 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anisotropic highly-conductive films of poly(3-methylthiophene) from epitaxial electropolymerization on oriented poly(vinylidene fluoride)†
Dianming
Sun
a,
Yongxiu
Li
a,
Zhongjie
Ren
*a,
Martin R.
Bryce
*b,
Huihui
Li
a and
Shouke
Yan
*a
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: skyan@mail.buct.edu.cn; renzj@mail.buct.edu.cn
bDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 13th June 2014
Abstract
Electrochemical polymerization of 3-methylthiophene on highly oriented poly(vinylidene fluoride) (PVDF) film was achieved by cyclic voltammetry to yield well-ordered poly(3-methylthiophene) (P3MT) thin films with anisotropic structural and conductivity properties. The conductivity of P3MT along the direction perpendicular to the chain direction of PVDF, after electrochemical dedoping, is 59 ± 3 S cm−1, while that along the PVDF chain direction is 1.2 ± 0.4 S cm−1. The high conductivity of the P3MT is attributed to the well-ordered structure with its flat-on single crystals as confirmed by electron diffraction and Reflection Absorption Infra-red Spectroscopy (RAIRS). The data are consistent with P3MT chains aligned with π–π stacking perpendicular to the chain direction of the PVDF substrate. Epitaxial electropolymerization is an unusual method of preparing highly ordered thin films of organic semiconductors.
Introduction
The strategies for improving the performance of organic thin film electronic devices are diverse. Research is especially focused on synthesizing new active materials, device interface control, device structure modification and film morphology regulation.1–3 The control of morphology of the thin active layers in organic electronic devices by exploiting new deposition techniques is an essential and rational way towards optimization.4,5 An effective direction of charge transport is along a π–π stacking axis.6–8 This leads to anisotropic charge transport in crystals.9,10 Therefore, large-area well-ordered organic semiconductor thin films are expected to have excellent charge transport behavior in the specific π–π stacking direction.Electropolymerization (EP) with concurrent polymer film deposition has proved to be an especially useful method for the in situ preparation of electroactive and conducting polymer films.11–13 During the EP process, the precursor monomers are oxidized electrochemically; coupling reactions occur at the electrode surface, resulting in the formation of a polymer thin film on the electrode. The growth rate and thickness of the polymer films can be easily modulated by controlling the applied potential (or current density) and the total amount of charge passed through the cell, respectively. The morphology and properties of the EP films can also be optimized through choice of the EP conditions including preparation techniques and experimental parameters.14,15 For example, Ma et al. have recently developed a route utilizing electrochemical copolymerization and layer-by-layer polymerization to construct cross-linked networks, which are very promising candidates for applications in color-stable electroluminescent devices and solar cells.16–20
Electrochemical polymerization of thiophene and its derivatives has been researched extensively because of the outstanding optoelectronic properties of polythiophenes.21–25 However, the irregular molecular coupling and branching which usually occurs during traditional electrochemical deposition, is detrimental to the regularity of the conjugated polymer structure and prevents long-range π–π stacking. Thus the charge transport performance of the polymer is considerably reduced. Moreover, the crystalline domains in the electrochemically deposited polymer thin films are randomly oriented. It is therefore difficult to obtain thin films with optimal conductivity by electropolymerization.
Epitaxy is an efficient method for preparing well-defined thin films with controlled crystal structure and molecular orientation.26–29 Significant progress has been made in the epitaxial growth of organic semiconductive molecules on highly oriented polymer substrates.30–32 For example, in our previous work, vapour phase epitaxial growth of perylo[1,12-b,c,d]thiophene (PTH) on highly oriented polyethylene (PE) gave large-area well-arranged films of PTH with a unique crystal structure.33 From this background we considered that introducing epitaxy into an electropolymerization process could provide an unusual method for preparing highly ordered thin films of semiconductive materials. Goto et al. demonstrated that electrochemical polymerization of 2,7-di(2-furyl)fluorene in a macroscopically aligned nematic liquid crystal yields a uniaxially ordered polymer film.34 Electrochemical polymerization of bithiophene in a crystalline electrolyte has also been described.35 Sakaguchi et al. reported electrochemical epitaxial polymerization of 3-butoxy-4-methylthiophene on an iodine-covered gold electrode by applying voltage pulses. A surface-propagation mechanism gave single polythiophene wires which were observed by scanning tunnelling microscopy imaging.36 These studies34–36 did not report conductivity data for the aligned polymers. We are not aware of any previous studies on epitaxial electropolymerization induced by standard voltammetric cycling.
