
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long-lived charge-separated states produced in supramolecular complexes between anionic and cationic porphyrins†
Nathan L.
Bill
a,
Masatoshi
Ishida
bc,
Yuki
Kawashima
d,
Kei
Ohkubo
d,
Young Mo
Sung
b,
Vincent M.
Lynch
a,
Jong Min
Lim
b,
Dongho
Kim
*b,
Jonathan L.
Sessler
*a and
Shunichi
Fukuzumi
*de
aDepartment of Chemistry, The University of Texas at Austin, 105 E. 24th Street-Stop A5300, Austin, Texas 78712-1224, USA. E-mail: sessler@cm.utexas.edu
bDepartment of Chemistry, Yonsei University, Seoul 120-749, Korea. E-mail: dongho@yonsei.ac.kr
cEducation Centre for Global Leaders in Molecular Systems for Devices, Kyushu University, Fukuoka, 819-0395, Japan
dDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370
eDepartment of Bioinspired Chemistry (WCU), Ewha Womans University, Seoul 120-750, Korea
First published on 29th May 2014
Abstract
A new supramolecular approach to generating a long-lived photoinduced charge separated state is described. It is predicated on the use of tetra-anionic sulfonated porphyrins (1-M4−: M = H2 and Zn) that form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 supramolecular complexes with dicationic zinc(II) porphyrinato tetrathiafulvalenes (2-Zn2+) via strong electrostatic interactions. The X-ray crystal structure of the complex 1-Zn4−/(2-Zn2+)2 reveals a slipped sandwich-type interaction wherein 1-Zn4− is covered on both its top and bottom faces by two separate 2-Zn2+ porphyrins. Upon photoexcitation of the supramolecular ensemble, efficient photoinduced electron transfer from 1-M4− to the triplet excited state [2-Zn2+]* occurs to afford the triplet charge-separated (CS) states, as revealed by laser flash photolysis and EPR measurements. The CS state was found to decay via intramolecular back electron transfer within the supramolecular complex. This was evidenced by the observation that the CS state decay of this three-component system obeyed first-order kinetics and afforded the same long lifetimes irrespective of the initial concentrations of the CS state (e.g., 83 ms for the 1-H24−/(2-Zn2+)2 complex in benzonitrile at 298 K). Such an extremely long CS lifetime is thought to result from the spin-forbidden back electron transfer and the small electron coupling term, as inferred from temperature dependent studies of the CS lifetime. Decay of the CS state via intermolecular back electron transfer between two separate CS species of the type [1-M˙3−/(2-Zn˙+)(2-Zn2+)] is not observed, as revealed by the absence of second order decay kinetics. The absence of appreciable bimolecular decay processes and consequently the long-lived nature of the CS state is attributed to the central radical trianionic porphyrin (1-M˙3−) being protected from close-contact interactions with other species, precluding bimolecular decay processes. This supramolecular effect is thought to be the result of the radical species, 1-M˙3−, being sandwiched between two cationic porphyrins (2-Zn˙+ and 2-Zn2+). These latter cationic entities cover the top and bottom of the anionic species thus providing both a physical and electrostatic barrier to intermolecular deactivation processes. These conclusions are supported by solution state binding studies, as well as solid state single crystal X-ray diffraction analyses.
:2 supramolecular complexes with dicationic zinc(II) porphyrinato tetrathiafulvalenes (2-Zn2+) via strong electrostatic interactions. The X-ray crystal structure of the complex 1-Zn4−/(2-Zn2+)2 reveals a slipped sandwich-type interaction wherein 1-Zn4− is covered on both its top and bottom faces by two separate 2-Zn2+ porphyrins. Upon photoexcitation of the supramolecular ensemble, efficient photoinduced electron transfer from 1-M4− to the triplet excited state [2-Zn2+]* occurs to afford the triplet charge-separated (CS) states, as revealed by laser flash photolysis and EPR measurements. The CS state was found to decay via intramolecular back electron transfer within the supramolecular complex. This was evidenced by the observation that the CS state decay of this three-component system obeyed first-order kinetics and afforded the same long lifetimes irrespective of the initial concentrations of the CS state (e.g., 83 ms for the 1-H24−/(2-Zn2+)2 complex in benzonitrile at 298 K). Such an extremely long CS lifetime is thought to result from the spin-forbidden back electron transfer and the small electron coupling term, as inferred from temperature dependent studies of the CS lifetime. Decay of the CS state via intermolecular back electron transfer between two separate CS species of the type [1-M˙3−/(2-Zn˙+)(2-Zn2+)] is not observed, as revealed by the absence of second order decay kinetics. The absence of appreciable bimolecular decay processes and consequently the long-lived nature of the CS state is attributed to the central radical trianionic porphyrin (1-M˙3−) being protected from close-contact interactions with other species, precluding bimolecular decay processes. This supramolecular effect is thought to be the result of the radical species, 1-M˙3−, being sandwiched between two cationic porphyrins (2-Zn˙+ and 2-Zn2+). These latter cationic entities cover the top and bottom of the anionic species thus providing both a physical and electrostatic barrier to intermolecular deactivation processes. These conclusions are supported by solution state binding studies, as well as solid state single crystal X-ray diffraction analyses.
Introduction
Extensive efforts have been devoted to developing mimics of the photosynthetic reaction centre. This complex biological ensemble supports a long-lived charge-separated (CS) state that is produced via photoexcitation of a chlorophyll dimer followed by a series of subsequent electron transfer events.1–6 Several synthetic systems have previously been reported with CS lifetimes comparable to, or even longer than, that of the naturally occurring photosynthetic reaction centre.7–10 Recently, supramolecular CS systems comprised of porphyrin donors and fullerene acceptors have received considerable attention. Interest in such systems reflects the fact that, in suitably designed systems, efficient photoinduced electron transfer occurs and long-lived CS states—whose decay rates parallel those observed for covalently bonded systems—are produced.11,12 However, to date photoinduced CS states of electron donor–acceptor linked molecules with lifetimes longer than milliseconds have only been attained under conditions where the positions of CS molecules are fixed through use of frozen glasses or heterogeneous media.7–10,13,14 In homogeneous solution, relatively fast diffusion rates dictate that bimolecular charge recombination processes predominate over what are expected to be slower intramolecular charge recombination pathways.7–10,13 One approach to preventing such bimolecular charge recombination processes would be to protect the electron donor moiety from interactions with other electron acceptor moieties (or vice versa). This is expected to give rise to bimolecular electron transfer processes that are highly non-adiabatic and thus significantly retarded. In the case of relatively planar systems, protection from unwanted bimolecular encounters could be achieved by covering both the top and bottom faces by large charged moieties. However, to our knowledge, supramolecular complexes that embody these design principles and which afford long-lived CS states have yet to be reported.We detail here a supramolecular ensemble composed of one tetra-anionic porphyrin and two dicationic porphyrins. Tetraphenylporphyrin tetrasulfonates (1-M4−: M = H2 and Zn) were chosen as electron donors,15 whereas a dicationic porphyrinato zinc complex (2-Zn2+), produced by the two-electron oxidation of a π-extended 1,3-dithiol-2-ylidene quinoidal porphyrin (porphyrin-bridged TTF) was used as the electron acceptor.16 It was found that 2-Zn2+ forms a 2:1 supramolecular complex with 1-M4− in benzonitrile (PhCN), wherein both the top and bottom faces are covered by large charged molecules (Scheme 1). In accord with design expectations, this system gives rise to an extremely long-lived CS state upon photoirradiation. The enhanced lifetime for the CS state relative to monomeric control systems is ascribed to an absence of bimolecular decay pathways, as discussed below.
