 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceObserving the oxidation state turnover in heterogeneous iridium-based water oxidation catalysts†
Alessandro
Minguzzi
*ac,
Ottavio
Lugaresi
ac,
Elisabetta
Achilli
b,
Cristina
Locatelli
ac,
Alberto
Vertova
ac,
Paolo
Ghigna
bc and
Sandra
Rondinini
ac
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20133, Milan, Italy. E-mail: alessandro.minguzzi@unimi.it
bDepartment of Chemistry, University of Pavia, Viale Taramelli 16, 27100, Pavia, Italy
cIstituto Nazionale di Scienza e Tecnologia dei Materiali, via Giusti 9, Firenze, Italy
First published on 19th May 2014
Abstract
In this work the oxidation states assumed by Ir in oxide systems used as heterogeneous catalysts for water oxidation are determined by means of in situ X-ray Absorption Spectroscopy (XAS). Using a highly hydrated iridium oxide film allows the maximum number of Ir sites to be involved in the electrochemical processes occurring at the catalysts during water oxidation (oxygen evolution reaction, OER). X-ray Absorption Near Edge Structure (XANES) spectra clearly indicate the co-existence of Ir(III) and Ir(V) at the electrode potentials where OER occurs. This represents a fundamental step both in the understanding of the water oxidation mechanism catalysed by heterogeneous Ir oxide systems, and in the possible tailoring of electrocatalysts for OER.
Introduction
One of the most discussed aspects of heterogeneous catalysis is whether the mechanisms of the catalytic reactions can be compared to similar cases in homogeneous catalysis. The same is true in electrocatalysis,1 where further complications arise from the need for a conducting support, on which the catalyst is deposited or chemically bound (permanently or just before charge transfer). Apart from very recent and innovative supports,2 the presence of a conducting support often restricts the available specific surface area, and therefore the number of sites that can be investigated by spectroelectrochemical methods, for example. Heterogeneous catalysts present a multiplicity of surface active sites that show different behaviour, which can be averaged and analysed to reach a general mechanistic understanding. This is much more difficult than for the catalytic cycle of a homogeneous catalyst, where at least the exact chemical nature of the initial state is well-known. Considering well-oriented single crystals,3 or combining experimental and computational approaches,4 can help to bridge the gap between the homogeneous and heterogeneous “worlds”. Another very promising approach is to study single nanoparticles, to avoid averaged measurements from a massive number of different sites on different surfaces.5,6In this work a new approach is undertaken, exploiting the fact that, at a given instant, site speciation is distributed on all the possible conditions that they can assume during the catalytic cycle. We will do this by considering iridium oxide, a system of great interest, being one of the most frequently used anode materials in electrochemical industrial processes7 (for water electrolysis,8 cathodic protection,9 and metal electrowinning10) and for the most advanced photoanodes.11 In spite of this, the turnover mechanism of IrO2 (as well as those of many other electrocatalysts) is not yet clarified. The most important mechanistic aspect of water oxidation catalysis is related to how the catalyst enters the reaction mechanism:12 it is more than likely that the catalytic site may act as a redox mediator between the current collector and the species in solution.13,14 A similar behaviour was recently observed using scanning electrochemical microscopy (SECM), in the case of the reaction of various reductants over oxidized platinum surfaces.15 In the particular case of water oxidation over oxide materials, this results in the following general mechanisms:
Higher valence oxide formation:
| Ir2xOx + H2O → Ir2(x+1)Ox+1 + 2H+ + 2e− | (1) |
Higher valence oxide decomposition:
| Ir2(x+1)Ox+1 → Ir2xOx + 1/2O2 | (2) |
