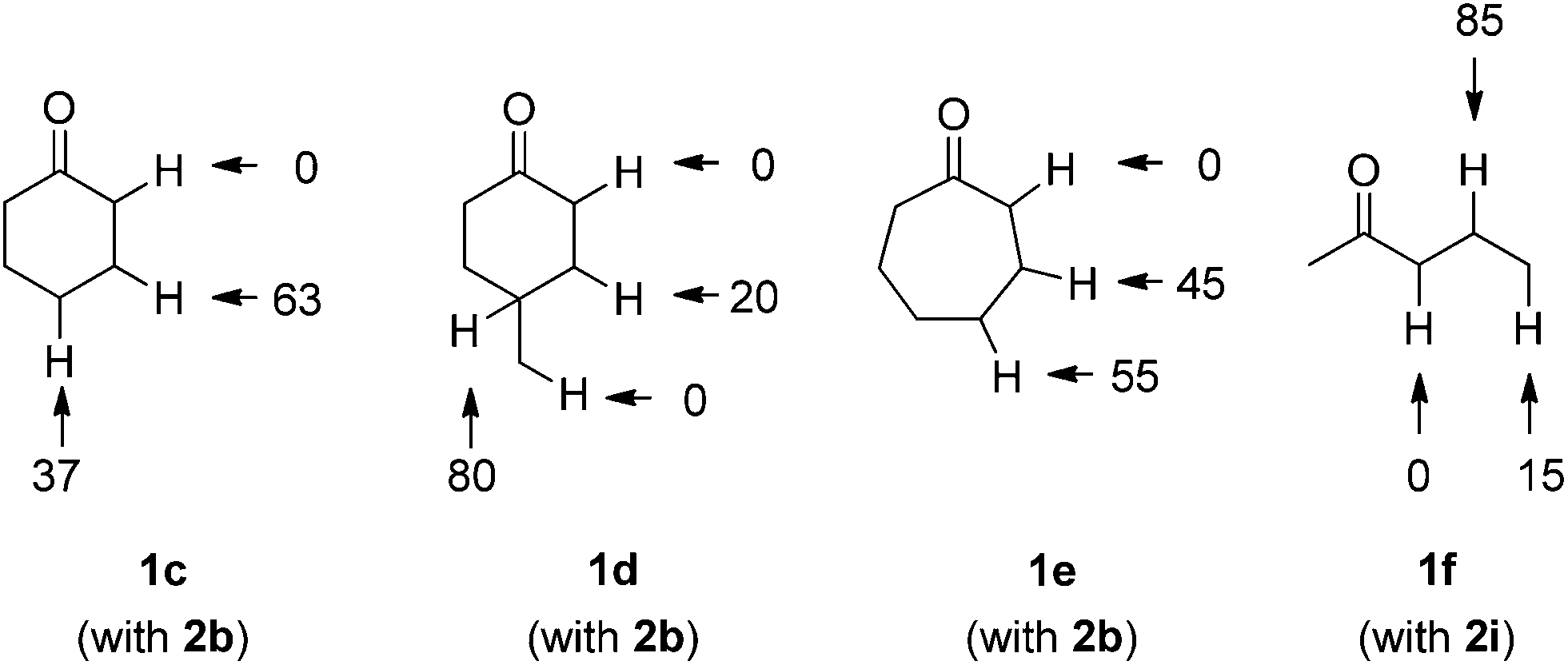 | ||
| Scheme 3 Selectivity in the C–H photocatalyzed cleavage in cyclohexanone (1c), 4-methylcyclohexanone (1d), cycloheptanone (1e) and 2-pentanone (1f). | ||
The approach described here compares favorably with related radical processes previously described for C–H functionalization in cycloalkanones. Sparse examples have been reported dealing with the reaction of ketones with hetero-atom radicals.14 Among them, there is only one report of selective β-chlorocarbonylation of carbonyl derivatives, which took place by irradiation in the presence of oxalyl chloride via hydrogen abstraction by chlorine (Cl˙) radicals.14a
A plausible mechanism is depicted in Scheme 4 for the photocatalyzed β-acylation of 1a. We expected that excited polyoxotungstate anion [W10O32]4−* would have an electronegative oxygen character as the reactive site,15 and therefore the observed selectivity is in favor of β-C–H abstraction (at least for 1a).16 The β-keto radical thus formed then undergoes consecutive addition to CO (when present) and to the electron-deficient olefin (e.g. acrylonitrile 2a) to form an adduct radical. Back-hydrogen atom transfer from the reduced form of the tungstate anion to the latter radical gives the desired product 4a, restoring the TBADT catalyst.