Herein, we report the preparation of highly oriented poly(3-methylthiophene) (P3MT) through epitaxial electrochemical deposition. To achieve this goal, highly oriented poly(vinylidene fluoride) (PVDF) ultrathin films were deposited on indium tin oxide (ITO) electrodes followed by electropolymerization of 3-methylthiophene on the PVDF modified ITO. In this process, epitaxial growth of an oriented P3MT layer is achieved on the PVDF thin film during electrochemical deposition; the film displays high conductivity with anisotropic charge transport. This is a new strategy for preparing anisotropic semiconductive polymer films.
Results and discussion
Electrodeposition and characterization
Highly oriented PVDF films were prepared according to a melt-draw technique introduced by Petermann et al.37,38Fig. 1a shows the polarized optical micrograph of a PVDF film, which shows weak birefringence since it is thin (30–50 nm). The drawing direction during film preparation is indicated by an arrow, i.e. the molecular chain direction of the PVDF is 45° relative to the polarization direction. When rotating the sample about the light beam axis by 45°, leading to a parallel alignment of the polarizer and PVDF drawing direction, extinction of the light takes place (see Fig. 1b) due to the high orientation of the PVDF film. | ||
| Fig. 1 (a) and (b) Polarized optical micrographs of a highly oriented PVDF film on ITO. The arrow indicates the drawing direction of the PVDF film during preparation, which is 45° (a) and 0° (b) relative to the polarizer direction. (c) Polarized optical micrograph of a P3MT film directly electrodeposited onto bare ITO glass and then dedoped. (d) The same sample shown in frame (c) but rotated by 45° about the light beam axis. | ||
P3MT was chosen as the semiconductive polymer as it is known to be readily obtained by standard electropolymerization of 3-methylthiophene.39–41 Electrochemical coupling of 3-methylthiophene was performed using cyclic voltammetry (CV).13 The PVDF layer is thin and combined with its dielectric properties electron transfer can occur readily to the electrode to enable the P3MT film to grow on the PVDF. A scanning electron microscopy (SEM) image of the glass-ITO-PVDF-P3MT assembly is shown in the ESI.† For comparison, 3-methylthiophene was first electropolymerized through potential sweeps between −0.2 and 1.0 V vs. Ag/Ag+ at 40 mV s−1 on bare ITO glass in a 0.5 mM chloroform–acetonitrile (3/2 v/v) solution containing 0.1 M Bu4NPF6 electrolyte. Then, the obtained P3MT film was reduced (dedoped) at 0 V for 2 h following reported procedures.42,43 The P3MT film changed from greenish-black to red upon dedoping. As shown in Fig. 1c and d, the resultant dedoped P3MT films exhibit an isotropic feature with birefringence that remains unchanged during the rotation of the sample about the light axis. Dedoping served to improve the crystallinity of the film; doped P3MT is known to form a disordered semicrystalline structure on ITO.42
The electrochemical polymerization potential of 3-methylthiophene on highly oriented PVDF covered ITO glass (1.22 V) is slightly higher than that on bare ITO glass (0.95 V) in the same electrolyte (Fig. S1†). Thus 3-methylthiophene was electropolymerized through potential sweeps between −0.2 and 1.2 V vs. Ag/Ag+ at 40 mV s−1 on PVDF modified ITO glass in a 0.5 mM chloroform–acetonitrile (3/2 v/v) solution. As shown in Fig. 2, both the anodic and cathodic peak currents increase in the successive cycles, indicating the coupling reaction of 3-methylthiophene units and the growth of the polymer film on the electrode.13,25,44 In the inset of Fig. 2a, the evolution of the oxidation peak current at ca. 0.6 V versus cycle number illustrates a linear increase of the polymerization of 3-methylthiophene on the electrode. In addition, the X-ray photoelectron spectrum (XPS) of dedoped P3MT on the PVDF film (Fig. 2b) shows the characteristic sulfur and fluorine peaks, with no detectable phosphorus (from the PF6− counterion at ca. 130 eV) or nitrogen (from the NBu4+ electrolyte at ca. 400 eV) indicating the successful deposition and dedoping of P3MT.