More recently, a combined set of experimental and computational results have led to a bi-nuclear model that also takes into account the interesting role of pH in a possible mechanism for OER.23 Note that none of the studies mentioned include any experimental evidence of the central role of Ir in the OER catalytic cycle, and therefore the co-presence of different Ir charge states under OER conditions. This is also due to the fact that determining the oxidation states of the metals in solid compounds under working conditions is a difficult task. A probe is needed that is sensitive to the local electronic structure and can be used in both in situ or in operando environments. X-ray Absorption Spectroscopy (XAS) is the technique of choice here, as hard X-rays can have a large penetration depth in matter, and therefore have the capability to work in in operando environments. In addition, electronic transitions to the bounded state are found close to an absorption edge. Due to the Δl = ±1 selection rule, direct access to the d states is obtained by the use of the LII or LIII edge, where the initial states are of p character, the transition to the s final states being of negligible intensity. In this work, in situ XAS at the Ir-LIII edge is used to observe the changes in the oxidation state of iridium, during the oxygen evolution reaction by electrolytic water oxidation. XAS is particularly suited for monitoring oxidation states and changes thereof as the edge energy position is affected by the well-known chemical shift. Schematically, the edge position is determined by the coulombic interaction between the nucleus of the photoabsorber and the electron involved in the photoionization process. This interaction is in turn screened by all the other electrons around the photoabsorber and therefore increases with increasing oxidation state. Thus, the edge shifts at a higher energy with an increasing oxidation state. A similar approach for Ir oxides has been adopted previously24,25 but, to the authors' best knowledge, the present work represents the first study carried out under oxygen evolution conditions. In order to obtain a good correlation between the bias imposed at the electrode and the X-ray absorption, we used Electrodeposited Iridium Oxide Films (EIROFs). These materials are of increasing interest,26 as they have the peculiar property of being highly hydrated (which is why they are sometimes called hydrous iridium oxide films, HIROFs), thus allowing an easy ion mobility within the film. This in turn causes all the Ir centres to undergo the electrochemical phenomena (i.e. oxidation state transitions) caused by an external perturbation (e.g. a potential change).27 Indeed, while on iridium oxide particles/layers (such as in dimensionally stable anodes, DSA®) only a small fraction of Ir surface sites (1–2%)28,29 participate in any redox event, on highly hydrated films all material is involved in the electrochemical phenomena. This is the basis of the relevant peculiarities previously observed in EIROFs: for example, they are electrochromic materials, being colourless in the reduced form and blue in the oxidized form.
In situ XAS measurements24 and FEXRAV16 allowed us to address the nature of the two forms: the reduced, colourless form is attributed to a prevalence of Ir(III) and has semiconducting properties, while the oxidized one is attributed to Ir(IV) and it is a metallic conductor. This was well demonstrated in the presence of a fast redox couple in solution: the charge transfer chain is limited by the low conductivity of the Ir(III) form but behaves as a metallic conductor in the Ir(IV) form. This transition occurs between 0.6 and 1 V (ref. 16) while the shift from semiconductivity to metallic conductivity occurs at around 0.6–0.8 V vs. RHE.30,31
At potentials higher than 1 V, the behaviour of Ir oxide is not well understood, partly because of the lack of suitable methods for analysing the surface conditions of the material in operando. Notwithstanding previous investigations which highlighted the formation of a higher oxidation state, most likely Ir(V),16,23 the behaviour of Ir sites under water oxidation conditions (in principle, for E > E0 = 1.23 V vs. RHE, at pO2 = 1 atm and aH2O = 1) is still unknown and leads to an exciting discussion, as previously summarised. In addition, is not clear whether the formation of a higher oxide is correlated to OER onset and other kinetic features (Tafel slope, exchange current), i.e. the parameters typically extrapolated and evaluated to judge the activity of a catalyst.32
EIROFs, because of their high level of hydration, allow at least the observation of the distribution of the oxidation states, in order to treat heterogeneous catalysts as similar to homogeneous catalysts: the XAS result in this case acts as as a “photograph” of the Ir speciation in the electrode at any applied potential.