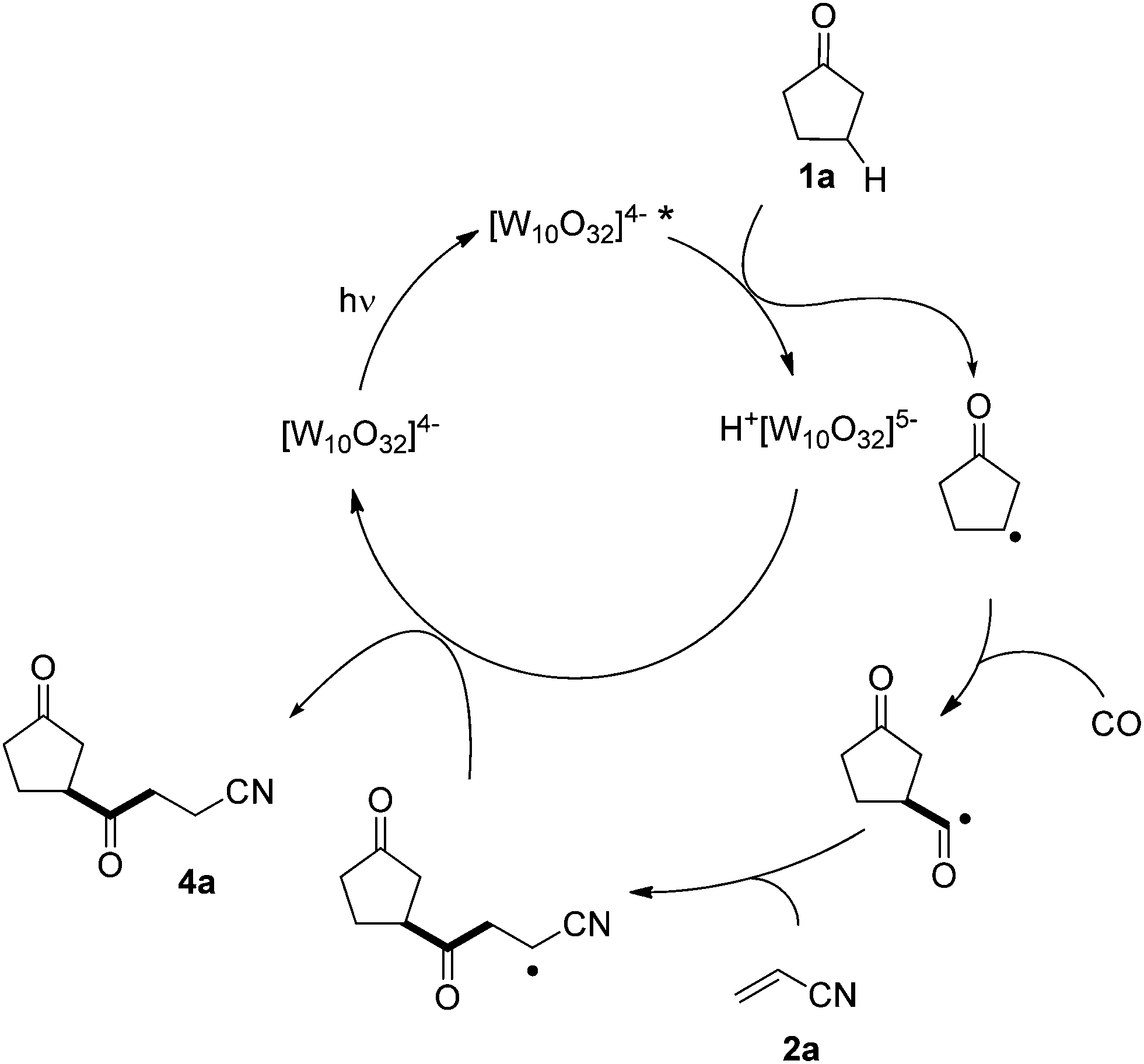 | ||
| Scheme 4 Proposed mechanism for the photocatalyzed β-C–H/C–C conversion in cyclopentanone and ensuing carbonylation. | ||
We can then assume that α-C–H abstraction in cyclic ketones is inefficient, as previously demonstrated in related studies on the reactivity of excited TBADT with acetone.17 In fact, acetone was safely used as a solvent in TBADT photocatalyzed reactions.8e,17
The simplicity of the procedure is another advantage of the method, since it requires merely exposing the solution to the sun in a glass vessel on a window ledge (Fig. 1).8e This 100% atom economical process has several benefits from the environmental point of view, because no artificial energy is required for irradiation, heating, cooling or stirring in order to carry out selective C–H/C–C conversion.18
 | ||
| Fig. 1 Pyrex glass vessel (20 mL capacity) used for the sunlight photocatalyzed β-C–H/C–C conversion of 1a. | ||
Conclusions
In summary, we have developed a simple straightforward strategy for β-alkylation and acylation of cyclopentanones using electron-deficient alkenes as alkylating reagents under irradiation in the presence of TBADT as the catalyst. Interestingly, sunlight irradiation gave good results. The differentiation of favorable and unfavorable polar radical transition states has been exploited successfully in order to direct the regioselectivity. β-Acylation of cyclopentanone, which uses a three-component system comprising cyclopentanone, CO and electron-deficient alkenes, was also achieved. We believe that the radical polar effect provides a powerful strategy to induce selective catalytic C–H functionalization and we are now pursuing further studies along this line.Experimental section
Typical procedure for β-alkylation of cyclopentanone (method A)
A magnetic stir bar and tetrabutylammonium decatungstate (TBADT, 66.4 mg, 0.02 mmol) were placed in a Pyrex flask. CH3CN (10 mL), cyclopentanone (1a, 420.6 mg, 5 mmol) and acrylonitrile (2a, 53.1 mg, 1.0 mmol) were added to the reaction flask. The resulting solution was purged with nitrogen, then screw-capped and irradiated by a SolarBox (xenon lamp, 500 W m−2, 53 °C) under stirring for 24 h. After the reaction, the solvent was evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel (hexane/ethyl acetate = 3/1) to give 83.7 mg of 3-(3-oxocyclopentyl)propanenitrile 3a (61%).Typical procedure for sunlight-induced β-alkylation of cyclopentanone (method B)