 | ||
| Fig. 2 (a) CV for the polymerization of 3-methylthiophene at a scan rate of 40 mV s−1. The first scan is shown in red. (b) XPS spectrum of the dedoped P3MT on PVDF film; the inset shows an expansion of the range from 50 to 250 eV. | ||
Fig. 3a and b present the polarized optical micrographs of a P3MT film electrochemically deposited onto oriented PVDF covered ITO glass and then dedoped. A regular structure with lathlike P3MT crystals of microns in length can be observed with strong birefringence. The crystals exhibit a well-ordered structure with their long axes aligned along the drawing direction of the PVDF film, indicating the high orientation of the P3MT crystals, i.e. the occurrence of epitaxial crystallization of P3MT on the highly oriented PVDF during electropolymerization, and not simply the growth of nanofibres. This is further confirmed by the occurrence of light extinction when the sample was rotated about the beam axis (see Fig. 3b). A possible driving force for epitaxial growth of the P3MT could be intermolecular hydrogen-bonding H(thiophene)⋯F(PVDF) interactions45 which align the growing polymer chains on the PVDF surface. This mechanism is conceptually similar to the proposal of Sakaguchi et al. that the oriented substrate-lattice structure of iodine adsorbed onto Au(111) facilitates an iodine–thiophene interaction leading to epitaxial polymerization.36 Roncali et al. also showed by SEM the existence of “parallel grooves” in P3MT films grown under standard voltammetric conditions on platinum electrodes.40 However, unlike the present study, these authors did not report anisotropy in the optoelectronic properties of the films.
 | ||
| Fig. 3 (a)–(c) polarized optical micrographs of P3MT films electrodeposited onto ITO glass covered with oriented PVDF film and then dedoped. The arrows indicate the stretching directions of the PVDF films during preparation, which is 45° (a), 0° (b) and 45° (c) apart from the polarizer direction. The deposition conditions were 40 mV s−1, 50 cycles for (a) and (b); and 10 mV s−1, 10 cycles for (c). An AFM image corresponding to (c) is shown in (d). An XRD profile of sample (a) is presented in (e) with an inset of the corresponding electron diffraction pattern of P3MT detached from the PVDF. | ||
The morphologies of the P3MT film can be well tuned by changing the condition of electrochemical deposition. As shown in Fig. 3c, with a low scan rate and few scan cycles, lathlike crystals with a width of 10–20 μm are obtained. These crystals do not cover the whole PVDF substrate because of the short scan time. The thickness of the P3MT crystals is ca. 0.6 μm, as determined by the atomic force microscopy (AFM) image shown in Fig. 3d. With increasing numbers of cycles, a two-dimensional close packed film composed of lathlike P3MT crystals is obtained, similar to that shown in Fig. 3a. The thickness of the P3MT crystals in Fig. 3a is also ca. 0.6 μm. P3MT has very poor solubility in organic solvents, so the degree of polymerization of the bulk material cannot be accurately determined by GPC. The degree of polymerization of the soluble portions of the P3MT film in tetrahydrofuran was found by GPC to be 16, with polydispersity ca. 1.3. The UV-Vis absorption spectrum of the dedoped film (λmax 510 nm) is consistent with previous data for electrochemically dedoped P3MT films,39 whereas the spectrum of the THF solution (λmax 400 nm) is assigned to oligomer chains with a lower degree of polymerization, based on literature precedents46,47 (see ESI†). Even thicker films of P3MT can also be produced by further increasing the number of CV cycles.
To probe the chain arrangement of P3MT in the lathlike crystals, X-ray and electron diffraction data were obtained. In the X-ray diffraction profile, Fig. 3e, except for the weak reflection peaks corresponding to the thin PVDF substrate and ITO glass (indicated by the asterisks), several peaks are observed for the P3MT with a main sharp peak located at 2θ = 11.5°. This reflects the successful deposition of P3MT on the PVDF surface. By detaching the P3MT from the PVDF modified ITO glass (see ESI†) the electron diffraction profile of the P3MT was obtained. As inserted in Fig. 3e, sharp and well-defined diffraction spots can be observed. All of these spots can be accounted for by a hexagonal unit cell with a-axis parameter of 0.886 nm. The appearance of only (hk0) diffraction spots indicates a flat-on orientation of P3MT crystals with molecular chains perpendicular to the substrate surface. Due to the difficulty in detaching the P3MT/PVDF double layers together from the ITO glass, no superimposed electron diffraction has been obtained. In this case, the mutual orientation between the P3MT and PVDF crystals cannot be determined.