Results and discussion
Fig. 1 shows the Ir-LIII X-ray Absorption Near Edge Structure (XANES) spectra collected for the EIROF electrode under different applied potentials. Some of these spectra have been already reported in ref. 16. For better reference, the spectra of powdered IrCl3 and IrO2 are added for Ir(III) and Ir(IV), respectively. | ||
| Fig. 1 Normalized Ir-LIII edge XANES spectra of the electrode material under different applied potentials (A). The spectra of bulk IrO2 and IrCl3 are shown as a reference (B). The second derivatives are also shown (C). For better clarity, the spectra are shifted along the y axis, and vertical lines marking the energies of the White Lines of IrCl3, IrO2 and Ir(V) (predicted) are drawn. | ||
Transition from the Ir 2p levels to continuum states and localized 5d states gives rise to a step (edge) hereafter modelled with an arctangent function, and to peaks (usually known as White Lines, WLs), modelled with a Lorentzian function.33 The widths of both the arctan and Lorentzian functions are controlled by the core hole lifetime via an indetermination relationship.34 Other effects can contribute to the broadening of the spectral shape, the most significant being the spread of 5d orbitals into a band, and the crystal field splitting. Crystal field split transitions are expected for compounds with isolated [IrO6]n− octahedra. When the octahedra interact, the 5d Ir states on different sites form crystal orbitals: this is equivalent to stating that when the bandwidth of the Ir 5d states is larger than the crystal field splitting, the latter effect is no longer observed. This rationale is in agreement with what has been reported in ref. 35.
With this in mind, and assuming a single charge (oxidation) state for Ir at a given applied potential value, we expect to observe two possible types of WL: (i) “narrow”, in the case of non-interacting octahedra and partially filled Ir 5d orbitals (such as for Ir(III), with fully occupied t2g states) and (ii) “broad”, in which the transition from the Ir 2p levels to the split (t2g + eg) 5d states is evident.
With this in mind, we can now discuss the spectra shown in Fig. 1, starting with the energy position of the WLs. For an applied potential of 0.2 V, the energy position of the WL is very close to that of IrCl3, indicating a state near to Ir(III). When the potential is increased up to 1 V, the gradual shift of the WL towards a higher energy indicates a progressive oxidation of the Ir. At 1 V, the spectrum is almost identical to that of IrO2, indicating that at this potential the electrode material is composed of this, and thus the Ir is in the Ir(IV) oxidation state. These initial observations, that have been the basis of the first application of FEXRAV,16 confirm the expected peculiar behaviour of the highly hydrated iridium oxide film adopted in this work: all the Ir sites present in the deposit respond homogeneously to the potential bias applied to the electrode.
When the applied potential is >1 V, the WL gradually shifts towards even higher energies: in detail, (i) a small shift is observed at 1.2 V; (ii) at 1.3 V the WL shows the maximum amplitude and the peak appears to have been almost replaced by a plateau; (iii) at 1.5 V the WL has a broad maximum at ca. 11222.1 eV; (iv) at 1.6 V the maximum at ca. 11222.1 eV shows a clear tail at a lower energy.
In addition to variations of the energy positions, the spectra of Fig. 1 also show remarkable variations in the amplitude and width of the WLs. As said before, crystal field splitting is one of the phenomena that can contribute to the spectral shape of the Ir-LIII WL. This effect is better discussed by looking at the second derivative of the spectra.35,36 The contribution of the transitions to the continuum states, as described by the arctangent, is in this case negligible. For a perfect octahedral environment, two negative peaks are expected, corresponding to the transitions to the t2g and eg states. The intensity of these transitions does not directly reflect the statistical count of the holes in the t2g and eg states, as solid state effects (phonon coupling) and the covalency of the Ir–O bonds also have to be taken into account.36 Again, it should be noted that Ir(III) in its low spin state (the most stable state for 5d transition metals) has a t2g6 electronic configuration and thus only the transition to the eg states takes place. Solid state effects can even mask crystal field splitting via band formation, as in the case of pure IrO2.36 In addition, distortions of the [IrO6]n− octahedra result in some degeneracy reduction, the latter effect being expected to be larger for the eg than for the t2g states.