Tetrabutylammonium decatungstate (TBADT, 66.4 mg, 0.02 mmol) was placed in the Pyrex flask (Fig. 1). CH3CN (10 mL), cyclopentanone (1a, 420.6 mg, 5 mmol) and methyl vinyl ketone (2b, 70.1 mg, 1.0 mmol) were added to the reaction flask. The resulting solution was purged with nitrogen, then screw-capped and irradiated by exposing the reaction vessel to solar light on a window ledge of the Department of Chemistry of Pavia (Italy; 45° 10′ 00′′ North, 9° 10′ 00′′ East) in the July–September 2013 period for 8 h per day (from 9 a.m. to 5 p.m.). After the reaction, the solvent was evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel (hexane/ethyl acetate = 5/1) to give 89.4 mg of 3-(3-oxobutyl)cyclopentanone 3b (58%).Typical procedure for β-acylation of cyclopentanone
A magnetic stir bar, CH3CN (10 mL), TBADT (66.4 mg, 0.02 mmol), cyclopentanone (1a, 1262 mg, 15 mmol) and acrylonitrile (2a, 53.1 mg, 1 mmol) were placed in a stainless steel autoclave equipped with a Pyrex glass liner to permit irradiation of the contents. The autoclave was closed, purged three times with carbon monoxide, pressurized with 200 atm of CO and then irradiated by a xenon arc lamp (300 W) with stirring for 20 h. After the reaction, excess CO was discharged at room temperature. The solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel (hexane/ethyl acetate = 1/2) to afford 95.8 mg of 4-oxo-4-(3-oxocyclopentyl)butanenitrile 4a (58%).Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research from the MEXT and the JSPS. We thank Prof A. Albini (University of Pavia) for fruitful discussions.Notes and references
- For general reviews on C–H functionalization, see:
(a) J.-Q. Yu and Z. Shi, Top. Curr. Chem., 2010, 292, 1–380 Search PubMed
; (b) G. Dyker, Handbook of C–H Transformations, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed
- For recent reviews on transition metal-catalyzed C(sp3)–H functionalization, see:
(a) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed
- For recent reports on radical mediated C(sp3)–H/C–C functionalization, see:
(a) T. Hoshikawa and M. Inoue, Chem. Sci., 2013, 4, 3118 RSC