To further confirm the arrangement of molecular chains of P3MT in the crystals, Reflection Absorption Infra-red Spectroscopy (RAIRS) was used. For RAIRS the resultant electric field vector is perpendicular to the metal surface. Therefore, if molecules are adsorbed onto the substrate with a preferred orientation, vibrational modes having transition moments perpendicular to the surface will appear with greater intensity than modes having transition moments parallel to the surface.48,49Fig. 4 displays the characteristic bands of P3MT. Herein, we focus on the out-of-plane deformation modes of thiophene Cβ–H at 825 cm−1 for which the transition moments are perpendicular to the P3MT backbones, and the methyl deformation mode at 1378 cm−1 which is used sometimes as the internal standard because the frequency and intensity of this vibration mode is not sensitive to structural changes.50,51 Comparing the FTIR (Fig. 4a) with RAIR spectra (Fig. 4b), the band of Cβ–H shifts to the higher wavenumber in FTIR. It has been reported that reflectance spectra clearly show large changes in both peak position and shape compared to transmission spectra.52,53 To gain qualitative information about the molecular orientation, we avoid the disturbance of the band distortions by comparing the peak area of bands at 825 and 1378 cm−1 (A825/A1378) in the same spectra. The value of A825/A1378 is greater in the FTIR (Fig. 4a) than that in RAIR spectra (Fig. 4b). On the basis of the mutually perpendicular direction of the electric field vector for transmission vs. p-polarized RAIR modes, we conclude that the main-chain of P3MT in the crystalline film is aligned perpendicular to the substrate. These data corroborate the polarized optical microscopy data in Fig. 3 and further establish the epitaxial orientation of P3MT at the molecular level, rather than a macroscopic fibre-like structure.
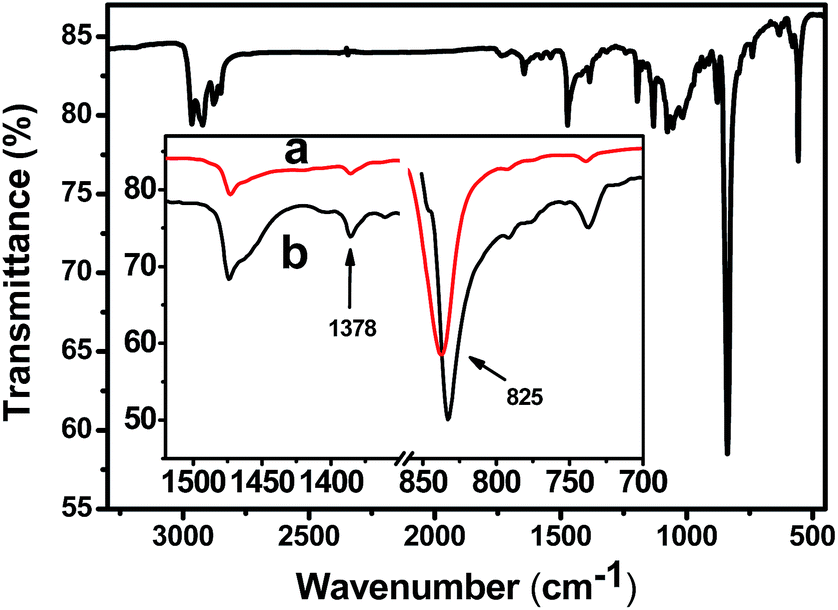 | ||
| Fig. 4 FTIR of dedoped P3MT on PDVF–ITO; the inset shows expanded spectra of (a) FTIR and (b) RAIR spectra. | ||
Conductivity measurements
The I–V characteristics of the dedoped P3MT film in different directions were measured using a two-probe method. Au electrodes were placed onto the films through mask deposition. As shown in Fig. 5A and B, two types of devices were fabricated in order to compare the conductivity of P3MT in the direction parallel and perpendicular to the drawing direction of the PVDF films. Fig. 5C shows the I–V properties of the oriented P3MT films in two measuring directions. It is clear that the slope of the I–V profiles decreased dramatically when measured in the direction parallel to the drawing direction of PVDF. On the other hand, the electric currents in the direction perpendicular to the stretching direction