The second derivatives of the spectra are reported in Fig. 1C. For all the applied potentials, excluding 0.7 and 1.3 V, crystal field split transitions are not observed, while for 0.7 and 1.3 V a double peak structure is found, which likely corresponds to the transition from the Ir 2p levels to the split (t2g + eg) 5d states (crystal field splitting). According to previous results, the splitting observed at 0.7 and 1.3 V – that is, 3.1 and 4 eV – is consistent with the presence of Ir(IV) and Ir(V), respectively.35,36
This experimental evidence means that, at 0.7 V, Ir(IV) is already formed at least partially, but that it has a structure composed of non-interacting octahedra: a sort of “pre-rutile” structure.
This could be in line with the existence of the pre-peak observed in the cyclic voltammetry (CV) curve between 0.5 and 0.6 V (RHE), whose origin is still unclear.23,24
In a similar way, at 1.3 V, Ir assumes the highest oxidation state (V) and the structure deviates again from that of the Ir(IV)-rutile.
At higher potentials, i.e. for E = 1.5 and 1.6 V, the single-peak structure in the second derivative and the presence of a shoulder in the WL strongly indicates that the WL broadening cannot be attributed to crystal field splitting.
The only reasonable explanation for this experimental evidence is the co-existence of more than one oxidation state of Ir.
In order to validate this rationale, the XANES spectra were fitted with a combination of Lorentzian and arctan functions.37 In detail: (i) a single arctan plus Lorentzian couple, when a single oxidation state is present and no crystal field splitting is observed; (ii) two arctan and Lorentzian couples when crystal field splitting is not observed and the WL is too large to account for a single oxidation state, and (iii) a single arctan and two Lorentzian functions at 0.7 and 1.3 V, according to the discussion above. The three combinations of Lorentzian and arctan functions are shown in Fig. 2 for the representative spectra recorded at 0.2, 1.3 and 1.6 V.
 | ||
| Fig. 2 Fits of the normalized Ir-LIII edge XANES spectra of the electrode material under two applied potentials; A: 1.6 V; B: 1.3 V; C: 0.2 V. | ||
As said before, the energy positions of the many different spectral features in the XANES spectra depend on the charge state of the photoabsorber. In particular, the maximum of the absorption coefficient μ,16 and therefore the energy position of each Lorentzian peak, can be used as a measure of the oxidation state of the Ir at the different working conditions of the electrode material.
Fig. 3 shows data obtained by assuming an energy shift of 1.3 eV per unit change of oxidation state,25 a figure that is in agreement with the difference in energy positions of the maxima for μ in the spectra of IrCl3 and IrO2. It should be noted that the presence of Ir in different oxidation states for working potentials ≥1.3 V is directly supported by the larger full width at half maximum (FWHM) of the WL found for these applied potentials, and as such must be seen as a direct experimental finding. As a final remark, we can note that the integrated area of the WL is, in principle, proportional to the number of holes in the d states. This correlation, however, is non-linear, because of the presence of multiple oxidation states of the Ir; these have different electronic configurations, in different amounts, for different applied potentials (the intensity of the WL depends on the pertinent matrix element for the electronic transition). At applied potentials larger than 1.4 V, bubbles of gaseous oxygen form at the interface between the electrode and the electrolyte, preventing some of the Ir atoms in the electrode from participating in the reaction. In particular, band formation broadens the d states and reduces the amplitude of the WL.36
 | ||
| Fig. 3 EIROF cyclic voltammetry (blue line, left axis) recorded at 2 mV s−1 in aqueous 0.5 M H2SO4 superimposed to the Ir oxidation state values, as derived from the XANES WL peak energies spectra (yellow to magenta dots, right axes). The light blue areas indicate the potential regions in which a crystal field split was observed: at these potentials, the charge state is derived by the (t2g + eg) 5d orbital splitting values. | ||
Indeed, when plotting the integrated areas of the WL as a function of the applied potential (see ESI, Fig. S1†), a non-linear increase is found up to 1.3 V, followed by a decrease in intensity with a further increase in the potential. It should be noted that, indirectly, this is an assessment of the reliability of our approach to data analysis: from the above discussion, the integrated area is indeed expected to reach a maximum at 1.3, when the number of holes in the d states is at a maximum and the presence of crystal field split transitions points towards a state with non-interacting octahedra, with a small bandwidth for the d states.
