 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVolcano-shape glycerol oxidation activity of palladium-decorated gold nanoparticles†
Zhun
Zhao
a,
Joni
Arentz
b,
Lori A.
Pretzer
c,
Pongsak
Limpornpipat
a,
James M.
Clomburg
a,
Ramon
Gonzalez
a,
Neil M.
Schweitzer
e,
Tianpin
Wu
e,
Jeffrey T.
Miller
e and
Michael S.
Wong
*acdf
aDepartment of Chemical and Biomolecular Engineering, Rice University, 6100 S. Main Street, Houston, TX 77005, USA. E-mail: mswong@rice.edu; Fax: +1-713-348-5478; Tel: +1-713-348-3511
bDepartment of Chemical Engineering, University of Groningen, The Netherland
cDepartment of Chemistry, Rice University, USA
dDepartment of Civil and Environmental Engineering, Rice University, USA
eChemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, IL 60439, USA
fDepartment of Materials Science and NanoEngineering, Rice University, USA
First published on 2nd June 2014
Abstract
Bimetallic PdAu catalysts are more active than monometallic ones for the selective oxidation of alcohols, but the reasons for improvement remain insufficiently detailed. A metal-on-metal material can probe the structure–catalysis relationship more clearly than conventionally prepared bimetallics. In this study, Pd-on-Au nanoparticles with variable Pd surface coverages (sc%) ranging from 10 to 300 sc% were synthesized and immobilized onto carbon (Pd-on-Au/C). Tested for glycerol oxidation at 60 °C, pH 13.5, and 1 atm under flowing oxygen, the series of Pd-on-Au/C materials showed volcano-shape catalytic activity dependence on Pd surface coverage. Increasing surface coverage led to higher catalytic activity, such that initial turnover frequency (TOF) reached a maximum of ∼6000 h−1 at 80 sc%. Activity decreased above 80 sc% mostly due to catalyst deactivation. Pd-on-Au/C at 80 sc% was >10 times more active than monometallic Au/C and Pd/C, with both exhibiting TOF values less than ∼500 h−1. Glyceric acid was the dominant primary reaction product for all compositions, with its zero-conversion selectivity varying monotonically as a function of Pd surface coverage. Glyceric acid yield from Pd-on-Au/C (80 sc%) was 42%, almost double the yields from Au/C and Pd/C (16% and 22%, respectively). Ex situ X-ray absorption near edge structure analysis of two Pd-on-Au/C materials with comparable activities (60 sc% and 150 sc%) showed that the former had less oxidized Pd ensembles than the latter, and that both catalysts were less oxidized compared to Pd/C. That Au stabilizes the metallic state of surface Pd atoms may be responsible for activity enhancement observed in other PdAu-catalyzed oxidation reactions. Decorating a Au surface with Pd generates a catalyst that has the deactivation resistance of Au, the higher glyceric acid selectivity of Pd, and the synergistically higher activities that neither metal has.
1. Introduction
Glycerol, a polyalcoholic compound with uses ranging from cosmetics, soaps, and pharmaceuticals to a chemical precursor for polyethers, polyols, polyesters, and alkyd resins manufacturing,1–3 has recently become an abundant and inexpensive compound due to its generation as a byproduct in biodiesel, oleo-chemical and bioethanol production processes.4–6 The tremendous growth of these industries has led to a significant decrease in crude glycerol prices during recent years (from $0.25 per lb in 2004 to ∼$0.08 per lb in 2011), leading to interest in the development of new and improved methods to generate higher-value products from glycerol.7Over the last 10 years, the selective oxidation of glycerol using supported precious metal catalysts, such as Pt, Pd, and Au, has been extensively studied.8–13 Investigated for the selective oxidation of mono alcohols and diols under mild conditions,14–17 Au catalysts were shown by Carrettin et al. to be very active for glycerol oxidation at high-pH conditions, with 100% selectivity to glyceric acid.11,13 Other researchers reported that Au catalysts were more active than Pd catalysts at high pH, but also less selective to glyceric acid.18–23 The inconsistency in literature probably stems from differences in preparation method, nanoparticle size, support composition, and reaction conditions (e.g., pH, temperature, initial concentration of glycerol, and O2 pressure).20–26 Nevertheless, the presence of base (typically NaOH) is required for Au catalysis to deprotonate glycerol.19,27,28 Davis and co-workers showed that oxygen atoms originate from hydroxyl groups instead of molecular oxygen during glycerol oxidation by Au and Pt.19
In comparison, supported Pd catalysts are active for glycerol oxidation under a wide range of pH values, with maximum glycerol oxidation activities found at high-pH conditions.13,19,29 At neutral or acidic conditions, they favor the formation of dihydroxyacetone (from the oxidation of secondary alcoholic group of glycerol) and C1/C2 molecules (from cleavage of a glycerol C–C bond).19,30 However, Pd catalysts can become over-oxidized, reducing their glycerol oxidation activity.10,14,31,32 Pd is also susceptible to poisoning by in situ-generated carbon monoxide.
PdAu catalysts have been studied by several research groups for glycerol oxidation. Hutchings and co-workers prepared Pd–Au alloyed nanoparticles (NPs) immobilized on activated carbon, and showed they were significantly more active than supported monometallic Pd and Au NPs.21 They concluded that higher surface fraction of Au contributed to higher activity and selectivity to glyceric acid. They further showed that alloyed Pd–Au NPs immobilized on MgO were active for glycerol oxidation under base-free conditions with glyceric acid as the main product.33
Prati and co-workers have extensively examined the effects of reaction conditions (e.g., catalyst amount, temperature, pressure, and NaOH amount) and catalyst preparation methods (e.g., sequence of metal salt reduction, reducing and stabilizing agent, particle size, and support).20,22–26,34,35 They reported that simultaneous reduction of Pd and Au salts led to more active and selective catalysts, compared to catalysts resulting from sequential reductions.25 They observed a decrease in activity and an increase in glyceric acid as particle size increased (for Pd, Au, and PdAu compositions).22 The bimetallic catalysts were more active than either monometallic Pd and Au catalysts; the most active composition was a Pd/Au atomic ratio of 10/90 with 1 wt% total metal loading on activated carbon.23,26
Davis and co-workers synthesized Au-covered Pd NPs through Au salt reduction onto activated carbon-supported Pd NPs.12 The resulting AuPd bimetallic catalysts were less active than monometallic Au but more selective to glyceric acid. They proposed that Pd kept the Au metal in a highly dispersed form which decreased the formation of H2O2 that was responsible for C–C bond cleavage.12
Despite these efforts, there remains limited understanding about how the bimetallic structure affects glycerol oxidation. We propose Pd-coated Au nanoparticles (Pd-on-Au NPs) can be used to address this issue, by allowing for a systematic study of Pd surface coverage effects. The synthesis involves the reduction of palladium salt species onto colloidal Au NPs at ambient conditions using H2 gas, and surface coverage is readily controlled through the amount of Pd precursor added. Previously, we investigated the structure–property relationship of Pd-on-Au NPs for the catalytic hydrodechlorination (HDC) of trichloroethene (TCE) and other organochlorides.36–42 The catalytic NPs showed a volcano-shaped TCE HDC activity dependence on Pd surface coverage, and had significantly higher activity and greater deactivation resistance to chloride and sulfide ions than monometallic Pd catalysts. XAS (X-ray absorption spectroscopy) analysis confirmed the Au-core, Pd-shell structure of the NPs.39,40 Pd-on-Au NPs can carry out HDC reduction reactions, but have not been studied in oxidation reactions before.
This study reports the catalytic behavior of carbon-supported bimetallic Pd-on-Au NPs for the water-phase oxidation of glycerol as a function of Pd surface coverage. Au NPs with a diameter of 4 nm were decorated with Pd metal and immobilized onto carbon support. The catalytic activity of these materials was tested using a semi-batch reactor, and activity was reported in terms of metal-normalized first-order rate constant kcat and initial turnover frequency values (corrected for mass transfer effects and normalized to surface metal atoms). Catalytic activity, selectivity to glyceric acid, and deactivation resistance were quantified in relation to Pd surface coverage. Selectivity values were extrapolated to zero glycerol conversion to differentiate the primary and secondary reaction products as a function of catalyst composition also. Ex situ XAS spectroscopy analysis of selected samples before and after reactions was carried out to assess the oxidative reaction conditions on the state of the two metals.
2. Experimental
2.1. Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The carbon slurry was collected and dried in a vacuum oven at 70 °C overnight until no further mass loss from evaporated water was observed. The material was then ground into powder form and stored in the dark at ambient conditions. Activated carbon, in the untreated (“as-is” carbon) and treated forms (“as-processed” carbon), was used for control experiments.