of PVDF are much larger. This demonstrates unambiguously an anisotropic electrical conduction of the dedoped P3MT films. The electrical conductivity of dedoped P3MT along the direction perpendicular to the drawing direction of PVDF is σrt 59 ± 3 S cm−1, for several different samples. This value is comparable to that reported for P3MT doped with FeCl3.54 By contrast, the electrical conductivity of P3MT along the stretching direction of PVDF is reproducibly 1.2 ± 0.4 S cm−1. The anisotropy is ca. 50. For comparison, the electrical conductivity of the as-prepared (doped) P3MT on bare ITO displays isotropic conductivity values of 1840 ± 25 S cm−1 and 0.08 S cm−1 for doped and dedoped films, respectively. This data is consistent with a previous value (σmax 1975 S cm−1) for electrochemically doped P3MT films supported on adhesive tape.39 In the present study the comparative higher conductivity (1.2 ± 0.4 S cm−1) of the dedoped P3MT along the stretching direction of PVDF is explained by a regular arrangement of P3MT molecular chains on the PVDF. The high conductivity of our samples of dedoped P3MT can be attributed to the well-ordered structure of its single crystals related to stereoregular conjugated molecular chains, rather than branched chains. Moreover, the excellent conductivity of the P3MT film suggests efficient π–π stacking of the P3MT molecules along the direction perpendicular to the drawing direction of PVDF. The proposed structural ordering is represented in Fig. 5D. We note that conductivity values as high as σrt 400 S cm−1 have been reported for crystals of single-component molecular metals based on metal-dithiolenes which are non-doped (neutral) π-stacked species.55,56 | ||
| Fig. 5 Sketch of the two-probe methods: (A) conductivity measurement along the direction perpendicular to the drawing direction of the PVDF thin film. (B) Conductivity measurement along the direction parallel to the drawing direction of the PVDF film. (C) The I–V properties of the oriented dedoped P3MT film in two different directions. Plots A and B are the data for diagrams (A) and (B), respectively. Inset shows an expanded y axis for plot B. (D) Proposed structural ordering. | ||
To establish that our strategy is versatile for obtaining anisotropic highly conductive polythiophene derivatives, the analogous polymerization of pure thiophene and 3-hexylthiophene on oriented PVDF was shown to give anisotropic films. Polarized optical micrographs are shown in Fig. S5.† The resulting dedoped PT and P3HT films display anisotropic conductivity, with values of 70 ± 4 S cm−1 and 50 ± 2 S cm−1, respectively, along the direction perpendicular to the drawing direction of PVDF, and 1.6 ± 0.3 S cm−1 and 0.9 ± 0.1 S cm−1 along the stretching direction of PVDF.