As represented in Fig. 3, for E >1.3 V, Ir assumes two distinct oxidation states, namely (III) and (V). This is in line with the participation of the Ir sites in the heterogeneous Ir oxide catalyst in the catalytic cycle, as is well established for homogeneous catalysis, and is described in Scheme 1.
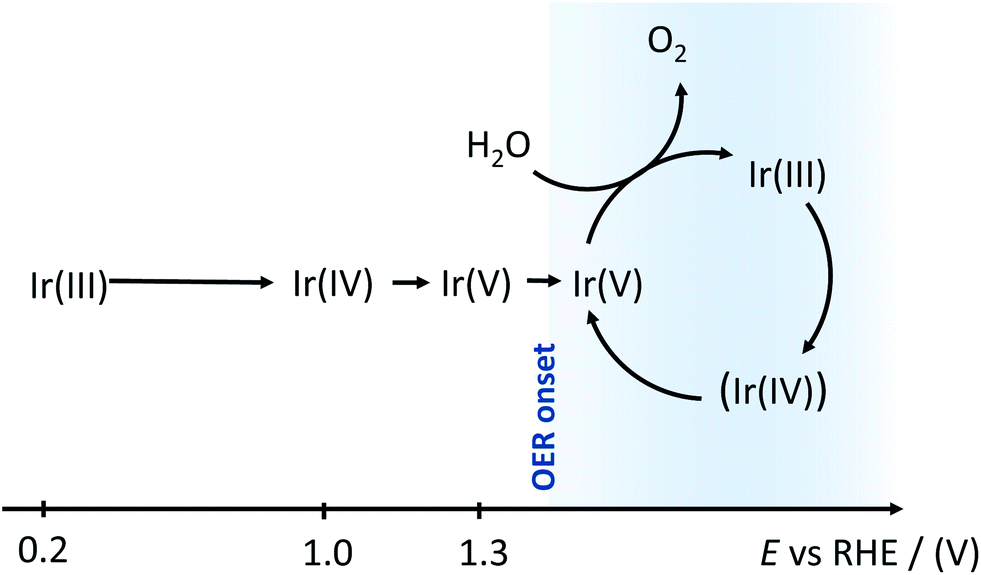 | ||
| Scheme 1 Model of the Ir oxidation states in the potential window considered up to water oxidation conditions, as derived from the XANES spectra fittings. | ||
For completeness, we report that the XANES fitting at 1.5 and 1.6 V was also tried using three Lorentzian peaks to take into account the presence of Ir(IV) in the catalytic cycle (Fig. S2†). However, the fit quality did not improve (for 1.6 V, the sum of squares is 1.274 and 1.190 respectively, versus 1.52 obtained using only one Lorentzian) and nothing could be reliably assessed about the presence of Ir(IV) in the catalytic cycle. This is why, in Scheme 1, we prefer to indicate Ir(IV) in parenthesis.
Concerning the mechanism suggested in Scheme 1, it is noteworthy that Steegstra et al.23 recently proposed, on the basis of CV and DFT calculations, a binuclear mechanism for OER that involves the formation of neighbour Ir(V) sites as a necessary condition. At the same time, at a low pH (i.e. under our working conditions), the stabilizing effects of hydrogen bonding might lead to the accumulation of a non-catalytic form of Ir(V) that shifts the observed OER onset to higher potentials. We believe that this hypothesis can satisfactorily explain the XANES data collected at 1.3 V, which indicate the presence of an Ir(V) oxide. Unfortunately, we have not been able to carry out similar experiments under higher pH values, because of the instability of EIROF under these conditions for the timespan of the experiment (recording one spectrum requires a few hours).