000 rpm. The carbon slurry was collected and dried in a vacuum oven at 70 °C overnight until no further mass loss from evaporated water was observed. The material was then ground into powder form and stored in the dark at ambient conditions. Activated carbon, in the untreated (“as-is” carbon) and treated forms (“as-processed” carbon), was used for control experiments.
For carbon-supported Pd-on-Au catalysts, the immobilization procedure was the same except that the as-synthesized Pd-on-Au sols were used in place of the Au sol. The Au loading for all Pd-on-Au/C catalysts was kept constant at 1 wt%, while the Pd loading varied according to the Pd surface coverage. Specifically, 10, 30, 50, 60, 80, 100, 150 and 300 sc% Pd-on-Au/C catalysts have calculated Pd loadings of 0.025, 0.074, 0.123, 0.147, 0.196, 0.245, 0.400, and 0.938 wt%, respectively. Carbon-supported with 1 wt% Pd (Pd/C) was prepared in the same manner by mixing 314 mL of Pd sol (31.8 mg Pd per L) with 1.0 g of activated carbon.
2.2. Catalyst characterization
Synthesized catalyst samples were characterized with transmission electron microscopy, nitrogen physisorption, inductively coupled plasma-atomic emission spectroscopy and X-ray absorption spectroscopy.37,39Catalysts were treated in a continuous-flow reactor, which consisted of a quartz tube (1 inch OD, 10 inch length) sealed with Kapton windows by two Ultra-Torr fittings. Ball valves were welded to each Ultra-Torr fitting and served as the gas inlet and outlet. An internal K type thermocouple (Omega) was placed against the catalyst sample holder to monitor temperature. Catalyst samples were pressed into a cylindrical sample holder consisting of six wells, forming a self-supporting wafer. The catalyst amount used was calculated to give an absorbance (μ×) of approximately 1.0. XAS spectra were collected on the reduced samples and in air at room temperature. A minimum metal loading of 1 wt% for both Pd and Au was needed for a satisfactory signal-to-noise ratio in the collected XAS spectra. Two compositions representative of <100 sc% and >100 sc% NPs were chosen: 60 sc% Pd-on-Au and 150 sc% Pd-on-Au. For the 60 sc% Pd-on-Au/C sample, 1407 mL of sol were mixed with 1 g of C, followed by the rest of the immobilization procedure described in Section 2.1.3. The resulting solid was calculated to have 1 wt% Pd and 6.8 wt% Au. For the 150 sc% Pd-on-Au/C sample, 542 mL of sol were mixed with 1 g of C; the calculated metal contents were 1 wt% Pd and 2.5 wt% Au. Pd/C and Au/C used for the glycerol oxidation reaction (with the appropriate 1 wt% metal loading) were also used for XAS analysis.
Each of the 4 catalyst materials (Au/C, Pd/C, 60 sc% Pd-on-Au/C, and 150 sc% Pd-on-Au/C) were treated in two different ways, generating 8 treated samples and 4 untreated samples (as references) for XAS analysis. The “0 h rxn” treatment refers to catalysts charged to the reaction medium and recovered, without performing the glycerol reaction. The “3 h rxn” treatment refers to the catalysts charged to the reaction medium and recovered after 3 h of glycerol reaction time. The reaction conditions were the same as those used for catalytic testing (Section 2.3). Catalysts were recovered after immersing the bottle reactor in an ice bath to quench the reaction and bubbling the reaction medium with flowing He (>99.99%, Matheson) for 15 min. The reaction medium was cooled to 4 °C and centrifuged at 14000 rpm for 40 min. After decanting, the carbon slurry was re-suspended in DI water and centrifuged. This wash step was repeated, and the collected samples were dried in a vacuum oven at 70 °C until no further mass loss from evaporated water was observed. XAS spectra of each sample were collected under air at room temperature.
To assess the level of any oxidation of Pd and Au, all 12 samples were then reduced in flowing 3.5% H2 in He (50 cm3(STP) min−1) at 200 °C, purged with flowing He for 10 min and then cooled to room temperature. Traces of oxygen and moisture in the gases were removed by means of a purifier (Matheson PUR-Gas Triple Purifier Cartridge). Based on our previous work, this treatment method fully reduces oxidized Pd without altering the bimetallic nanostructure.39 These reduced samples were analyzed under He at room temperature.
2.3. Catalytic testing
All NP compositions were studied for catalysis in the supported form. Glycerol oxidation was performed in a screw-cap bottle (250 mL, Alltech) sealed with a Teflon–silicone septum as a semi-batch reactor. A magnetic stirrer, 101.65 mL of DI water, 4.28 mL of NaOH solution (10 M; prepared by dissolving 40 g NaOH (≥99.5%, Sigma-Aldrich) in DI water for a final volume of 100 mL), and 1.07 mL of glycerol solution (10 M; prepared by dissolving 92.1 g of glycerol (≥99.5%, Sigma-Aldrich) in DI water for a final volume of 100 mL) were placed in the reactor such that the final liquid volume was 107 mL and the concentrations of NaOH and glycerol in the reactor were 0.4 M and 0.1 M, respectively. The reactor content was heated to 60 °C (±0.5 °C) in a water bath, bubbled with O2 gas (99.99%, Matheson) for 15 min, and continuously stirred at 1000 rpm. After this, O2 was bubbled into the reaction medium at 120 mL min−1 and 0.2 g of catalyst was charged, marking the start of the reaction.Aliquots of the reaction fluid (500 μL) were periodically withdrawn via a stainless steel needle, and passed through a 0.2 μm microfiber syringe filter (25 mm, VWR) and analyzed through ion-exclusion high-performance liquid chromatography (HPLC). A Shimadzu Prominence SIL 20 system (Shimadzu Scientific Instruments, Inc., Columbia, MD, USA) equipped with an HPX-87H organic acid column (Bio-Rad, Hercules, CA, USA) and a refractive index detector (RID) was used. The column was operated at 315 K, with 30 mM H2SO4 mobile phase flowing at 0.3 cm3 min−1. The retention times and concentration calibration curves were determined for the eight compounds of glycerol, glyceric acid, oxalic acid, glycolic acid, tartronic acid, formic acid, acetic acid, and lactic acid. A slight loss in liquid volume due to water evaporation was observed during the reaction (∼3 mL volume loss after 3 h), for which the measured concentrations were corrected.
Selectivity to a reaction product (Si) was calculated as the percentage of glycerol that converted into that product, i.e., product concentration Ci divided by the sum of the concentrations of the seven product compounds detected (glyceric acid, oxalic acid, glycolic acid, tartronic acid, formic acid, acetic acid, and lactic acid). The products of CO and CO2 were not monitored but they were inferred to form in small amounts (Section 3.4), allowing Si ≈ Ci/(Cgly,0 − Cgly), where Cgly,0 is the initial concentration of glycerol and Cgly is glycerol concentration. Selectivity values were also extrapolated to zero glycerol conversion by fitting the experimentally determined selectivity values to a monotonic, third-order polynomial function.
Glycerol oxidation kinetics was modeled as a first-order reaction (eqn (1)) with respect to glycerol (Fig. S1†) and zero-order with respect to O2 (Fig. S2†). The apparent initial first-order reaction rate constant kmeas (with units of h−1) was calculated from fitting eqn (2) to the first 2 h of the concentration–time profiles, where t is the reaction time. For catalysts observed with deactivation, kmeas was also calculated by fitting eqn (2) to the first 1 h of the concentration–time profiles as a comparison.
| −dCgly/dt = kmeas × Cgly | (1) |
| Cgly = Cgly,0 × exp(−kmeas × t) | (2) |
Glycerol conversion X was calculated as (Cgly,0 − Cgly)/Cgly,0, such that product yield is Yi = Si × X.