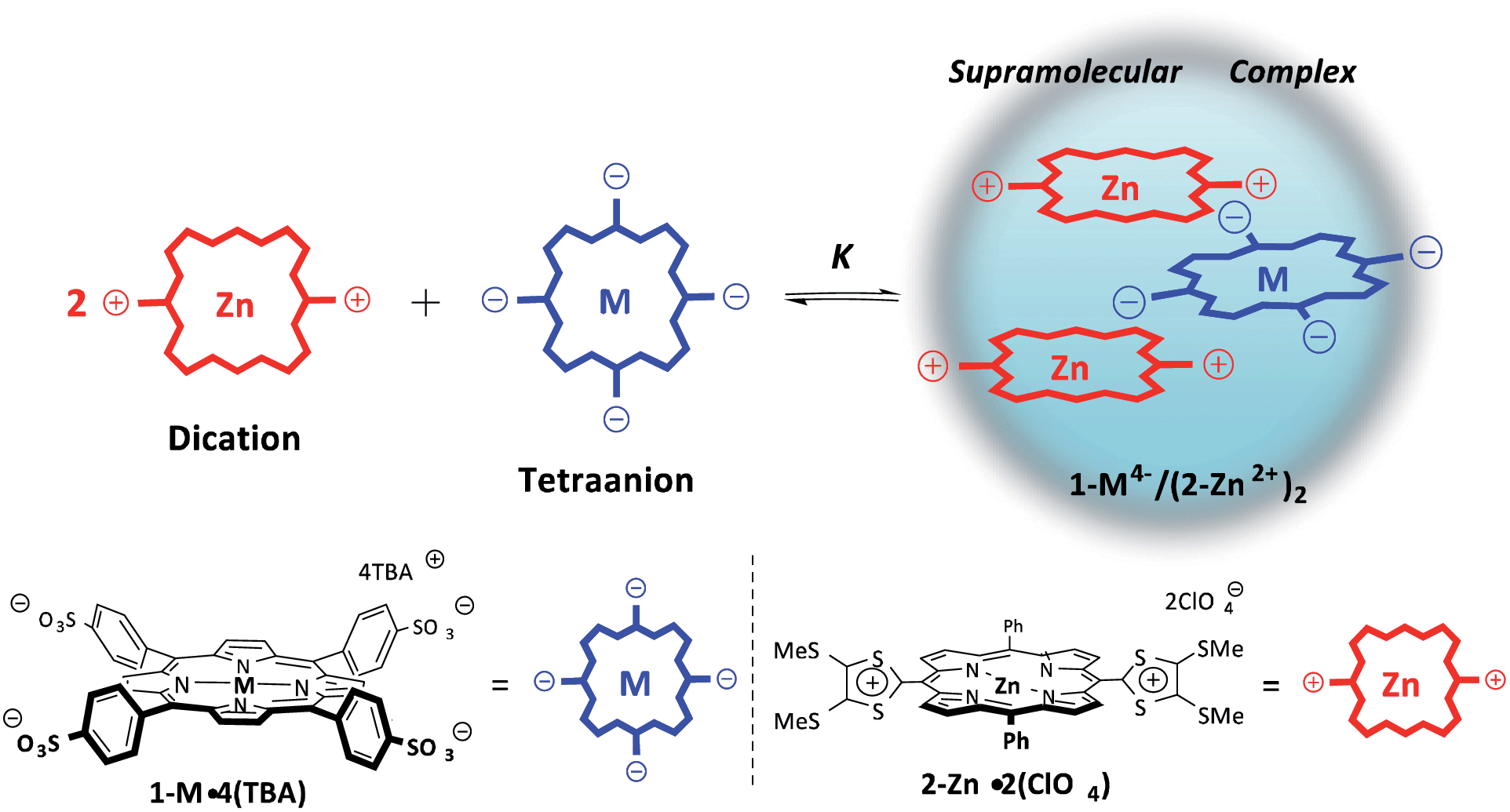 | ||
| Scheme 1 Formation of supramolecular porphyrin complexes, 1-M4−/(2-Zn2+)2; M = H2 or Zn. | ||
Results and discussion
Photocatalytic formation of the supramolecular complexes
The synthesis and characterization of 1-M·4TBA (M = H2 and Zn) and 2-Zn·2ClO4 used in this study have been previously reported.15,16 The formation of putative supramolecular assemblies of 1-H24−/(2-Zn2+)2 was first examined by means of 1H NMR spectral titration experiments carried out in DMSO-d6 (cf. Fig. S1 in the (ESI)†). Upon addition of 1-H24− to 2-Zn2+, a broadening, and slight upfield shift in the aromatic peaks of 2-Zn2+ was observed. These changes were taken as initial evidence for the formation of supramolecular complexes in DMSO-d6. However, the recorded shifts were not sufficiently large to fit the data to a standard binding profile and thus deeper insights into the nature of the complexes produced could not be drawn from this data.17Upon addition of 1-Zn4− to 2-Zn2+ in PhCN, changes in the absorption spectrum of 2-Zn2+ were observed (Fig. 1a). Isosbestic behaviour is seen, as would be expected for the intraconversion between two absorbing species. Job plot analysis is consistent with the formation of a 2:1 supramolecular ensemble involving 2-Zn2+ and 1-Zn4− (Fig. 1a). The net neutral nature of the putative ensemble leads us to suggest that its formation may be driven by electrostatic interactions. The formation constant of the supramolecular ensemble (K) is estimated by using a one-step binding model with a 1:2 overall stoichiometry according to eqn (1):
| K = [1-M4−/(2-Zn2+)2]/([2-Zn2+]2[1-M4−]) | (1) |
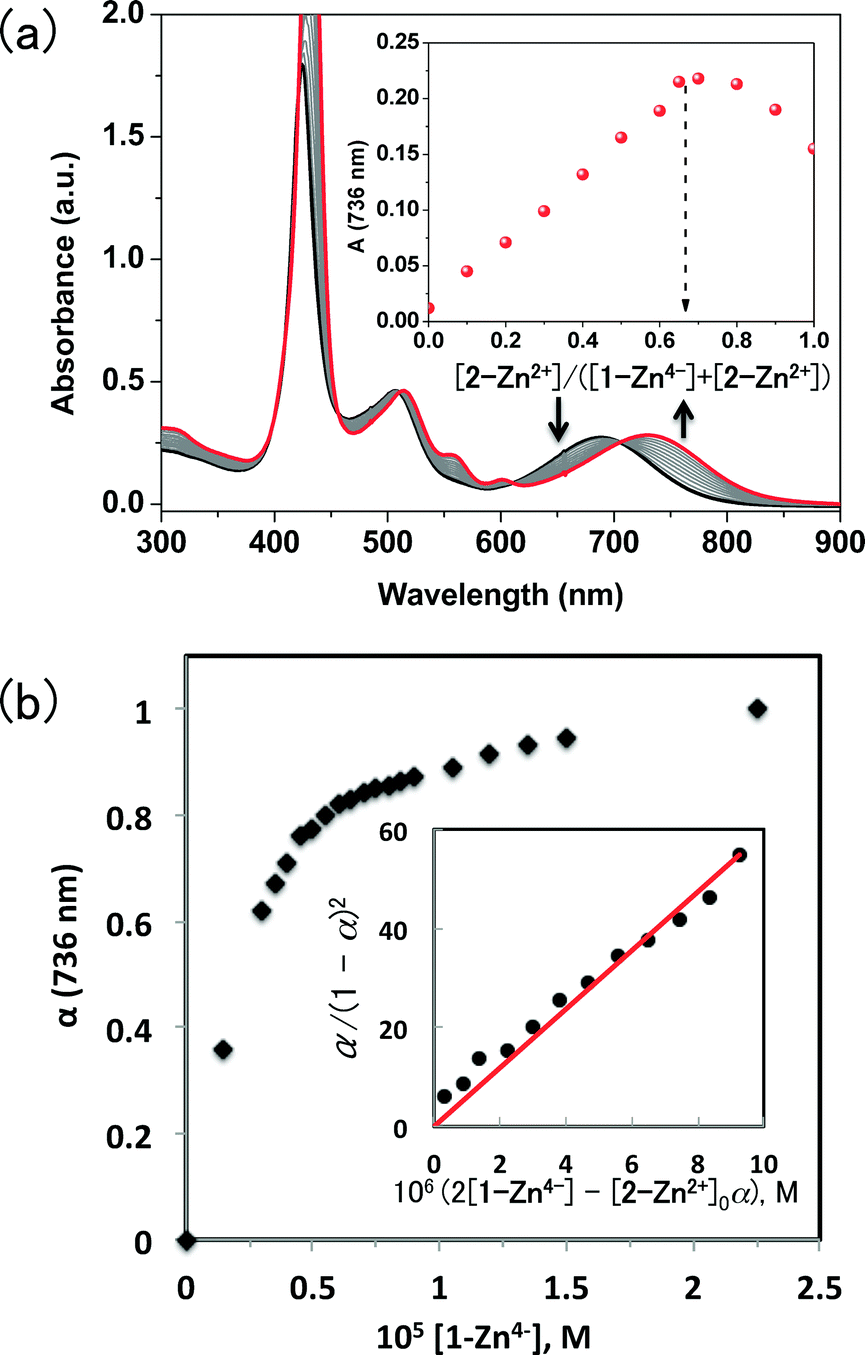 | ||
| Fig. 1 (a) Absorption spectral changes observed upon the treatment of a 10 μM PhCN solution of 2-Zn2+ with 1-Zn4− at 298 K. The original spectrum is shown with the bolded black line whereas the final spectrum is highlighted in red. The inset shows a Job plot for the UV-vis absorption at 736 nm constructed using various molar ratios of 2-Zn2+ and 1-Zn4−. (b) Plot of the ratio of the extent of complex 1-Zn4−/(2-Zn2+)2 formed (α) vs. the concentration of 1-Zn4−. The inset shows a plot of α/(1 − α)2vs. 2[1-Zn4−] − α[2-Zn2+]0; this latter plot was used to determine the association constant, K, according to eqn (3). | ||