Conclusions
In summary, highly oriented P3MT films with lathlike crystals were successfully prepared via electropolymerization of 3-methylthiophene on a highly oriented PVDF surface by cyclic voltammetry. The molecular chains in the crystals are aligned perpendicular to the film plane. These P3MT films after electrochemical dedoping exhibit anisotropic electrical conductivity with the anisotropy of conductivity of ca. 50. The value in the direction perpendicular to the long axis of the P3MT crystals is σrt 59 ± 3 S cm−1, which is remarkably high for a dedoped polymer film. The high conductivity should be attributed to the well-ordered structure of P3MT single crystals. Many chemical coupling methods can control the regioregularity of a single molecular chain of poly(3-alkylthiophene) derivatives, but cannot control the arrangement of all the polymer chains within the film.57 We have demonstrated that a main advantage of epitaxial electrochemical deposition is that structure can be ordered at both of these levels (intramolecular and intermolecular). This technique provides a new way to prepare high conductivity films of semiconductive polymers with anisotropic charge transport and could open new applications in optoelectronic devices.Experimental section
Materials
All reactants and solvents were purchased from commercial sources and used without further purification. Anhydrous and deoxygenated solvents were obtained by distillation over sodium benzophenone complex.Electrochemical polymerization to yield P3MT
ITO-coated glass substrates were cleaned in an ultrasonic bath with toluene, acetone, ethanol and deionized water, respectively, and then dried with nitrogen. Highly oriented PVDF film was then pasted onto the ITO by electrostatic forcing. Electrodeposition of P3MT was performed using a standard one-compartment, three-electrode electrochemical cell attached to a CHI 660E Electrochemical Workstation. The Ag/Ag+ nonaqueous electrode was used as reference electrode. ITO (1 cm2) was used as the working electrode and titanium metal was used as the counter electrode (area: 3 cm2). A mixture of 3-methylthiophene (0.5 mM) and Bu4NPF6 (0.1 M) in chloroform and acetonitrile (3/2 v/v) was the electrolyte solution. The electrodeposited films were prepared by CV with these experimental parameters: scan range from −0.20 to +1.20 V, scan rate of 5–100 mV s−1. After the electrodeposition process, the obtained P3MT film was dedoped at 0 V for 2 h. Finally, the resulting films were washed with a mixture of chloroform and acetonitrile (3/2 v/v) to remove unreacted precursors and supporting electrolytes, and then dried in a vacuum oven. All measurements were carried out under ambient conditions.Conductivity measurements
The current–voltage (I–V) characteristics of the sandwich devices were recorded with a Keithley 4200 SCS semiconductor parameter analyzer (Keithley, Cleveland, OH) equipped with a Micromanipulator 6150 probe station in a clean and metallically shielded box in an ambient environment. The P3MT together with PVDF film were detached from the ITO by HF solution (5% aqueous) and placed on a silicon wafer. Gold electrodes were then deposited onto the films as shown in Fig. 5A. The conductivity (σ) extracted from the linear region of the I–V profiles was calculated using the equation σ = Id/(VS), where d is the distance between the electrodes, and S is the cross section of the samples.Acknowledgements
The financial supports of NSFC (no. 21104002 & 51221002) and Beijing Higher Education Young Elite Teacher Project (YETP0491) are gratefully acknowledged. Z. R. thanks the China Scholarship Council for funding a visit to Durham University.Notes and references
- A. Facchetti, M. H. Yoon and T. J. Marks, Adv. Mater., 2005, 17, 1705–1725 CrossRef CAS.
- P. F. Baude, D. A. Ender, M. A. Haase, T. W. Kelley, D. V. Muyres and S. D. Theiss, Appl. Phys. Lett., 2003, 82, 3964–3966 CrossRef CAS PubMed.
- H. Meng, F. P. Sun, M. B. Goldfinger, F. Gao, D. J. Londono, W. J. Marshal, G. S. Blackman, K. D. Dobbs and D. E. Keys, J. Am. Chem. Soc., 2006, 128, 9304–9305 CrossRef CAS PubMed.
- D. I. James, J. Smith, M. Heeney, T. D. Anthopoulos, A. Salleo and I. McCulloch, Organic Semiconductor Materials for Transistors, in Organic Electronics II: More Materials and Applications, Wiley-VCH, Weinheim, 2012, pp. 1–155 Search PubMed.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- H. Moon, R. Zeis, E. J. Borkent, C. Besnard, A. J. Lovinger, T. Siegrist, C. Kloc and Z. Bao, J. Am. Chem. Soc., 2004, 126, 15322–15323 CrossRef CAS PubMed.
- M. D. Curtis, H. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318–4328 CrossRef CAS PubMed.
- L. Jiang, H. L. Dong and W. P. Hu, Soft Matter, 2011, 7, 1615–1630 RSC.
- A. F. Lv, Y. Li, W. Yue, L. Jiang, H. L. Dong, G. Y. Zhao, Q. Meng, W. Jiang, Y. D. He, Z. B. Li, Z. H. Wang and W. P. Hu, Chem. Commun., 2012, 48, 5154–5156 RSC.
- R. J. Li, L. Jiang, Q. Meng, J. H. Gao, H. X. Li, Q. X. Tang, M. He, W. P. Hu, Y. Q. Liu and D. B. Zhu, Adv. Mater., 2009, 21, 4492–4495 CrossRef CAS.