The metal-normalized rate constant kcat (with units of L per gmetal per h) was defined as kmeas divided by the total metal content charged to the reactor (Ctotal):
| kcat = kmeas/Ctotal | (3) |
To represent catalytic activity at the particle surface, initial turnover frequency (TOF) was defined as
| TOF = kcorr × Cgly,0/Csurf | (4) |
Apparent activation energy values were determined for Au/C, 30 sc%, 60 sc%, 150 sc% and 300 sc% Pd-on-Au/C, and Pd/C catalysts by running glycerol oxidation reaction at different temperatures. Reaction was conducted at 40, 50, 60, 70, and 80 °C for each catalyst with the following reaction conditions: 0.2 g catalyst, 1000 rpm stirring rate, 107 mL, 0.1 M glycerol, 0.4 M NaOH, and 120 mL min−1 O2 flow. Apparent activation energy Ea was calculated from the Arrhenius equation:
| kmeas = A × exp(−Ea/RT) | (5) |
Deviation of concentration profiles from first-order reaction kinetics was observed for several catalyst compositions, which was attributed to catalyst deactivation. When deactivation is caused by poisoning, it is often modeled as a power-law decay process.50–53 To quantify the extent of deactivation, we modeled the process as first-order in glycerol concentration:
| −dCgly/dt = a × k0meas × Cgly | (6) |
| a = exp(−kd × t) | (7) |
| Cgly = Cgly,0 × exp(k0meas/kd × (exp(−kd × t) − 1)). | (8) |
For a catalyst sample then, kd was determined from a ln(a)–time profile, with the activity function determined from the natural log of the fractional glycerol concentration (ln(Cgly/Cgly,0)) versus time using a second-order polynomial fitting. Each kd value was based on 3 experimental runs for each sample.
3. Results and discussion
3.1. Catalyst structure
Typical TEM images for Au NPs, 60 sc% Pd-on-Au NPs, 150 sc% Pd-on-Au NPs, Pd NPs and their immobilized forms are shown in Fig. 1. The mean diameters (and relative standard deviations) for NPs were measured to be 4.1 nm (21%), 4.2 nm (22%), 4.2 nm (24%), and 4.2 nm (32%) for Au NPs, 60 sc% Pd-on-Au NPs, 150 sc% Pd-on-Au NPs and Pd NPs, respectively, consistent with our previous studies.37,39,54,55 The mean diameters (and relative standard deviations) for the carbon-supported NPs were measured to be 4.1 nm (23%), 4.2 nm (24%), 4.3 nm (26%) and 4.2 nm (35%) for Au/C, 60 sc% Pd-on-Au/C, 150 sc% Pd-on-Au/C, and Pd/C, respectively. Thus, this nanoparticle immobilization method has a negligible effect on the mean size and size distribution of our NPs. The immobilization procedure did not modify the NP atomic structure, as shown in our previous studies.39,40 The BET surface areas for Au/C, 60 sc% Pd-on-Au/C, 150 sc% Pd-on-Au/C, and Pd/C were determined to be 859, 800, 900, and 744 m2 g−1, respectively. These values were somewhat close to the BET surface area of as-processed carbon (872 m2 g−1) and lower than that of as-is carbon (1013 m2 g−1), indicating some surface area loss after the immobilization method was carried out. | ||
| Fig. 1 TEM images of (a) Au NPs, (b) 60 sc% Pd-on-Au NPs, (c) 150 sc% Pd-on-Au NPs, and (d) Pd NPs, and their immobilized forms as (e) Au/C, (f) 60 sc% Pd-on-Au/C, (g) 150 sc% Pd-on-Au/C, and (h) Pd/C. | ||
3.2. Mass transfer resistances analysis
Mass transfer analysis during our glycerol oxidation reaction was performed to ensure kinetics controlled regime and to quantify mass transfer resistances for more accurate TOF calculations. The solubility of O2 in 60 °C DI water is 28 mg L−1 (=0.875 mM)56 much lower than the 100 mM initial glycerol concentration in the reactor, requiring a continuous feed of O2 gas through the reaction medium. Without the O2 flow, the reaction was much slower and glycerol conversion was low (Fig. S2†). Our reactor consisted of three phases: the headspace gas, liquid water, and solid catalyst. To ensure that the reaction rates were quantified properly, i.e., to account for any mass transfer resistances, we carried out the 3-phase reactor mass transfer test.47 Specifically, we determined the observed reaction rate constant kmeas of one of the most active catalysts (60 sc% Pd-on-Au/C catalyst) at different charges and at three different stirring rates (Fig. 2). | ||
| Fig. 2 The relationships between (a) observed reaction rate constant kmeas and catalyst mass concentration, (b) 1/kmeas and 1/(mass of catalyst) at 350, 700, and 1000 rpm stirring rates. The red circles mark the default catalyst charge and stirring rate for the glycerol oxidation reaction. The line in panel (a) was fitted with catalyst mass concentration from 0 to 1.87 g L−1; the red and black lines in panel (b) were fitted with data points only within linearity (solid points). Reaction conditions: 60 sc% Pd-on-Au/C, 60 °C, 107 mL, 0.1 M glycerol, 0.4 M NaOH, and 120 mL min−1 O2. | ||
At a stirring rate of 1000 rpm, kmeas values varied linearly with catalyst mass concentration from 0 to 1.87 g L−1 (catalyst mass from 0 to 0.2 g) (Fig. 2a), with the slope corresponding to kcat = 57.4 L per gmetal per h. This value was within 10% error of kcat calculated using the typical catalyst charge of 0.2 g (=21.4 mg-metal per L) for 60 sc% Pd-on-Au/C (61.0 L per gmetal per h, Table 1). A higher catalyst charge resulted in a sub-linear rate constant, indicating that the typical glycerol oxidation semi-batch reaction conditions (red circle, Fig. 2) were appropriately chosen to avoid external mass transfer limitation. Lower stirring rates reduced the kmeas values, as a result of poor external mass transfer (Fig. 2a). In fact, a stirring rate of 350 rpm was too low to keep the catalyst particles suspended in solution.
| Catalyst type | Glycerol/metal mol ratio | Glycerol conversion after 3 h reaction (%) | Carbon balance after 3 h reaction (%) | Reaction rate constantsa | ||||
|---|---|---|---|---|---|---|---|---|
| Au | Pd | k meas (h−1) | k cat (L per gmetal per h) | k corr (h−1) | TOFb (h−1) | |||
| a First-order reaction rate constants were calculated using data points in the first 2 h. Reaction rate constants in parentheses were calculated from the first 0.5 h of the concentration–time profile for Pd/C, and for the first 1 h of the concentration–time profile for 100, 150, and 300 sc% Pd-on-Au/C catalysts. b Units of initial TOF are mol-glycerol per mol-surface-atom per h. | ||||||||
| Creceived | — | — | 0 | — | 0 | 0 | 0 | 0 |
| Cprocessed | — | — | 0 | — | 0 | 0 | 0 | 0 |
| Au/C | 1054:1 |
— | 36.1 | 91.8 | 0.15 | 7.7 | 0.13 | 445 |
| 10 sc% | 1054:1 |
23230:1 |
58.7 | 92.7 | 0.30 | 15.5 | 0.32 | 809 |
| 30 sc% | 1054:1 |
7743:1 |
82.3 | 79.7 | 0.58 | 28.3 | 0.63 | 1582 |
| 50 sc% | 1054:1 |
4646:1 |
96.7 | 76.7 | 1.04 | 49.0 | 1.25 | 3036 |
| 60 sc% | 1054:1 |
3872:1 |
98.0 | 75.7 | 1.32 | 61.0 | 1.68 | 4038 |
| 80 sc% | 1054:1 |
2904:1 |
99.4 | 76.3 | 1.83 | 81.0 | 2.59 | 6076 |
| 100 sc% | 1054:1 |
2323:1 |
98.4 | 75.9 | 1.60 | 67.8 | 2.14 | 4915 |
| (1.65) | (69.8) | (2.23) | (5122) | |||||
| 150 sc% | 1054:1 |
1423:1 |
95.2 | 79.3 | 1.59 | 60.1 | 2.13 | 4084 |
| (2.07) | (78.2) | (3.07) | (5893) | |||||
| 300 sc% | 1054:1 |
607:1 |
94.7 | 78.1 | 1.43 | 39.1 | 1.86 | 2728 |
| (1.99) | (54.4) | (2.90) | (4254) | |||||
| Pd/C | — | 569:1 |
41.2 | 79.1 | 0.22 | 12.0 | 0.23 | 424 |
| (0.76) | (41.6) | (0.87) | (1601) | |||||
The values of mass transfer resistances were determined utilizing a method previously developed for TCE HDC reaction catalyzed by Pd-on-Au NPs.47 The gas–liquid mass transfer resistance (1/kglagl) was determined to have a significant effect on the observed rate constants, with corrected rate constants kcorr calculated using 1/kcorr = 1/kmeas − 1/kglagl (ESI†).