The ratio of the complex (α) was determined from the absorbance change associated with the formation of the 1:2 self-associated ensemble 1-M4−/(2-Zn2+)2 (Fig. 1b) as given by eqn (2), where A0 and A∞ are absorbance of 2-Zn2+ and 1-M4−/(2-Zn2+)2, respectively. From eqn (1) and (2), eqn (3) is derived when the concentration of 2-Zn2+ is fixed and the concentration of 1-M4− is varied.
| α = (A − A0)/(A∞ − A0) | (2) |
| α/(1 – α)2 = K[2-Zn2+]0(2[1-M4−] – α[2-Zn2+]0) | (3) |
A near-linear plot was obtained when α/(1 − α)2 was plotted vs. 2[1-Zn4−] − α[2-Zn2+]0 as shown in Fig. 1b. From the slope, the K value of 1-Zn4−/(2-Zn2+)2 was determined to be (5.9 ± 0.5) × 1011 M−2. In the same manner, the association constant of 1-H24−/(2-Zn2+)2 was determined to be (6.6 ± 0.5) × 1012 M−2 (cf. Fig. S2b in ESI†).
Upon addition of 2-Zn2+ to 1-Zn4− in PhCN under the conditions where the 1:2 supramolecular complex 1-Zn4−/(2-Zn2+)2 is expected to dominate, an attenuation of the fluorescence spectra of 1-Zn4− was observed (Fig. 2a). This finding leads us to suggest that, upon irradiation, either photoinduced electron- or energy transfer occurs from the anionic porphyrin subunit 1-Zn4− to the 2-Zn2+ moieties in the supramolecular assemblies. In such a case, the ratio of the complex (α) can be determined from the fluorescence intensity change due to formation of the complex 1-Zn4−/(2-Zn2+)2, as per eqn (4), where I0 and I are the initial fluorescence intensity of 1-Zn4− and the fluorescence intensity in the presence of excess 2-Zn2+, respectively. From eqn (1) and (2), eqn (5) is derived when the concentration of 1-Zn4− is fixed and the concentration of 2-Zn2+ is varied.
| α = (I0 – I)I0 | (4) |
| α/(1 – α) = K([2-Zn2+] – 2α[1-Zn4−]0)2 | (5) |
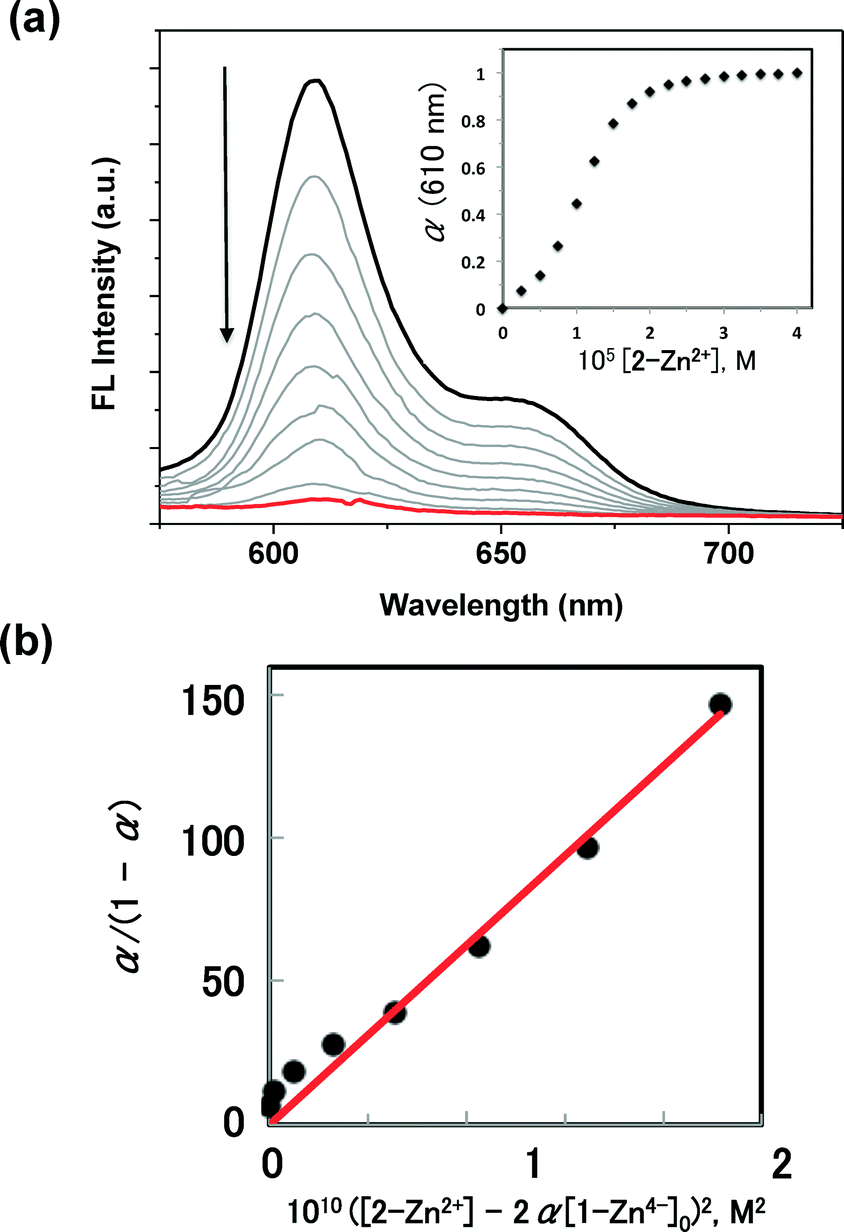 | ||
| Fig. 2 (a) Fluorescence spectral changes observed upon the treatment with a PhCN solution of 1-Zn4− (10 μM) with 2-Zn2+ at 298 K. Excitation wavelength: 430 nm. Inset shows a plot of the ratio of the complex (α) vs. the concentration of 2-Zn2+. (b) Plot of α/(1 − α) vs. ([2-Zn2+] − 2α[1-Zn4−]0)2, which was used to determine the association constant, K, according to eqn (5). | ||
An essentially linear plot was obtained when α/(1 − α) was plotted vs. ([2-Zn2+] −2α[1-Zn4−]0)2, as shown in Fig. 2b. From the slope, the K value corresponding to the formation of 1-Zn4−/(2-Zn2+)2 was determined to be (6.3 ± 0.5) × 1011 M−2; this K value agrees with that determined from the absorption spectral titration (vide infra). Such agreement provides support for the suggestion that the fluorescence quenching of 1-Zn4− observed upon treating with 2-Zn2+ reflects formation of the supramolecular complex 1-Zn4−/(2-Zn2+)2.