- G. Rydzek, L. Jierry, A. Parat, J. S. Thomann, J. C. Voegel, B. Senger, J. Hemmerle, A. Ponche, B. Frisch, P. Schaaf and F. Boulmedais, Angew. Chem., Int. Ed., 2011, 50, 4374–4377 CrossRef CAS PubMed.
- C. Jerôme and R. Jerôme, Angew. Chem., Int. Ed., 1998, 37, 2488–2490 CrossRef.
- J. Heinze, B. Frontana and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- M. Li, S. Ishihara, M. Akada, M. Liao, L. Sang, J. P. Hill, V. Krishnan, Y. Ma and K. Ariga, J. Am. Chem. Soc., 2011, 133, 7348–7351 CrossRef CAS PubMed.
- Y. Lv, L. Yao, C. Gu, Y. Xu, D. Liu, D. Lu and Y. Ma, Adv. Funct. Mater., 2011, 21, 2896–2900 CrossRef CAS.
- C. Gu, T. Fei, Y. Lv, T. Feng, S. Xue, D. Lu and Y. Ma, Adv. Mater., 2010, 22, 2702–2705 CrossRef CAS PubMed.
- C. Gu, T. Fei, L. Yao, Y. Lv, D. Lu and Y. Ma, Adv. Mater., 2011, 23, 527–530 CrossRef CAS PubMed.
- C. Gu, W. Dong, L. Yao, Y. Lv, Z. Zhang, D. Lu and Y. Ma, Adv. Mater., 2012, 24, 2413–2417 CrossRef CAS PubMed.
- C. Gu, Z. Zhang, S. Sun, Y. Pan, C. Zhong, Y. Lv, M. Li, K. Ariga, F. Huang and Y. Ma, Adv. Mater., 2012, 24, 5727–5731 CrossRef CAS PubMed.
- C. Gu, Y. Chen, Z. Zhang, S. Xue, S. Sun, K. Zhang, C. Zhong, H. Zhang, Y. Pan, Y. Lv, Y. Yang, F. Li, S. Zhang, F. Huang and Y. Ma, Adv. Mater., 2013, 25, 3443–3448 CrossRef CAS PubMed.
- P. Bauerle, U. Segelbacher, A. Maier and M. Mehring, J. Am. Chem. Soc., 1993, 115, 10217–10223 CrossRef.
- J. A. E. H. van Haare, L. Groenendaal, H. W. I. Peerlings, E. E. Havinga, J. A. J. M. Vekemans, R. A. J. Janssen and E. W. Meijer, Chem. Mater., 1995, 7, 1984–1989 CrossRef CAS.
- J. Roncali, Chem. Rev., 1992, 92, 711–738 CrossRef CAS.
- Z. Cai and C. R. Martin, J. Am. Chem. Soc., 1989, 111, 4318–4319 CrossRef.
- P. Blanchard, A. Cravino, E. Levellain, in Handbook of Thiophene-Based Materials, ed. I. F. Perepichka and D. F. Perepichka, Wiley, Chichester, 2009, vol. 2, pp. 419–453 Search PubMed.
- C. Tu, S. Jiang, H. Li and S. Yan, Macromolecules, 2013, 46, 5215–5222 CrossRef CAS.
- H. Zhou, S. Jiang and S. Yan, J. Phys. Chem. B, 2011, 115, 13449–13454 CrossRef CAS PubMed.
- J. Wu, H. X. Zhou, Q. Liu and S. Yan, Chin. J. Polym. Sci., 2013, 31, 841–852 CrossRef CAS PubMed.
- C. Yan, L. Guo, H. Chang and S. Yan, Chin. J. Polym. Sci., 2013, 31, 1173–1182 CrossRef CAS.
- D. Guo, K. Sakamoto, K. Miki, S. Ikada and K. Saiki, Phys. Rev. Lett., 2008, 101, 236103–236106 CrossRef.
- M. Brinkmann, S. Pratontep, C. Chaumont and J. C. Wittmann, Macromolecules, 2007, 40, 9420–9426 CrossRef CAS.
- S. J. Kang, Y. Y. Noh, K. J. Baeg, J. Ghim, J. H. Park, J. S. Kim, J. H. Park and K. Cho, Appl. Phys. Lett., 2008, 92, 052107 CrossRef PubMed.