3.3. Glycerol oxidation activity of carbon-supported Au NPs and Pd NPs
Monometallic Au/C and Pd/C catalysts were firstly tested for glycerol oxidation. The Au/C catalyst was active for glycerol oxidation (Fig. 3a and b). Glycerol reached a conversion of ∼36% after 3 h. Glyceric acid was the major product, resulting from the oxidation of one of the two terminal OH groups. Other C3 compounds detected were lactic acid, and tartronic acid. The C2 compounds of oxalic acid, glycolic acid, and acetic acid and the C1 compound of formic acid were found in smaller amounts, consistent to previous reports.20,21,57 The carbon balance, determined by summing the carbons of the detected C1, C2 and C3 compounds and dividing by initial carbon content of glycerol was ∼92% at the end of the 3 h reaction. The ∼8% difference was attributed to the generation of mostly CO and CO2 formation, which was not monitored in this study but was observed in previous studies.13,57 The pH at the end of the reaction (13.4) did not change significantly from the initial pH of 13.5. The reaction rate constants kcat and initial TOF of Au/C were calculated to be 7.67 L per gmetal per h, and 445 h−1, respectively. This TOF was somewhat similar to what previous studies have reported: 321 h−1 for Au/graphite by Prati and coworkers,25 and 500 h−1 for Au/C by Hutchings and coworkers.21 Prati and coworkers reported TOFs of 845 and 1090 h−1 for Au/C catalysts in their later studies,20,23,24 and Davis and coworkers reported a TOF value of 17 s−1 (=61200 h−1) for their Au/C catalyst.12,27,57 These variations are attributable to differences in catalyst preparation methods, Au particle size, and reaction conditions. Table S1† compares reaction rate constants from various published reports. Au/C prepared through sol–gel immobilization method was more active than from wet impregnation; and smaller Au NPs, higher NaOH/glycerol mol ratio, and higher reaction temperature favored higher TOF values.
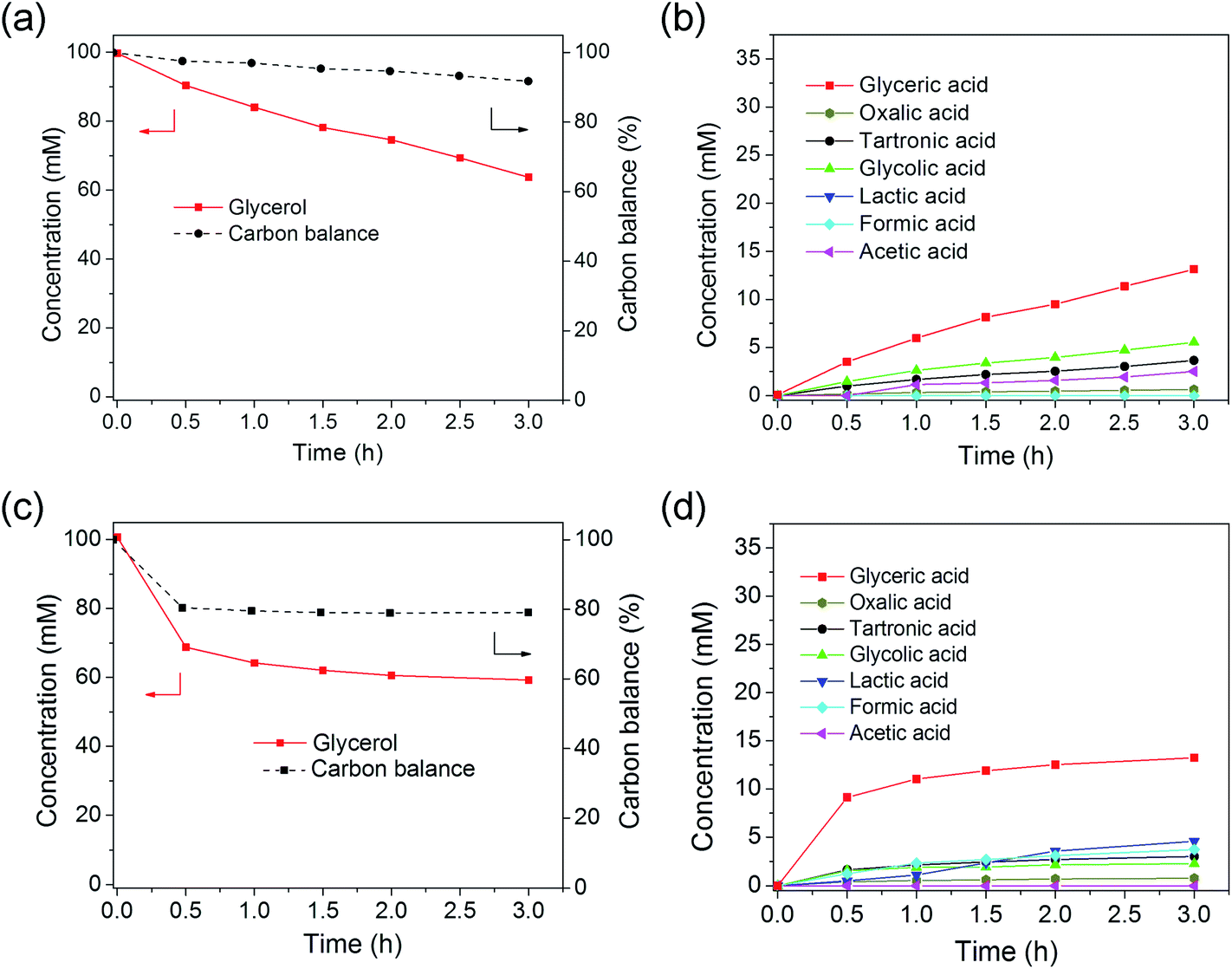 | ||
| Fig. 3 Glycerol oxidation using (a and b) Au/C and (c and d) Pd/C catalysts: (a and c) glycerol concentration and carbon balance, and (b and d) reaction products. Reaction conditions: 0.2 g Au/C or Pd/C, 60 °C, 1000 rpm stirring rate, 107 mL reaction volume, 0.1 M glycerol, 0.4 M NaOH, and 120 mL min−1 O2 flow. | ||
The Pd/C catalyst was more active for glycerol oxidation, reaching a conversion of ∼41% at the end of 3 h (Fig. 3c and d). The rapid drop in glycerol concentration followed by a slower decrease with time suggested an initially high reaction rate constant, followed by deactivation, as observed by others.12,23,24 Pd/C was estimated to have an initial TOF of 424 h−1, close to that of Au/C, if deactivation was not taken into account (Table 1). Considering only the rapid drop in glycerol concentration within the first 0.5 h gave a higher initial TOF of 1601 h−1. This value is somewhat close to that of commercial Pd/C catalyst and of Pd/C (2–3 nm) synthesized from sol–gel immobilization, but greater than that of sol–gel immobilized Pd/C with bigger Pd NPs size (5 nm) (Table S1†).
The deactivation of platinum group metal catalysts in the liquid phase oxidation of alcohols has been known for decades, and is one of the major obstacles in scale-up. Besson et al. and Mallat et al. concluded that deactivation of Pt-group metal catalysts in selective oxidation of alcohols mainly came from the degradation of catalyst structure (metal particle growth or metal leaching), poisoning of the metal sites by irreversibly adsorbed species (products, by-products, or impurities), and/or over-oxidation of the surface.31,32,58 Prati and coworkers proposed that Pd catalyst deactivation in ethylene glycol and glycerol oxidation was due to competitive adsorption with O2 which led to more oxidized, less active Pd metal sites.10,14 They also observed a significant loss of Pd metal after recycle tests in the ethylene glycol oxidation reaction.14 Zope and Davis recently showed that strong adsorption of ketone intermediates and their condensation products contributed mostly to the inhibition of Pt catalysts for glycerol oxidation.59 The carbon balance using the Pd/C catalyst was only 79% at the end of the reaction, correlating to catalyst deactivation by undetected reaction products.