In an effort to evaluate the thermodynamic behaviour of the complexation processes between the ionic porphyrins, isothermal titration calorimetry (ITC) was performed in PhCN (Fig. 3).
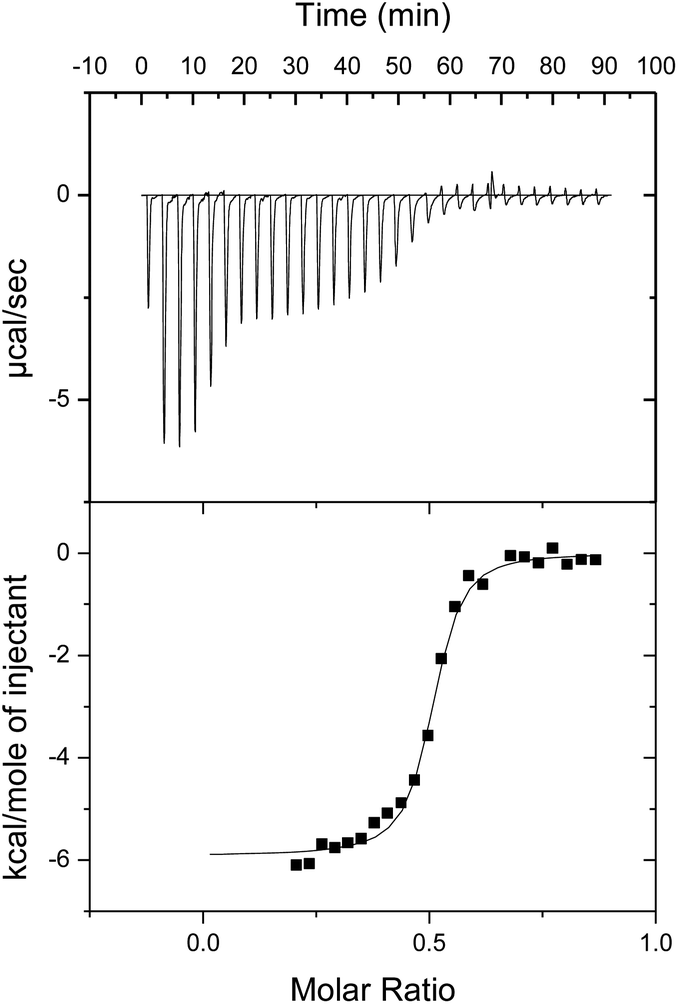 | ||
| Fig. 3 ITC plot showing the titration of 1-H24− into a PhCN solution of 2-Zn2+ at 298 K. | ||
In Fig. 3, the titration of 1-H24− into 2-Zn2+ is shown. An intense exothermic heat signature is observed over the first 6 injections. This thermodynamic process is ascribed to initial deaggregation of 2-Zn2+. Therefore these points were removed from the data set for the purpose of analyzing the binding parameters. Following the presumed initial deaggregation event, additional injections of 1-H24− led to a sigmoidal heat signal with an n-value, or equivalence point, near 0.5 molar equivalents, supporting a 1:2 1-H24− to 2-Zn2+ binding stoichiometry. Similar curves were obtained and identical conclusions were drawn from titrations of 1-Zn4− into 2-Zn2+ (Fig. S4 in ESI†). The values of heat of formation (ΔH) of 1-Zn4−/(2-Zn2+)2 and 1-H24−/(2-Zn2+)2 were determined to be −5.5 and −5.9 kcal mol−1, respectively.
Diffraction grade single crystals of the supramolecular complex 1-Zn4−/(2-Zn2+)2 suitable for X-ray analysis were obtained by layering an MeCN solution of 1-Zn·4TBA onto a THF solution of 2-Zn·2ClO4 and allowing the solutions to diffuse slowly over several weeks. The resulting structure revealed a 2:1 complex in a slipped-sandwich arrangement (Fig. 4).18 The observed structure is consistent with the presence of electrostatic interactions between the positively charged dithio lidinium rings in 2-Zn2+ and the negatively charged O-atom of the sulfonate groups in 1-Zn4−. There is no evidence of a significant contribution to the binding from π–π donor–acceptor interactions between porphyrin rings or from coordination of the sulfonate anions to the zinc-centres. The crystal lattice was characterized by an absence of external counter ions, as expected for an electronically neutral 1:2 supramolecular complex.
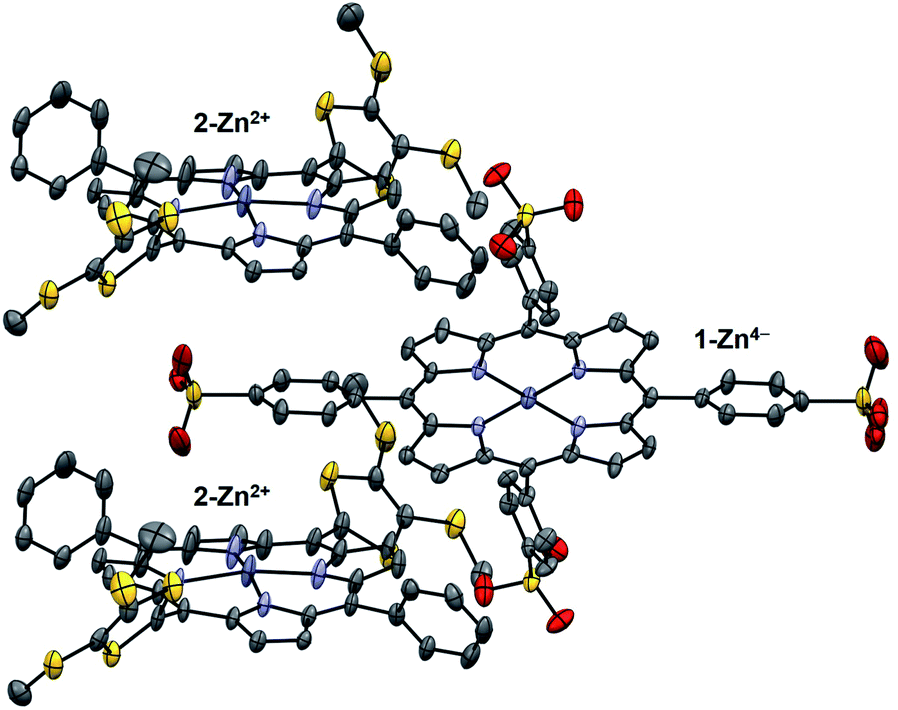 | ||
| Fig. 4 X-ray crystal structure of the supramolecular complex 1-Zn4−/(2-Zn2+)2. Thermal ellipsoids are scaled to the 50% level. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
In order to gain insight into the electronic structure of the complex, the molecular orbital distributions (e.g., HOMO and LUMO) were analyzed by single point HF/LanL2DZ calculations on the basis of the crystal structure. The electron density of the HOMO is fully localized on the 1-Zn4− moiety, whereas the LUMO are distributed among the 2-Zn2+ groups (Fig. S5 in ESI†). The intermolecular dipolar charge transfer character of the 1-Zn4−/(2-Zn2+) ensemble supports the assertion that photoinduced charge transfer involves both the 1-Zn4− donating electron density and the 2-Zn2+ acceptor moieties.