Fig. 4 represents a quasi steady-state E/log(j) curve of EIROF recorded under the same experimental conditions as adopted for the in situ XAS experiments. The onset of water oxidation is easily detected by drawing interpolating straight lines (thin-dashed lines), and it is apparent that it lies at about 1.38 V (which is an overpotential of 0.15 mV). Moreover, the current density needed to reach an overpotential of 0.25 V is 1.9 mA cm−2. These data are quite comparable with those reported for other types of highly hydrated IrO2 films,22 taking into account that the loading of the electrodes considered in the present work is 8 × 10−8 mol cm−2.
 | ||
| Fig. 4 Steady-state E vs. log(j) curve for EIROF in 0.5 M H2SO4. | ||
Note that the Tafel slope of the characteristics is equal to 50.2 mV dec−1, which is also in line with previous results.38
From the observed OER onset it follows that, while at 1.3 V no water oxidation occurs (in line with the “accumulation” of Ir(V)), at 1.5 and 1.6 V the reaction does proceed with fast kinetics. According to the in situ results described in this work, a rapid turnover is guaranteed by the fast cycling of the Ir(V)/Ir(III) couple.
Experimental section
Electrodeposited iridium oxide films (EIROFs) were prepared, following a modified version of a previously reported procedure:27,39 briefly, 0.0151 g of IrCl3·3H2O (Alfa Aesar) is dissolved in Milli-Q water (10 ml). After 30 min stirring, 100 μl of H2O2 (30%) is added and the resulting solution is kept under stirring conditions for 30 min. Then, 0.0518 g of oxalic acid is added. The solution is stirred for 10 min. Finally, dried K2CO3 is added until the pH reaches about 10.5. This procedure leads to the formation of a yellow solution, that turns blue/violet after 3 days at room temperature.The blue colloid is used as a deposition bath, from which IrOx is easily deposited onto a conductive support (a carbon disk deposited onto a 175 μm thick polyethylene terephthalate lamina, supplied by Dropsens) at a constant current density. We obtained durable and XAS-suitable deposits by applying 0.1 mA cm−2 for 600 s, using a Pt plate as the counter electrode.
Spectroelectrochemical cell
This consists of a poly-tetrafluoroethylene (PTFE) cell that contains the electrolyte solution (0.5 M aqueous H2SO4), a Pt foil counter electrode and the reference electrode (AgCl/Ag in 0.1 M KCl). The reference electrode is separated from the solution by a salt bridge consisting of a glass pipette filled with agar, containing 0.2 M aqueous KClO4.One side of the cell includes a hole that matches with the working electrode area. The working electrode is held between the PTFE cell and a polypropylene plate that also includes a hole for the X-ray beams. The complete experimental setup is fully described in ref. 16.
Electrochemistry
All the electrochemical measurements were carried out using a CHI potentiostat/galvanostat (model 633D) and a spectroelectrochemical cell. CV curves are recorded at 2 mV s−1, while quasi steady-state E/log(j) curves are recorded at 1 mV s−1.XAS
Fluorescence XAS (X-ray Absorption Spectroscopy) data were collected at the GILDA beamline (European Synchrotron Radiation Facility, ESRF, Grenoble) at the Ir-LIII edge.40 A Si(311) double crystal monochromator was used; the harmonic rejection was realised by Pd mirrors, having a cut-off energy of 20 keV, and a 13-element Ge fluorescence detector. The energy calibration was made by measuring the absorption spectrum of a metallic Hf foil at the Hf LI edge (Hf-LI: 11271 eV; Ir-LIII: 11215 eV). The reproducibility in energy was checked from time to time, typically after 4 or 5 scans, by measuring the absorption spectrum of an IrO2 standard, and