3.4. Glycerol oxidation activity of carbon-supported Pd-on-Au NPs
In comparison, the bimetallics showed very different catalysis behavior. After 3 h, glycerol conversion reached ∼98% with 60 sc% Pd-on-Au/C, indicating a much more active catalyst compared to Au/C and Pd/C (Fig. 4 and S5a†). This sample had the same Au loading as Au/C (1.0 wt% Au loading) and the reactions were carried out under the same conditions (glycerol:Au = 1054:1). The presence of surface Pd atoms (0.025 wt% Pd loading) greatly increased the TOF (4038 h−1) by 9.1 times over that of Au/C (445 h−1) and Pd/C (424 h−1). The pH decreased from 13.5 to 13.3 over the course of the reaction, indicating negligible pH change (Fig. S3†).
 | ||
| Fig. 4 Reaction profile of glycerol oxidation reaction using 60 sc% Pd-on-Au/C catalyst (a) glycerol and carbon balance, and (b) reaction products. Reaction conditions: 0.2 g 60 sc% Pd-on-Au/C, 60 °C, 1000 rpm stirring rate, 107 mL, 0.1 M glycerol, 0.4 M NaOH, and 120 mL min−1 O2 flow. | ||
The reaction rates of Pd-on-Au NPs with varying surface coverages were quantified in the same fashion (Table 1). The conversion–time profiles for all catalyst compositions followed first-order kinetics well (Fig. S4†). The effect of Pd surface coverage on catalytic activity was strong, as seen by the volcano plots of kcat and initial TOF (Fig. 5a and b). Since the kcat values are based on total Au and Pd metal content, the initial TOF values (normalized to total surface Au and Pd atoms) more realistically reflects the catalytic activity of the materials. Observed previously for reduction reactions, i.e., hydrodechlorination (HDC) of chlorinated ethenes,37,40,55,60 the volcano dependence on Pd surface coverage is seen clearly for an oxidation reaction for the first time. The most active catalyst was 80 sc% Pd-on-Au/C, which was 13.7× and 14.3× more active than Au/C and Pd/C catalysts, respectively, based on initial TOF's. In fact, all Pd-on-Au compositions were more active than the monometallic forms.
 | ||
| Fig. 5 Plots of (a) kcat (b) initial TOF, (c) apparent activation energy, and (d) deactivation constant values with Pd surface coverage. Reaction conditions for (a), (b), and (d): 0.2 g catalyst, 60 °C, 1000 rpm stirring rate, 107 mL, 0.1 M glycerol, 0.4 M NaOH, and 120 mL min−1 O2 flow. Reaction temperature range for (c): 40–80 °C. | ||
3.5. Glycerol oxidation selectivity
Selectivity profiles to various products were extensively analyzed for all catalysts to understand reaction mechanism better and to infer its dependence on Pd surface coverage. For all compositions, the main reaction product was glyceric acid, determined at 30% glycerol conversion. The next largest quantity was lactic acid, followed by tartronic acid (Table S2 and Fig. S5b†). Au/C produced less glyceric acid and more tartronic acid compared to the rest of the catalysts. Pd/C was more selective towards glyceric acid than Au/C (consistent with published values) and Pd-on-Au/C catalysts.18–23 Glyceric acid selectivity for Pd-on-Au catalysts was higher than Au/C, and it varied as a function of Pd surface coverage, with the highest selectivity detected at ∼60 sc% (Fig. 6b). Glyceric acid selectivity for all catalysts decreased with glycerol conversion, as glyceric acid oxidized further to form tartronic acid (Fig. S6a†). Lactic acid selectivity did not change with conversion (Fig. S6b†). As glycerol conversion approached 100%, the glyceric acid selectivity decreased precipitously as glyceric acid out-competed glycerol for the active sites. | ||
| Fig. 6 (a) Scheme of glycerol oxidation reaction and its products; plots of (b) glyceric acid selectivity vs. glycerol conversion and (c and d) selectivity values extrapolated to zero conversion (“initial selectivity”) for Au/C, Pd/C, and Pd-on-Au/C catalysts. Reaction conditions: 0.2 g catalyst, 60 °C, 1000 rpm stirring rate, 107 mL, 0.1 M glycerol, 0.4 M NaOH, and 120 mL min−1 O2 flow. | ||
Product yields (Yi = Si × X) can be calculated from selectivity–conversion plots readily (Fig. 6b). With Pd/C, glyceric acid yield was maximum (∼22%) at a glycerol conversion of ∼41%. With Au/C, glyceric acid yield was maximum (∼16%) at a glycerol conversion of ∼36%. Maximum yields for Pd-on-Au/C were 17%, 36%, 41%, 43%, 42%, 42%, 40%, and 38%, at 10, 30, 50, 60, 80, 100, 150, and 300 sc%, respectively.
Clearer trends were gained after estimating the product selectivity values at zero glycerol conversion. The zero-conversion selectivities to glyceric acid, lactic acid and tartronic acid (C3 products) were non-zero for Au, Pd, and Pd-on-Au of all Pd surface coverages, indicating these products were primary reaction products of glycerol oxidation (Fig. 6c). Glyceric acid selectivity increased with Pd surface coverage from 45% (for Au/C) to 60% (for Pd/C), whereas selectivities to all other products (if formed) generally decreased. The multiple primary products are concluded to be the result of multiple active site types activating different glycerol reaction pathways, with a majority of the active site population being glycerol-forming. The gradual change in zero-conversion selectivities must reflect the subtle structural changes as the catalyst transitions from monometallic Au to monometallic Pd, via the Pd-on-Au nanostructure.
Some products were not formed at zero glycerol conversion, indicating they resulted from a secondary reaction. The detected C1 (formic acid) and C2 (glycolic acid, acetic acid, and oxalic acid) species were primary or secondary products, depending on the catalyst (Fig. 6d). Au/C and Pd/C generated formic and glycolic acids as primary products; and oxalic and acetic acids are primary products for Au/C and secondary ones for Pd/C. For Pd-on-Au/C with Pd surface coverages ≤ 80 sc%, formic, glycolic and oxalic acids were primary. Above 80 sc%, all detected C1 and C2 species were generally secondary products.
The C1 and C2 products come from undesirable carbon–carbon bond cleavage. The oxidative glycol cleavage of glyceric acid can yield glyoxylic acid and formaldehyde, which then oxidize into the detected oxalic acid and formic acid, respectively. Carbon–carbon bond cleavage in C2 and C3 compounds can occur to generate other products like glycolic acid, acetic acid, and CO2. Davis and co-workers found that H2O2 formed during glycerol oxidation with monometallic Pd and Au, and PdAu bimetallic catalysts. They showed that H2O2 contributed to C–C cleavage reactions and that Pd metal helped decompose H2O2 such that C–C cleavage products were lessened.12,27,57 They concluded that Au had a higher H2O2 formation rate than Pd, and that Pd had much higher H2O2 decomposition rate that Au.
The presence of H2O2 introduces into the selective glycerol oxidation reaction system a set of secondary, non-selective oxidation reactions. For the Pd-on-Au NP catalysts, the H2O2 formation and decomposition rates are unknown with relation to Pd surface coverage. It is difficult to relate the surface coverage effect on product distribution, though some trends with carbon balance can be seen. The carbon balance for all compositions decreased with increasing conversion, when comparing values at 30% conversion (Table S2†) to values at >30% conversion (i.e., end of 3 h reaction, Table 1). This loss in carbon can be attributed to increased formation of undetected CO, CO2 and possibly adsorbed ketone-like products.59 With increasing Pd surface coverage, the carbon balance decreased, likely for the same reasons. This carbon loss may be responsible for the increasing deactivation observed at >80 sc% via CO poisoning of the Pd metal or surface fouling by adsorbed products (Fig. 5d).
These zero-conversion and high-conversion selectivity data can be summarized into a reaction scheme (Scheme S1†). Glycerol reversibly deprotonates at high pH at one of its primary alcohol groups, forming glycerolate that presumably binds to, and oxidizes on, the catalyst surface into glyceraldehyde (via oxidative dehydrogenation). It would then form a hydrated surface species that oxidizes into glyceric acid, via a mechanism analogous to solution-phase aldehyde oxidation. Glyceraldehyde and its isomer dihydroxyacetone were not detected in our reaction system, but others have reported detecting both species using Pd and Pt catalysts at low pH.8,29,30 The significant amounts of lactic acid detected in this study and in other studies using Au/C57 infer the presence of glyceraldehyde, because lactic acid can form via the hydration of acrolein–methylglyoxal tautomers (which result from glyceraldehyde dehydration). Glyceric acid is converted into tartronic acid via oxidation of its primary alcohol group. All catalyst types contain active sites that lead to glyceric, lactic, and tartronic acids formation; lactic acid does not undergo further oxidation whereas glyceric acid does. Most catalyst types contain active sites that lead to minor amounts of C1, C2, and CO/CO2 products with concentrations increasing at the expense of glyceric acid. They also contain active sites that lead to H2O2 which, in turn, generate these products.