Redox potentials
Electrochemical studies, carried out using cyclic and differential pulse voltammetric (CV and DPV, respectively) methods, were performed to determine the redox potentials of the supramolecular complexes (Fig. 5). For 2-Zn·2ClO4 a two electron reduction process is observed at 0.21 V vs. SCE in PhCN. This value matches that previously reported for the oxidation of a neutral analogue of 2-Zn2+ in PhCN.16 In the supramolecular complexes, the two-electron reduction waves of 2-Zn2+ become quasi-reversible and a slight shift in the halfwave potentials is seen (to 0.17 V vs. SCE for 1-H24−/(2-Zn2+)2 and 0.19 V for 1-Zn4−/(2-Zn2+)2). The one-electron oxidation peaks of the sulfonated porphyrins, observed at 0.99 V and 0.69 V (vs. SCE, as estimated by DPV), are also shifted in the ensemble in comparison with those of free 1-H24− and 1-Zn4−, respectively. Based on these redox potentials, the thermodynamic CS energies were determined to be 0.82 eV for 1-H24−/(2-Zn2+)2 and 0.50 eV for 1-Zn4−/(2-Zn2+)2. The energy of the triplet and singlet excited states of 1-H24− (1[1-H24−]* (1.80 eV)), 3[1-H24−]* (1.48 eV) and 1-Zn4− (1[1-Zn4−]* (2.05 eV)) and 3[1-Zn4−]* (1.43 eV) are all greater than the thermodynamic CS state energy of the complexes. This provides support for the suggestion that photoinduced electron transfer processes from the excited states will be exergonic in the supramolecular complexes 1-H24−/(2-Zn2+)2 and 1-Zn4−/(2-Zn2+)2. The energy diagram for photoinduced electron transfer in 1-M4−/(2-Zn2+)2 is shown in Scheme 2.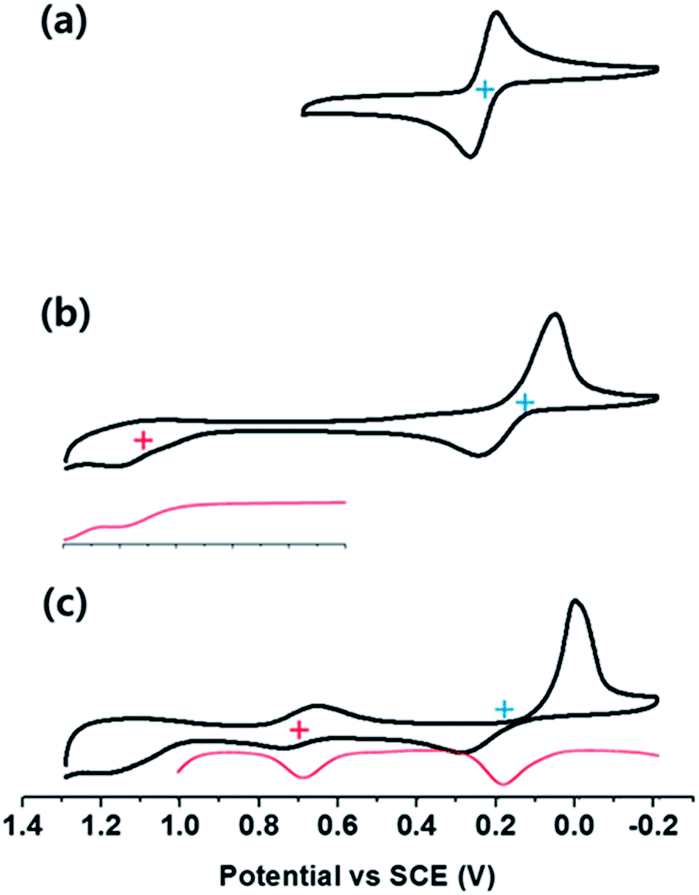 | ||
| Fig. 5 Cyclic voltammetric (CV) and differential pulse voltammetric (DPV) curves derived from electrochemical studies of (a) a 2.0 mM solution of 2-Zn·2ClO4, (b) a mixture of a 2.0 mM solution of 2-Zn·2ClO4 and a 1.0 mM solution of and 1-H2·4TBA, (c) a mixture of a 2.0 mM solution of 2-Zn·2ClO4 and a 1.0 mM solution of 1-Zn·4TBA. All spectra were performed in PhCN containing 0.1 M TBAPF6 as the supporting electrolyte. | ||
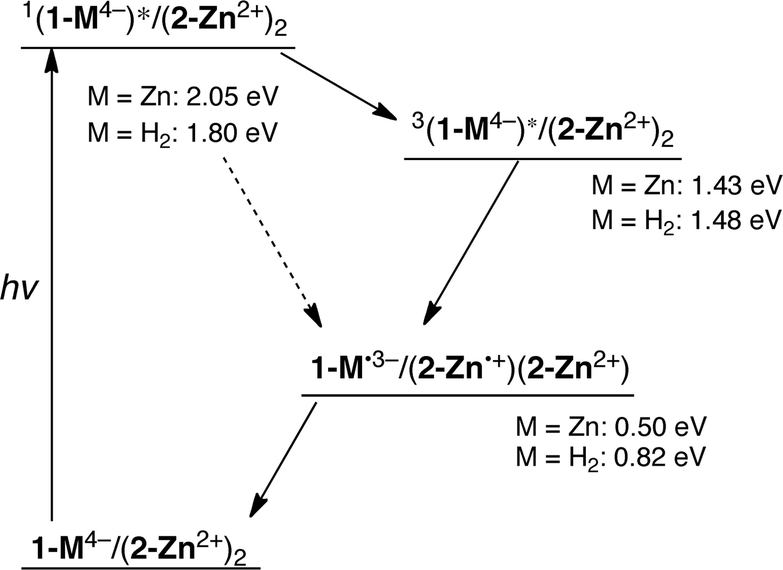 | ||
| Scheme 2 Energy diagram for the supramolecular complex 1-M4−/(2-Zn2+)2. | ||
Photodynamics