- For C(sp3)–H functionalization at β-position of carbonyl compounds via enamine formation using organocatalyst, see:
(a) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef CAS PubMed
- For indirect approaches via α,β-unsaturated carbonyl compounds, see:
(a) Z. Huang and G. Dong, J. Am. Chem. Soc., 2013, 135, 17747 CrossRef CAS PubMed
- For selected recent reviews on photocatalysis, see:
(a) M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727 CrossRef
- For reviews on decatungstate photocatalysis, see:
(a) M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609 RSC
- For selected examples of synthetic applications using decatungstate photocatalyst, see:
(a) H. Qrareya, D. Ravelli, M. Fagnoni and A. Albini, Adv. Synth. Catal., 2013, 355, 2891 CrossRef CAS
- For TBADT photocatalyzed carbonylation of alkanes, see:
(a) I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, S. Montanaro and M. Fagnoni, Org. Lett., 2013, 15, 2554 CrossRef CAS PubMed
- F. G. Bordwell, T. Gallagher and X. Zhang, J. Am. Chem. Soc., 1991, 113, 3495 CrossRef CAS
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25 RSC
- B. T. Gröbel and D. Seebach, Synthesis, 1977, 357 CrossRef
- For recent examples of radical carbonylation, see:
(a) T. Fukuyama, N. Nakashima, T. Okada and I. Ryu, J. Am. Chem. Soc., 2013, 135, 1006 CrossRef CAS PubMed
-
(a) A. Bashir-Hashemi and J. R. Hardee, J. Org. Chem., 1994, 59, 2132 CrossRef CAS
- D. Dondi, D. Ravelli, M. Fagnoni, M. Mella, A. Molinari, A. Maldotti and A. Albini, Chem.–Eur. J., 2009, 15, 7949 CrossRef CAS PubMed
- In the present system, α-C–H cleavage could occur in an equilibrium with back H-donation.
- C. Tanielian, F. Cougnon and R. Seghrouchni, J. Mol. Catal. A: Chem., 2007, 262, 164 CrossRef CAS PubMed
- The quantum yield of the process was estimated to be 0.04 based on an average flux of 30 W m−2 in the 295–400 nm range where the absorption of the photocatalyst is complete. This value is in agreement with that previously reported in the UV light-promoted activation of C–H hydrogens in cyclohexane (0.06, see: D. Dondi, M. Fagnoni and A. Albini, Chem.–Eur. J., 2006, 12, 4153 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, and analytical data for all new compounds. See DOI: 10.1039/c4sc01072h |
| This journal is © The Royal Society of Chemistry 2014 |