3.6. Activation energy and deactivation analysis
The apparent activation energy Ea was determined in the temperature range of 40–80 °C for several compositions: 45 ± 5, 40 ± 2, 39 ± 2, 40 ± 3, 41 ± 3, and 51 ± 3 kJ mol−1, for Au/C, 30 sc%, 60 sc%, 150 sc%, and 300 sc% Pd-on-Au/C, and Pd/C catalysts, respectively. The measured value for Au/C was somewhat close to published values of Ea (50 ± 5 kJ mol−1 from experiment61 and 49.6 ± 2.9 kJ mol−1 from kinetic modeling62). The Ea values of Pd and PdAu bimetallic catalysts have not been reported for glycerol oxidation before. All these values exceeded 25 kJ mol−1, a value below which mass transfer processes (i.e., gas–liquid or liquid–solid mass transfer, or intraparticle diffusion) have their apparent activation energies.63 This corroborates our mass transfer analysis that the reaction runs were carried out in the kinetically controlled regime.In the classical oxidative dehydrogenation mechanism model of alcohol oxidation over transition metals (e.g., Pt, Pd, Ru, and Au), it is commonly accepted that β-C–H bond cleavage is the rate-limiting step (rls) during catalysis (Scheme S1†).58,64,65 The implication is that the activation energy values measured for the different catalytic structures correspond to the metal-mediated scission of the first β-C–H bond of glycerol molecule. The PdAu compositional effect on activation energy has been studied for reactions like acetylene hydrogenation,66 CCl2F2 hydrodechlorination,67 thiophene hydrodesulfurization,68 methane oxidation,69 and formic acid decomposition.70 In these reported cases, the presence of Pd lowered activation energies relative to monometallic Au. The various Pd-on-Au compositions had Ea's lower than that of Au/C by ∼5 kJ mol−1, correlating to their higher catalytic activities (Fig. 5c).
A compensation effect was observed in comparing the monometallic compositions with the bimetallic ones, in which ln(A) values varied linearly with Ea values (Table S3 and Fig. S7†). The Pd-on-Au NP/C catalysts had Ea values close to an average of ∼40 kJ mol−1 and an average ln(A) value of 13.6. Within experimental uncertainty, there was no difference among these bimetallics. The Au/C catalyst had higher values of Ea and ln(A), and Pd/C had the highest. This compensation effect implies that the Ea and ln(A) values are related due to a linear relationship between the rls activation energy and the adsorption energy of the surface reaction intermediate, i.e., the Brønsted–Evans–Polanyi relationship.71,72 At conditions where Langmuir-like adsorbate surface coverage is close to 100%, the observed activation energy equals the rls activation energy plus the heat of adsorption. Here, assuming the active sites are mostly occupied by the presumptive glycerolate surface intermediate, the Pd-on-Au NP/C samples are more active than Pd/C and Au/C due to lower rls activation energies and due to weaker adsorption strength of the glycerolate. Both are indicative of differences in active site population.
The deactivation could be seen to worsen with increasing Pd surface coverage, by comparing the rate constants determined after the first 2 h of reaction and those determined after the first 0.5–1 h (Table 1). The deactivation rate constant kd was determined for each of the catalysts, assuming a power-law model for poisoning (Fig. 5d). The Pd-on-Au NPs with Pd surface coverage from 0 sc% to 80 sc% had kd values very close to zero, indicating no or negligible deactivation was observed. Above 80 sc%, kd increased to 0.2 ± 0.1 h−1 (at 100 sc%) and further to 0.7 ± 0.2 and 0.7 ± 0.2 h−1 for 150 and 300 sc% Pd-on-Au/C respectively, approaching the kd for Pd/C (1.1 ± 0.2 h−1). Pd-on-Au catalysts with calculated sub-monolayer Pd coverages did not deactivate during glycerol oxidation, while Pd-on-Au with higher Pd coverages had increasingly poor deactivation resistance. Accounting for deactivation, rate constants and TOF values did not fall off from their peak values at 80 sc% as significantly, indicating there were other factors responsible for decreased activity above 80 sc%. Overall, the Pd-on-Au structure was both more active and more deactivation resistant than monometallic Pd/C. The Pd-on-Au catalysts with surface coverages below 80 sc% had the deactivation resistance of Au but had higher activity than Au.
3.7. Assessment of reaction environment on metal oxidation state through XANES characterization
In a previous study, we analyzed using EXAFS the bimetal structure of ∼3 nm (as well as ∼7 nm and ∼10 nm) Pd-on-Au NPs.40 Showing a Au-rich core and a Pd-rich surface, these NPs had the well-established volcano-shape dependence of TCE HDC activity on Pd surface coverage. At the volcano peak location of 50 sc%, the Pd atoms were metallic and in the form of two-dimensional (2-D) ensembles on the Au surface, based on coordination numbers derived from EXAFS fitting. At lower Pd surface coverages, Pd atoms were metallic and less coordinated to one another, on average, due to smaller ensembles and/or more isolated Pd atoms. At higher Pd surface coverages, the atoms tended towards higher Pd–Pd coordination numbers, on average, due to larger 2-D ensembles and 3-D Pd ensemble formation. A fraction of the Pd atoms was bound to oxygen, which we attributed to the topmost Pd atoms of the ensembles being oxidized.The ∼4 nm Pd-on-Au NPs of this work have similar metal structural features as described above, and so the detailed EXAFS data and coordination number (CN) analysis are not presented here.73 Briefly, Pd-on-Au/C (60 sc%) had a Pd–Pd CN of 2.0 and a Pd–Au CN of 7.2, indicating very small Pd ensembles (one Pd atom contacts ∼2 other Pd atoms, on average) bound directly to the Au surface (one Pd atom contacts ∼7.2 Au atoms, on average). Pd-on-Au/C with a higher Pd content (150 sc%) had Pd–Pd CN = 2.4 and Pd–Au CN = 5.4, indicating Pd ensembles were larger and were bound to the Au surface with less contact per Pd atom. These two samples were chosen for XAS analysis, because they had similar catalytic activity on either side of the volcano peak (Fig. 5a and b).
It was not known how the oxidation states of Pd-on-Au would be affected by the harsher conditions of glycerol oxidation (60 °C, O2 atmosphere, and pH ∼ 13.5) compared to those of TCE HDC (room temperature, H2 atmosphere, near-neutral pH).36–42 We thus performed ex situ XANES characterization on 4 samples (Au NPs, Pd NPs, and the two Pd-on-Au NP compositions) under 3 conditions: as-prepared, before the oxidation reaction, and after the oxidation reaction.
The 3 sets of XANES spectra for Au/C were essentially the same (Fig. 7a), indicating that the as-synthesized catalyst structure did not change after being charged to the reactor or after the reaction run. The entire spectrum was essentially the same as that for reduced Au/C, and the lack of change indicated the Au was in the metallic state in all cases. The oxidation state of the Au in the 60 sc% and 150 sc% Pd-on-Au NPs was also invariant, and Au stayed metallic for all samples (Fig. 7b and c).
 | ||
| Fig. 7 XANES spectra at the Au LIII edge for (a) Au/C and Pd-on-Au/C with (b) 60 sc% and (c) 150 sc%; and at the Pd K edge for (d) Pd/C and Pd-on-Au/C with (e) 60 sc% and (f) 150 sc%. | ||
In comparison, the Pd/C catalyst behaved differently. From the linear combination fitting of PdO and reduced Pd/C (treated at 200 °C under 4% H2/He for ∼30 min, purged with He and cooled to room temperature) spectra, the percentage of Pd atoms that was oxidized increased from 10% (for the as-synthesized case) to ∼20% (for the “0 h rxn” case), and remained at ∼20% after the reaction (Fig. 7d). Accounting for a calculated Pd dispersion of 34.8%, the corresponding percentages of oxidized surface Pd were estimated as ∼29%, ∼58%, and ∼58%, respectively.
The XANES spectra for 60 sc% Pd-on-Au/C were negligibly different under the three different conditions (Fig. 7e). The percentage of oxidized Pd stayed at ∼20% in the as-synthesized, pre-reaction and post-reaction samples; most of the Pd (∼80%) was metallic. In terms of surface Pd, a minimum of ∼20% would be oxidized if 100% Pd dispersion is assumed. In comparing the 60 sc% case with the monometallic Pd case, Au appears to suppress Pd oxidation greatly.
The XANES spectra for 150 sc% Pd-on-Au/C also did not vary much under the different conditions either (Fig. 7f). However, the 150 sc% Pd-on-Au/C contained a generally higher percentage of oxidized Pd: ∼35%, ∼40% and ∼30% for as-synthesized, pre-reaction and post-reaction samples. Assuming a calculated Pd dispersion of 73.4%, the corresponding percentages of oxidized surface Pd were ∼48%, ∼55% and ∼41%. Thus, the 150 sc% sample was more oxidized than the 60 sc% one in all three cases. The 150 sc% sample was more oxidized than Pd/C in the as-synthesized case, had comparable oxidation percentage before the reaction, and was less oxidized after the reaction.