The dynamics of the photoinduced excited state of 1-M4−/(2-Zn2+)2 (M = H2 and Zn) were examined by laser flash photolysis. Upon femtosecond photoexcitation of 1-H24−/(2-Zn2+)2 at 400 nm, fast energy transfer from the S2 excited state of the 1-H24− moiety to the previously characterized S1 excited state of 2-Zn2+ is observed with a rate constant of 1.7 × 1010 s−1 (Fig. 6a).19 This is followed by intersystem crossing to populate the triplet excited state, 3[2-Zn2+]*, a process that occurs with a rate constant of 7.1 × 108 s−1. Similar transient absorption changes were observed for the supramolecular ensemble containing 1-H24− (cf. Fig. S6 in ESI†). Thus, no electron transfer occurs from the singlet excited state of 2-Zn2+ to 1-H24− in competition with intersystem crossing to the triplet excited state. This absence of electron transfer holds in spite of the large driving force (1.55 and 0.98 eV for 1-Zn4−/(2-Zn2+)2 and 1-H24−/(2-Zn2+)2, respectively). The much slower rate of electron transfer as compared to intersystem crossing may reflect the relatively small orbital interactions between the 1-H24− and 2-Zn2+ moieties present in the complex. This observation is ascribed to the slipped-sandwich arrangement between the porphyrin components (Fig. 4), which precludes π–π interactions between the donor and acceptors that make up the complex.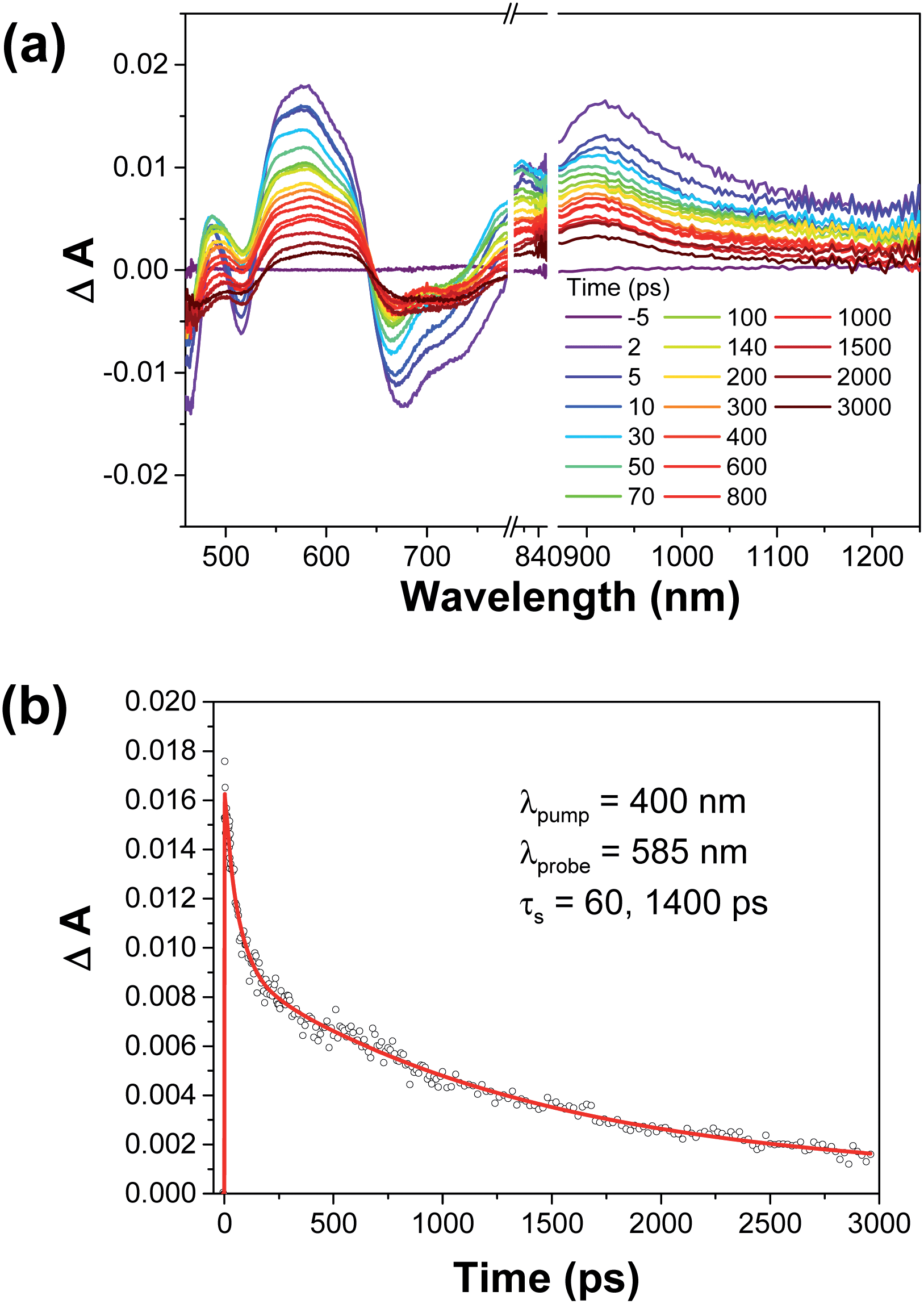 | ||
| Fig. 6 (a) Femtosecond transient absorption spectra of the supramolecular ensemble 1-H24−/(2-Zn2+)2. The spectra were recorded in situ in deaerated PhCN at 298 K following laser photoexcitation at 400 nm. (b) Time profile of the spectral traces in (a) at 585 nm. | ||
The transient absorption spectra obtained by nanosecond laser flash photolysis were consistent with photoinduced electron transfer from 1-H24− to 2-Zn2+ (Fig. 7a). For instance, a sharp absorption band at 870 nm, diagnostic of [2-Zn˙+]16 is seen. A faint absorption band originating from the radical trianion, (1-H2)˙3−, around 550 nm is also observed, although the absorption from the latter species overlaps with some of the absorption features from [2-Zn˙+].20 Based on the time course associated with the evolution of the spectral features, electron transfer from 1-H24− to 3[2-Zn2+]* is thought to occur within 3 ns and 1 μs.