every time it was found to be better than 0.1 eV. All measurements were carried out at room temperature. As the Ir content was as low as ca. 40 μg cm−2, self-absorption effects in the fluorescence spectra are in principle negligible. In any case, this was verified by comparing the spectra of standard IrO2 measured in transmission mode with that of the electrode material at 1 V, the two spectra being indistinguishable. In order to check the reproducibility of the results presented here, each experiment was repeated three times, at four different applied potentials, and with three electrodes prepared in different batches, obtaining identical results.Conclusions
In conclusion, in this work the electronic structure of Electrodeposited Iridium Oxide Films (EIROFs) has been investigated in situ using XAS at the Ir-LIII edge, at the applied potential where the water oxidation reaction takes place. In particular, it has been demonstrated that during the reaction Ir is present in both the Ir(III) and Ir(V) oxidation states. This represents the first experimental evidence concerning the turnover mechanism by which IrO2 acts as an electrocatalyst for the water splitting reaction. As already mentioned, the turnover between Ir(III) and Ir(V) is in agreement with the mechanisms proposed for Ir complexes in cases of homogeneous catalysis.Acknowledgements
We are thankful to Dr Netzahualcoyotl (Netz) Arroyo Curras and Prof. Allen J. Bard for fruitful discussions. BM08 (GILDA) @ESRF is acknowledged for the provision of beamtime, travel and accommodation expenses (Exp. CH-3511). Thanks are given to Dr Francesco d'Acapito for considerable help during the experiment setup and data collection. Financial support by Fondazione Cariplo (2010-0506), MIUR (Futuro in Ricerca 2013, project RBFR13XLJ9) and Università degli Studi di Milano is gratefully acknowledged.Notes and references
- Y.-F. Huang, D. Y. Wu, A. Wang, B. Ren, S. Rondinini, Z.-Q. Tian and Ch. Amatore, J. Am. Chem. Soc., 2010, 132, 17199–17210 CrossRef CAS PubMed.
- M. A. Méndez, L. Alibabaei, J. J. Concepcion and T. J. Meyer, ACS Catal., 2013, 3, 1850–1854 CrossRef.
- L. Kavan, M. Grätzel, S. E. Gilbert, C. Klemenz and H. J. Scheel, J. Am. Chem. Soc., 1996, 118, 6716–6723 CrossRef CAS.
- A. Wang, Y.-F. Huang, U. K. Sur, D. Y. Wu, B. Ren, S. Rondinini, Ch. Amatore and Z.-Q. Thian, J. Am. Chem. Soc., 2010, 132, 9534–9536 CrossRef CAS PubMed.
- X. Xiao and A. J. Bard, J. Am. Chem. Soc., 2007, 129, 9610–9612 CrossRef CAS PubMed.
- J. H. Park, S. N. Thorgaard, B. Zhang and A. J. Bard, J. Am. Chem. Soc., 2013, 135, 5258–5261 CrossRef CAS PubMed.
- A. Minguzzi, F.-R. F. Fan, A. Vertova, S. Rondinini and A. J. Bard, Chem. Sci., 2012, 3, 217–229 RSC.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS PubMed.
- J.-P. Gueneau de Mussy, J. V. Macpherson and J.-L. Delplancke, Electrochim. Acta, 2003, 48, 1131–1141 CrossRef CAS.
- P. Pedeferri, Constr. Build. Mater., 1996, 10, 391 CrossRef.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., 2010, 122, 6549–6552 CrossRef.
- H. Dau, C. Limberg, T. Reier, M. Risch, M. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- S. Fierro, T. Nagel, H. Baltruschat and C. Comninellis, Electrochem. Commun., 2007, 9, 1969–1974 CrossRef CAS PubMed.
- J. Rodriguez-Lopez, A. Minguzzi and A. J. Bard, J. Phys. Chem. C, 2010, 114, 18645–18655 CAS.
- A. Minguzzi, O. Lugaresi, C. Locatelli, S. Rondinini, F. D'Acapito, E. Achilli and P. Ghigna, Anal. Chem., 2013, 85, 7009–7013 CrossRef CAS PubMed.
- D. K. Bediako, C. Costentin, E. C. Jones, D. G. Nocera and J.-M. Savéant, J. Am. Chem. Soc., 2013, 135, 10492–10502 CrossRef CAS PubMed.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS PubMed.