- S. D. Jiang, H. L. Qian, W. Liu, C. R. Wang, Z. H. Wang, S. K. Yan and D. B. Zhu, Macromolecules, 2009, 42, 9321–9324 CrossRef CAS.
- K. Kawabata, H. Yoneyama and H. Goto, Polym. Chem., 2010, 1, 1606–1608 RSC.
- H. Goto, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 622–628 CrossRef CAS.
- H. Sakaguchi, H. Matsumura and H. Gong, Nat. Mater., 2004, 3, 551–557 CrossRef CAS PubMed.
- J. Petermann and R. M. Gohil, J. Mater. Sci., 1979, 14, 2260–2264 CrossRef CAS.
- J. Wang, H. Li, J. Liu, Y. Duan, S. Jiang and S. Yan, J. Am. Chem. Soc., 2003, 125, 1496–1497 CrossRef CAS PubMed.
- J. Roncali, A. Yassar and F. Garnier, J. Chem. Soc., Chem. Commun., 1988, 581–582 RSC.
- A. Yassar, J. Roncali and F. Garnier, Macromolecules, 1989, 22, 804–809 CrossRef CAS.
- J. Lukkari, M. Alanko, L. Heikkila, R. Laiho and J. Kankare, Chem. Mater., 1993, 5, 289–296 CrossRef CAS.
- Z. W. Sun and A. J. Frank, J. Chem. Phys., 1991, 94, 4600–4608 CrossRef CAS PubMed.
- G. A. dos Reis, I. F. L. Dias, H. de Santana, J. L. Duarte, E. Laureto, E. Di Mauro and M. A. T. da Silva, Synth. Met., 2011, 161, 340–347 CrossRef CAS PubMed.
- M. Li, S. Tang, F. Z. Shen, M. R. Liu, W. J. Xie, H. Xia, L. L. Liu, L. L. Tian, Z. Q. Xie, P. Lu, M. Hanif, D. Lu, G. Cheng and Y. G. Ma, Chem. Commun., 2006, 3393–3395 RSC.
- J. D. Dunitz, ChemBioChem, 2004, 5, 614–621 CrossRef CAS PubMed.
- J. Roncali, F. Garnier, M. Lemaire and R. Garreau, Synth. Met., 1986, 15, 323–331 CrossRef CAS.
- Y. Cao, D. Guo, M. Pang and R. Qian, Synth. Met., 1987, 18, 189–194 CrossRef CAS.
- S. A. Francis and A. H. Ellison, J. Opt. Soc. Am., 1959, 49, 131–133 CrossRef CAS.
- Y. Zhang, Y. Lu, S. Yan and D. Shen, Polym. J., 2005, 37, 133–136 CrossRef CAS.
- G. Zerbi and B. Chierichetti, J. Chem. Phys., 1991, 94, 4646–4658 CrossRef CAS PubMed.
- Y. Yuan, J. Zhang, J. Sun, T. Zhang and Y. Duan, Macromolecules, 2011, 44, 9341–9350 CrossRef CAS.
- D. L. Allara, A. Baca and C. A. Pryde, Macromolecules, 1978, 11, 1215–1220 CrossRef CAS.
- B. Schneider, J. Stokr, P. Schmidt, M. Mihajlov, S. Dirlikov and N. Peeva, Polymer, 1979, 20, 705–712 CrossRef CAS.
- Y. F. Nicolau and P. J. Moser, J. Polym. Sci., Part B: Polym. Phys., 1993, 31, 1529–1543 CrossRef CAS.
- H. Tanaka, Y. Okano, H. Kobayashi, W. Suzuki and A. Kobayashi, Chem. Rev., 2004, 104, 5243–5264 CrossRef PubMed.
- J. P. M. Nunes, M. J. Figueira, D. Belo, I. C. Santos, B. Ribeiro, E. B. Lopes, R. T. Henriques, J. Vidal-Gancedo, J. Veciana, C. Rovira and M. Almeida, Chem.–Eur. J., 2007, 13, 9841–9849 CrossRef CAS PubMed.
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc01068j |
| This journal is © The Royal Society of Chemistry 2014 |