While in situ XAS analysis would provide stronger evidence, there appears to be a correlation between the extent of Pd oxidation (assessed under ex situ conditions) and smaller glycerol oxidation rate constants, i.e., the more oxidized the Pd, the less active it is, consistent with literature. Baiker and co-workers examined the effect of Pd oxidation state on benzyl alcohol oxidation using O2 with Pd/Al2O3 under non-aqueous condition.74 Through XANES analysis, they showed that as-synthesized Pd/Al2O3 had its surface Pd fully oxidized and exhibited little activity for alcohol oxidation at 50 °C. After treatment under H2 at 50 °C, the surface Pd was fully reduced, resulting in much higher catalytic activity. The Pd over-oxidized during the reaction (leading to deactivation), if the O2 content was too high. They also examined the oxidation state of supported Au catalysts in the aerobic organic-phase oxidation of 1-phenylethanol at 80 °C. They concluded that metallic Au was the most active phase and oxidized Au was less active, through in situ XAS.75 Maclennan and coworkers studied the structural change of Pd–Au core–shell nanoparticles during crotyl alcohol oxidation using in situ XAS and observed that Au prevented the re-oxidation of surface Pd atoms.76
Additional insights into Pd metal oxidation can be understood in terms of metal corrosion. The Pourbaix diagram for Pd metal shows that Pd can be metallic or can form an oxidized passivating layer based on the reduction potential of the water environment, at a given pH.77 Under our reaction conditions (1 atm O2, 60 °C, and pH = 13.5), the reduction potential of the reaction fluid (quantified as pE) is calculated to be 5.14, corresponding to a voltage potential (Eh) of +0.30 V (ESI†). This is higher than the minimum voltage potential (∼0.01 V calculated at 60 °C and pH 13.5) at which metallic Pd oxidizes to form a PdO layer, indicating that the glycerol oxidation reaction conditions thermodynamically favor the oxidation of Pd metal. This point is consistent with surface Pd atoms of Pd/C becoming more oxidized (up to ∼60%) once in contact with the reaction fluid. That the surface Pd atoms of 60 sc% Pd-on-Au/C sample showed less oxidation (∼20%) indicated the supporting Au metal directly modified the oxidation potential of the Pd ensembles (i.e., electronic effect), such as to increase Pd resistance to oxidation under reaction conditions. If the Pd content was high enough, such as in the 150 sc% case, then the Au had less ability to increase oxidation resistance of the larger Pd ensembles.
4. Conclusions
Pd-on-Au nanoparticles comprising 4 nm Au particles decorated with Pd metal of varying amounts were synthesized and immobilized onto carbon. First-order glycerol oxidation kinetics and reaction product analyses showed that catalytic activity, selectivity, and deactivation resistance strongly varied with Pd surface coverage. All bimetallic compositions were more active than the monometallic counterparts, with maximum catalytic activity at 80 sc% such that near complete glycerol conversion can be achieved. The apparent activation energies for the bimetallics were similar in value, and at least 5 kJ mol−1 less than those for the monometallics. The observed compensation effect suggested the Pd-on-Au catalysts had lower adsorption energy and lower ‘true’ activation energies, reflecting the different reactivities of the surface active sites across the compositional spectrum. At surface coverages above 80 sc%, the active sites became susceptible to deactivation, perhaps by CO poisoning or by adsorbate fouling. The seven detected reaction products accounted for most of the carbon balance for all catalyst compositions, with glyceric, tartronic, and lactic acids as the three-carbon products. Pd-on-Au had glyceric acid selectivities between those of the monometallics at a given conversion, but gave the highest glyceric acid yields. The other products resulted from C–C cleavage, either catalyzed directly at the Pd-on-Au particle surface or promoted by reaction with in situ formed H2O2. The glycerol oxidation reaction conditions favor the oxidation of Pd metal, and ex situ XANES results suggest the possibility of Au usefully suppressing Pd oxidation during reaction. The metal-on-metal materials design allows Au to improve the stability of the supported metal, leading to a more robust and active bimetallic catalyst.Acknowledgements
We acknowledge financial support from the National Science Foundation (CBET-1134535), the Welch Foundation (C-1676), Sigma Xi Grants-in-Aid of Research (GIAR) program (G20111015157503), and Rice University. JTM was supported by the National Science Foundation (EEC-0813570) and as part of the Institute for Atom-efficient Chemical Transformations (IACT), an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences. We thank Prof. M. B. Tomson for use of the inductively coupled plasma optical emission spectrometer (ICP-OES), and Dr C. Yan for assistance. We also thank Mr J. C. Velazquez for help in BET experiment and analysis, Drs S. Gullapalli and Z. L. Schaefer for help in TEM imaging, and Drs K. N. Heck, J. C. Forsythe, and H.-F. Qian for helpful discussions. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, and Office of Basic Energy Sciences (DE-AC02-06CH11357). MRCAT operations are supported by the Department of Energy and MRCAT member institutions.References
- D. T. Johnson and K. A. Taconi, Environ. Prog., 2007, 26, 338–348 CrossRef CAS.
- M. Pagliaro, and M. Rossi, The Future of Glycerol, The Royal Society of Chemistry, 2nd edn, 2010 Search PubMed.
- R. Christoph, B. Schmidt, U. Steinberner, W. Dilla and R. Karinen, Glycerol, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- V. F. Wendisch, S. N. Lindner and T. M. Meiswinkel, Use of Glycerol in Biotechnological Applications, Biodiesel-Quality, Emissions and By-Products, InTech, 2011 Search PubMed.
- E. B. Board, 2009–2010: EU biodiesel industry restained growth in challenging times. Annual biodiesel production statistics, 2010 Search PubMed.
- F. O. Licht, World Ethanol and Biofuels Report, 2010 Search PubMed.
- J. M. Clomburg and R. Gonzalez, Trends Biotechnol., 2013, 31, 20–28 CrossRef CAS PubMed.
- H. Kimura, Appl. Catal., A, 1993, 105, 147–158 CrossRef CAS.
- P. Gallezot, Catal. Today, 1997, 37, 405–418 CrossRef CAS.
- F. Porta and L. Prati, J. Catal., 2004, 224, 397–403 CrossRef CAS PubMed.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin and G. J. Hutchings, Chem. Commun., 2002, 696–697 RSC.
- W. Ketchie, M. Murayama and R. Davis, J. Catal., 2007, 250, 264–273 CrossRef CAS PubMed.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1329–1336 RSC.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552–560 CrossRef CAS.
- C. Bianchi, F. Porta, L. Prati and M. Rossi, Top. Catal., 2000, 13, 231–236 CrossRef CAS.
- F. Porta, L. Prati, M. Rossi, S. Coluccia and G. Martra, Catal. Today, 2000, 61, 165–172 CrossRef CAS.
- L. Prati and G. Martra, Gold Bull., 1999, 32, 96–101 CrossRef CAS.
- E. Rodrigues, S. Carabineiro, X. Chen, J. Delgado, J. Figueiredo, M. Pereira and J. Órfão, Catal. Lett., 2011, 141, 420–431 CrossRef CAS PubMed.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- C. Bianchi, P. Canton, N. Dimitratos, F. Porta and L. Prati, Catal. Today, 2005, 102–103, 203–212 CrossRef CAS PubMed.
- N. Dimitratos, J. A. Lopez-Sanchez, J. M. Anthonykutty, G. Brett, A. F. Carley, R. C. Tiruvalam, A. A. Herzing, C. J. Kiely, D. W. Knight and G. J. Hutchings, Phys. Chem. Chem. Phys., 2009, 11, 4952–4961 RSC.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Lennon, F. Porta, L. Prati and A. Villa, Catal. Lett., 2006, 108, 147–153 CrossRef CAS PubMed.
- A. Villa, C. Campione and L. Prati, Catal. Lett., 2007, 115, 133–136 CrossRef CAS.
- L. Prati, A. Villa, C. Campione and P. Spontoni, Top. Catal., 2007, 44, 319–324 CrossRef CAS PubMed.
- N. Dimitratos, F. Porta and L. Prati, Appl. Catal., A, 2005, 291, 210–214 CrossRef CAS PubMed.
- D. Wang, A. Villa, F. Porta, L. Prati and D. Su, J. Phys. Chem. C, 2008, 112, 8617–8622 CAS.
- W. Ketchie, M. Murayama and R. Davis, Top. Catal., 2007, 44, 307–317 CrossRef CAS.
- K. N. Heck, B. G. Janesko, G. E. Scuseria, N. J. Halas and M. S. Wong, ACS Catal., 2013, 3, 2430–2435 CrossRef CAS.
- R. Garcia, M. Besson and P. Gallezot, Appl. Catal., A, 1995, 127, 165–176 CrossRef CAS.