- K. S. Joya, N. K. Subbaiyan, F. D'Souza and H. J. M. de Groot, Angew. Chem., Int. Ed., 2012, 51, 9601–9605 CrossRef CAS PubMed.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017–16029 CrossRef CAS PubMed.
- S. Fierro, A. Kapałka and Ch. Comninellis, Electrochem. Commun., 2010, 12, 172–174 CrossRef CAS PubMed.
- T. Nakagawa, C. A. Beasley and R. W. Murray, J. Phys. Chem. C, 2009, 113, 12958–12961 CAS.
- P. Steegstra, M. Busch, I. Panas and E. Ahlberg, J. Phys. Chem. C, 2013, 117, 20975–20981 CAS.
- Y. Mo, I. C. Stefan, W.-B. Cai, J. Dong, P. Carey and D. A. Scherson, J. Phys. Chem. B, 2002, 106, 3681–3686 CrossRef CAS.
- A. R. Hillman, M. A. Skopek and S. Gurman, Phys. Chem. Chem. Phys., 2011, 13, 5252–5263 RSC.
- J. D. Blakemore, N. D. Schley, G. W. Olack, Ch. D. Incarvito, G. W. Brudvig and R. H. Crabtree, Chem. Sci., 2011, 2, 94–98 RSC.
- M. A. Petit and V. Plichon, J. Electroanal. Chem., 1998, 444, 247–252 CrossRef CAS.
- A. Minguzzi, C. Locatelli, G. Cappelletti, C. L. Bianchi, A. Vertova, S. Ardizzone and S. Rondinini, J. Mater. Chem., 2012, 22, 8896–8902 RSC.
- S. Fierro, T. Nagel, H. Baltruschat and Ch. Comninellis, Electrochem. Commun., 2007, 9, 1969–1974 CrossRef CAS PubMed.
- I. A. Lervik, M. Tsypkin, L.-E. Owe and S. Sunde, J. Electroanal. Chem., 2010, 645, 135–142 CrossRef CAS PubMed.
- C. Locatelli and A. Minguzzi, in preparation.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- B. K. Agarwal, X-ray Spectroscopy: An Introduction (Springer Series in Optical Sciences), Springer-Verlag, Berlin, Heidelberg, New York, 1991 Search PubMed.
- L. Mandelstam and Ig. Tamm, J. Phys., 1945, 9, 249–254 Search PubMed.
- J.-H. Choy, D.-K. Kim, S.-H. Hwang, G. Demazeau and D.-Y. Jung, J. Am. Chem. Soc., 1995, 117, 8557–8566 CrossRef CAS.
- J.-H. Choy, D. K. Kim, G. Demazeau and D.-Y. Jung, J. Phys. Chem., 1994, 98, 6258–6262 CrossRef CAS.
- P. Ghigna, G. Spinolo, M. Scavini, U. A. Tamburini and A. V. Chad-wick, Physica C, 1995, 253, 147–155 CrossRef CAS.
- S. Gottesfeld and S. Srinivasan, J. Electroanal. Chem., 1978, 86, 89–104 CrossRef CAS.
- K. Yamanaka, Jpn. J. Appl. Phys., 1989, 28, 632–637 CrossRef CAS.
- F. D'Acapito, S. Colonna, S. Pascarelli, G. Antonioli, A. Balerna, A. Bazzini, F. Boscherini, F. Campolungo, G. Chini, G. Dalba, I. Davoli, P. Fornasini, R. Graziola, G. Licheri, C. Meneghini, F. Rocca, L. Sangiorgio, V. Sciarra, V. Tullio and S. Mobilio, ESRF Newsletter, 1998, 30, 42–44 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Integrated area of WLs as a function of the applied potential; XANES spectra fit using three Lorentzian + arctan. See DOI: 10.1039/c4sc00975d |
| This journal is © The Royal Society of Chemistry 2014 |