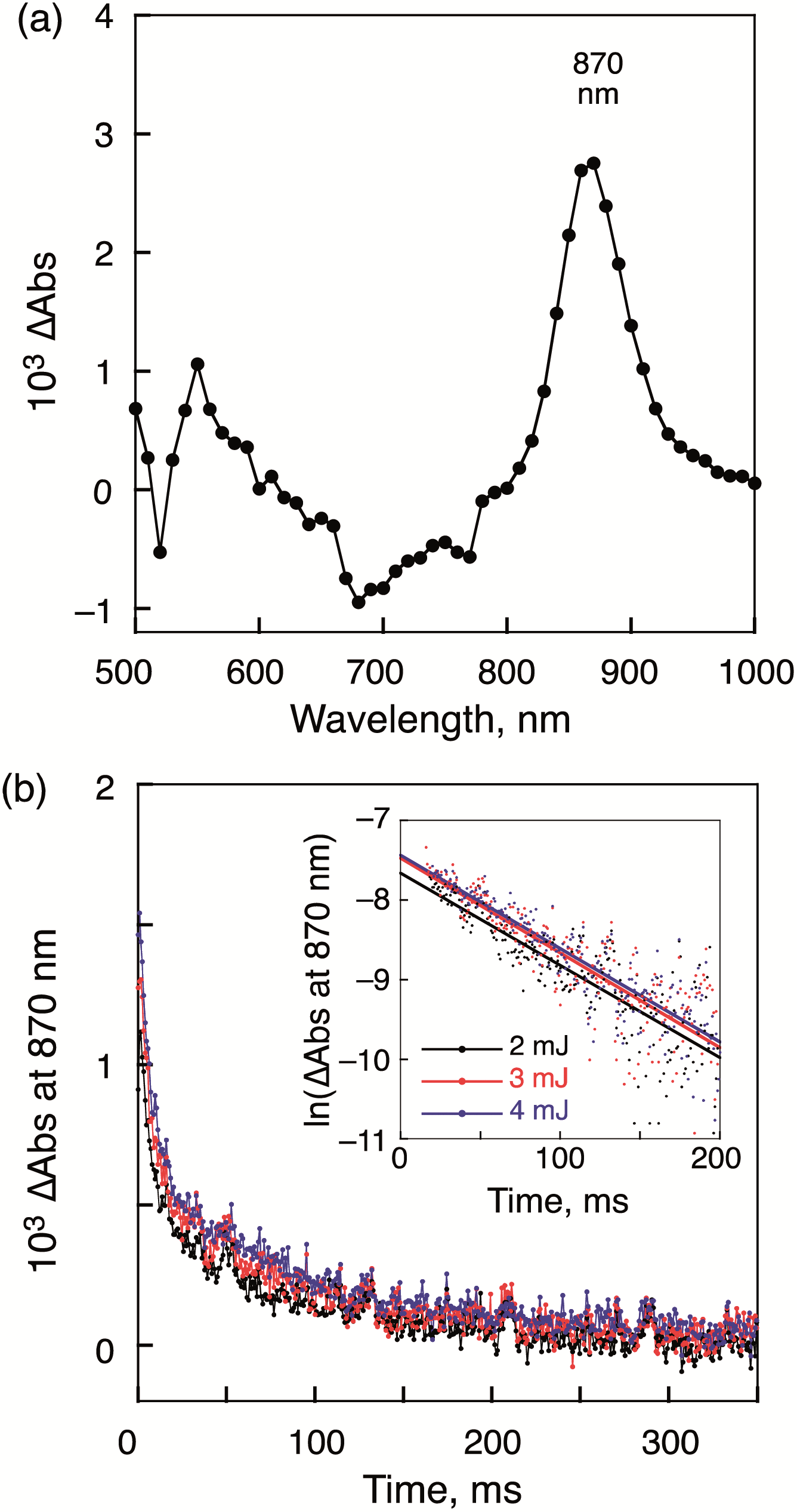 | ||
| Fig. 7 (a) Transient absorption spectra of the 1-H24−/(2-Zn2+)2 complex recorded at 1.0 ms after nanosecond laser excitation at 430 nm. [1-H2·4TBA] = 2.0 × 10−6 M, [2-Zn·2ClO4] = 4.0 × 10−6 M. (b) Decay time profiles corresponding to the change in absorbance at 870 nm recorded using various laser intensities (2–4 mJ per pulse). Inset: first-order plots. | ||
This conclusion is based on the observation that the spectral features assigned to the CS state were not evident at 3 ns, but present at 1 μs. The transient absorption signals associated with the CS state proved surprisingly persistent. The decay of the CS state obeys first-order kinetics and the slope of the first-order plots proved invariant to laser intensity and therefore the concentration of the photoinduced CS state. We take this as an indication that the charge recombination occurs within the supramolecular complex and there is no contribution from intermolecular charge recombination between supramolecular complexes. Such findings are consistent with our design expectations, namely that intermolecular charge recombination processes are attenuated as the result of electrostatic repulsion. Presumably, the two positively charged moieties, 2-Zn2+ and 2-Zn˙+, between which the radical trianionic porphyrin 1 M˙3− is sandwiched, provide a positively charged buffer layer that prevents close contact with either another supramolecular ensemble or a free electron donor moiety (i.e., 2-Zn˙+). The first-order decay rate constant, kCS of the CS state, 1-H2˙3−/(2-Zn˙+)(2-Zn2+), was determined to be 12 s−1 (Table 1). This corresponds to a CS lifetime, τCS of 83 ms. Similar measurements performed on the supramolecular complex, 1-Zn4−/(2-Zn2+)2, which also afforded millisecond long CS states (i.e., 43 ms) (cf. Fig. S7 in ESI†). No evidence of a bimolecular decay process was seen.
| Complexa | τ CS ms | k CR s−1 | λ probe nm | ||
|---|---|---|---|---|---|
| 2 mJ | 3 mJ | 4 mJ | |||
| a Generated in situ in PhCN. b CS lifetime determined by ns-transient absorption spectroscopy with 4 mJ laser power. c Rate constants of CR determined at different laser intensities. d Wavelength probed at the excited state absorption band. | |||||
| 1-Zn4−/(2-Zn2+)2 | 43 | 23 | 23 | 23 | 870 |
| 1-H24−/(2-Zn2+)2 | 83 | 12 | 12 | 12 | 870 |
According to the Marcus theory of non-adiabatic electron transfer, the temperature dependence of the first-order rate constant of electron transfer (kET) is given by eqn (6),
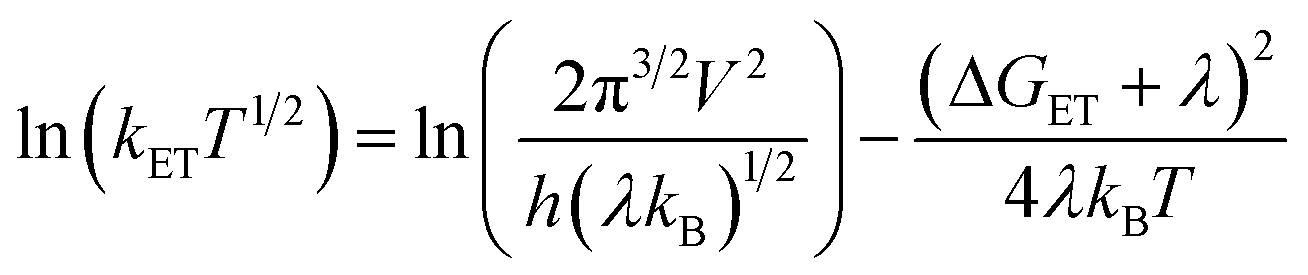 | (6) |
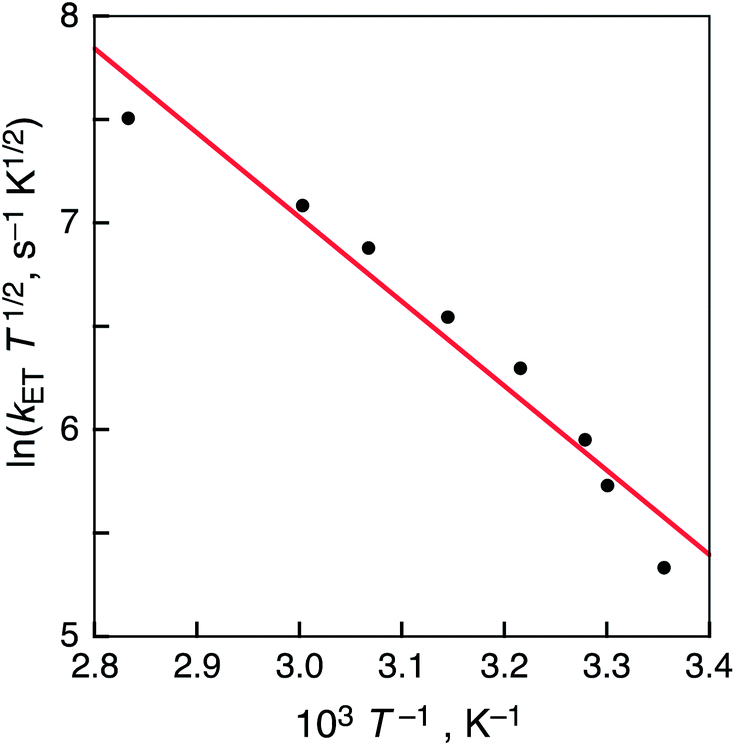 | ||
| Fig. 8 Plot of ln(kETT1/2) vs. T−1 for the charge recombination of 1-H2˙3−/(2-Zn˙+)(2-Zn2+) in PhCN. | ||
The CS states of the supramolecular ensembles 1-M4−/(2-Zn2+)2 (M = H2 and Zn) were observed by EPR measurements at 4 K upon subjecting a PhCN glass to photoirradiation with a high pressure Hg lamp (Fig. 9). EPR signals, containing broad shoulders, were observed at g = 2.0019 and 2.0934, respectively. This mixture of signals is as expected for a CS ensemble containing a sulfonated porphyrin radical trianion (g = 2.002)12d and a radical cation of a porphyrin-bridged TTF (g = 2.0036).16 A characteristic triplet signal was observed at g = 4.36.22 Considering the MO electron distribution (Fig. S5 ESI†), the triplet signal leads us to suggest that a small electronic coupling between the spatially separated donor and acceptor moieties in the ensemble may be required to achieve a long-lived CS state.23
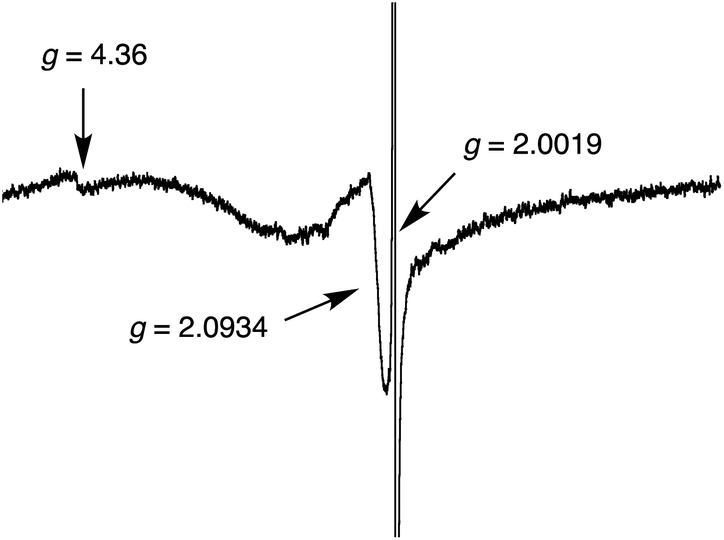 | ||
| Fig. 9 EPR spectrum of the CS state of 1-H24−/(2-Zn2+)2 complex in PhCN generated via photoirradiation with a high-pressure Hg lamp (1000 W) at 4 K. | ||
Conclusions
In summary, we have constructed two related supramolecular donor–acceptor ensembles, namely 1-H24−/(2-Zn2+)2 and 1-Zn4−/(2-Zn2+)2. These 1:2 complexes are formed in PhCN via electrostatic interactions involving tetraanionic porphyrins and dication porphyrin-bridged TTF moieties. Upon photoexcitation of the supramolecular complexes, efficient photoinduced electron transfer from 1-M4− to 2-Zn2+ occurs. The associated triplet CS states were found to be persistent in the order of milliseconds (e.g., up to 83 ms). This relatively long CS state lifetime is ascribed to the positively charged 2-Zn2+ species that flank the faces of 1-M4− and which preclude contacts with other potential donors, such as free 2-Zn2+ or an intact three-component ensemble. The present study thus illustrates a new, supramolecular-based approach to creating long-lived CS states.