- H. Kimura, K. Tsuto, T. Wakisaka, Y. Kazumi and Y. Inaya, Appl. Catal., A, 1993, 96, 217–228 CrossRef CAS.
- M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127–141 CrossRef CAS.
- T. Mallat and A. Baiker, Catal. Today, 1994, 19, 247–283 CrossRef CAS.
- G. L. Brett, Q. He, C. Hammond, P. J. Miedziak, N. Dimitratos, M. Sankar, A. A. Herzing, M. Conte, J. A. Lopez-Sanchez, C. J. Kiely, D. W. Knight, S. H. Taylor and G. J. Hutchings, Angew. Chem., Int. Ed., 2011, 50, 10136–10139 CrossRef CAS PubMed.
- N. Dimitratos, C. Messi, F. Porta, L. Prati and A. Villa, J. Mol. Catal. A: Chem., 2006, 256, 21–28 CrossRef CAS PubMed.
- N. Dimitratos, A. Villa and L. Prati, Catal. Lett., 2009, 133, 334–340 CrossRef CAS PubMed.
- M. O. Nutt, J. B. Hughes and M. S. Wong, Environ. Sci. Technol., 2005, 39, 1346–1353 CrossRef CAS.
- M. O. Nutt, K. N. Heck, P. J. J. Alvarez and M. S. Wong, Appl. Catal., B, 2006, 69, 115–125 CrossRef CAS PubMed.
- K. N. Heck, B. G. Janesko, G. E. Scuseria, N. J. Halas and M. S. Wong, J. Am. Chem. Soc., 2008, 130, 16592–16600 CrossRef CAS PubMed.
- Y. L. Fang, J. T. Miller, N. Guo, K. N. Heck, P. J. J. Alvarez and M. S. Wong, Catal. Today, 2011, 160, 96–102 CrossRef CAS PubMed.
- L. A. Pretzer, H. J. Song, Y.-L. Fang, Z. Zhao, N. Guo, T. Wu, I. Arslan, J. T. Miller and M. S. Wong, J. Catal., 2013, 298, 206–217 CrossRef CAS PubMed.
- H. Qian, L. A. Pretzer, J. C. Velazquez, Z. Zhao and M. S. Wong, J. Chem. Technol. Biotechnol., 2013, 88, 735–741 CrossRef CAS.
- H. Qian, Z. Zhao, J. C. Velazquez, L. A. Pretzer, K. N. Heck and M. S. Wong, Nanoscale, 2014, 6, 358–364 RSC.
- J. W. Slot and H. J. Geuze, Eur. J. Cell Biol., 1985, 38, 87–93 CAS.
- J. M. Thomas, Pure Appl. Chem., 1988, 60, 1517–1528 CrossRef CAS.
- L. Lewis, Chem. Rev., 1993, 93, 2692–2730 CrossRef.
- T. Teranishi and M. Miyake, Chem. Mater., 1998, 10, 594–600 CrossRef CAS.
- Y. L. Fang, K. N. Heck, P. J. J. Alvarez and M. S. Wong, ACS Catal., 2011, 1, 128–138 CrossRef CAS.
- ImageJ, v. 1.40. National Institutes of Health, 2008, http://rsb.info.nih.gov/ij/.
- N. Guo, B. R. Fingland, W. D. Williams, V. F. Kispersky, J. Jelic, W. N. Delgass, F. H. Ribeiro, R. J. Meyer and J. T. Miller, Phys. Chem. Chem. Phys., 2010, 12, 5678–5693 RSC.
- H. S. Fogler, Elements of Chemical Reaction Engineering, Pearson Education, Inc., Upper Saddle River, NJ 07458, 4th edn, 2005 Search PubMed.
- C. H. Bartholomew and R. J. Farrauto, Fundamentals of Industrial Catalytic Processes, John Wiley & Sons, Inc., Hoboken, New Jersey, 2nd edn, 2005 Search PubMed.
- Y. Schuurman, B. F. M. Kuster, K. van der Wiele and G. B. Marin, Appl. Catal., A, 1992, 89, 47–68 CrossRef CAS.
- J. H. Vleeming, B. F. M. Kuster and G. B. Marin, Ind. Eng. Chem. Res., 1997, 36, 3541–3553 CrossRef CAS.
- K. N. Heck, M. O. Nutt, P. J. J. Alvarez and M. S. Wong, J. Catal., 2009, 267, 97–104 CrossRef CAS PubMed.
- M. S. Wong, P. J. J. Alvarez, Y. L. Fang, N. Akcin, M. O. Nutt, J. T. Miller and K. N. Heck, J. Chem. Technol. Biotechnol., 2009, 84, 158–166 CrossRef CAS.
- D. Tromans, Hydrometallurgy, 1998, 48, 327–342 CrossRef CAS.
- W. C. Ketchie, Y.-L. Fang, M. S. Wong, M. Murayama and R. J. Davis, J. Catal., 2007, 250, 94–101 CrossRef CAS PubMed.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed.
- B. N. Zope and R. J. Davis, Green Chem., 2011, 13, 3484 RSC.
- Y.-L. F. Z. Zhao, P. J. J. Alvarez and M. S. Wong, Appl. Catal., B, 2013, 140–141, 468–477 CrossRef PubMed.
- S. Demirel-Gülen, M. Lucas and P. Claus, Catal. Today, 2005, 102-103, 166–172 CrossRef PubMed.
- S. Demirel, M. Lucas, J. Warna, D. Murzin and P. Claus, Top. Catal., 2007, 44, 299–305 CrossRef CAS.
- C. N. Satterfield, Mass transfer in heterogeneous catalysis, M.I.T. Press, Cambridge, MA, 1970 Search PubMed.
- N. Worz, A. Brandner and P. Claus, J. Phys. Chem. C, 2009, 114, 1164–1172 Search PubMed.
- K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2003, 9, 4353–4361 CrossRef CAS PubMed.
- C. Visser, J. G. P. Zuidwijk and V. Ponec, J. Catal., 1974, 35, 407–416 CrossRef CAS.
- M. Bonarowska, A. Malinowski, W. Juszczyk and Z. Karpiński, Appl. Catal., B, 2001, 30, 187–193 CrossRef CAS.
- A. M. Venezia, V. La Parola, G. Deganello, B. Pawelec and J. L. G. Fierro, J. Catal., 2003, 215, 317–325 CrossRef CAS.
- J. G. Firth, Trans. Faraday Soc., 1966, 62, 2566–2576 RSC.
- D. D. Eley and P. Luetic, Trans. Faraday Soc., 1957, 53, 1483–1487 RSC.
- P. J. Barrie, Phys. Chem. Chem. Phys., 2012, 14, 318–326 RSC.
- T. Bligaard, K. Honkala, A. Logadottir, J. K. Nørskov, S. Dahl and C. J. H. Jacobsen, J. Phys. Chem. B, 2003, 107, 9325–9331 CrossRef CAS.
- Z. Zhao, J. T. Miller, T. Wu, N. Schweitzer and M. S. Wong, Catal. Today, 2014 Search PubMed , to be submitted.
- J. D. Grunwaldt, M. Caravati and A. Baiker, J. Phys. Chem. B, 2006, 110, 9916–9922 CrossRef CAS PubMed.
- P. Haider, J. D. Grunwaldt, R. Seidel and A. Baiker, J. Catal., 2007, 250, 313–323 CrossRef CAS PubMed.
- A. Maclennan, A. Banerjee, Y. Hu, J. T. Miller and R. W. J. Scott, ACS Catal., 2013, 3, 1411–1419 CrossRef CAS.
- E. McCafferty, Introduction to Corrosion Science, Springer, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: detailed Au and Pd NP synthesis method; determination of mass transfer effects; Fig. S1 (plot of ln(1 − x) vs. time for 60 sc% Pd-on-Au/C); Fig. S2 (glycerol–time profiles for reaction with and without O2 flow); Table S1 (comparison of various catalysts in literature); Fig. S3 (pH of reaction medium–time profile); Fig. S4 (plot of ln(1 − x) vs. time for Au, Pd and Pd-on-Au/C catalysts); Fig. S5 (comparison of glycerol concentration profiles and selectivities); Fig. S6 (plot of selectivity to tartronic acid and lactic acid vs. conversion of glycerol); Table S2 (selectivities and carbon balance for all catalysts); Table S3 (list of apparent activation energies and pre-exponential factors); Fig. S7 (plot of natural logarithm of pre-exponential factor against apparent activation energy); calculation of voltage potential. See DOI: 10.1039/c4sc01001a |
| This journal is © The Royal Society of Chemistry 2014 |