Experimental section
Materials
Reagents and chemicals of the best available grade were purchased from commercial suppliers and were used without further purification. HPLC grade solvents were used in all steady-state and time-resolved spectroscopic studies. Benzonitrile (PhCN) was distilled over phosphorus pentaoxide (P2O5) immediately before use. The freebase and zinc(II) complex forms of tetra-n-butylammonium 5,10,15,20-tetrakis(phenyl-sulfonate)porphyrin (1-H2·4TBA, 1-Zn·4TBA) and zinc(II) 10,20-diphenyl-5,15-bis(1,3-dithiol-2-yl)porphyrinato perchlorate (2-Zn·2ClO4) were prepared using literature methods.12d,16 The tetra-n-butylammonium hexafluorophosphate (TBAPF6) used for electrochemical studies was recrystallised twice from ethanol and dried in vacuo prior to use.Instruments
1H NMR spectra were recorded on a Bruker Avance II (400 MHz) for 1H spectrometer. Chemical shifts (δ-scale) are given in reference to the peak of residual non-deuterated dimethyl sulfoxide (2.50 ppm for 1H). UV-vis absorption spectra were recorded on a Hewlett Packard 8453 diode array spectrophotometer. Fluorescent spectra were recorded on a Horiba FluoroMax-4 spectrophotometer. All electrochemical measurements were performed on a CH Instruments 630B potentiostat in dry benzonitrile containing 0.1 M TBAPF6 as a supporting electrolyte under N2-atmosphere, using a glassy carbon electrode (3.0 mm diameter) as a working electrode, a Ag/AgNO3 electrode as a reference electrode, and a Pt wire as an auxiliary electrode. The glassy carbon electrode was polished prior to each measurement with BAS polishing alumina suspension and rinsed with deionized water and acetone before use. All potentials (vs. Ag/Ag+) were converted to values vs. SCE by adding 0.29 V.24Isothermal titration calorimetry (ITC)
ITC measurements were performed on a VP-ITC made by MircroCal. All measurements were performed at 298 K, and a stir rate of 307 rpm. The calorimeter reference power was set at 25 μcal s−1. The injection volumes were 7 μL in both cases. The titrations were continued until a minimum of 1.5 molar equivalents of 1-M4− had been added. The data was analyzed using Origin software provided by MircoCal.X-ray crystallography
Crystals of 1-Zn4−/(2-Zn2+)2 were grown by layering an MeCN solution of 1-Zn·4TBA onto a THF solution of 2-Zn·2ClO4 with a minimal amount of EtOH added to ensure solubility (<5%). Over the course of three weeks, crystals of the complex grew as long dark green needles. The datum crystal was cut from a larger crystal and had approximate dimensions: 0.65 × 0.19 × 0.16 mm. The data were collected at 153 K on a Nonius Kappa CCD diffractometer using a Bruker AXS Apex II detector and a graphite monochromator with MoKα radiation (λ = 0.71075 Å). Reduced temperatures were maintained by use of an Oxford Cryosystems 600 low-temperature device. A total of 1282 frames of data were collected using θ-scans with a scan range of 1.1° and a counting time of 44 seconds per frame. Details of crystal data, data collection, and structure refinement can be found in the ESI in Table S1†. Data reduction were performed using SAINT V8.27B.25 The structure was solved by direct methods using SIR9726 and refined by full-matrix least-squares on F2 with anisotropic displacement parameters for the non-H atoms using SHELXL-97.27 Structure analysis was aided by use of the programs PLATON9828 and WinGX.29Two molecules of solvent, one an ethanol molecule and one a molecule of n-hexane, were badly disordered. Attempts to model the disorder were unsatisfactory. The contributions to the scattering factors due to these solvent molecules were removed by use of the utility SQUEEZE30 in PLATON98.
The function, Σw(|Fo|2 − |Fc|2)2, was minimized, where w = 1/[(σ(Fo))2 + (0.087P)2 + (0.109P)] and P = (|Fo|2 + 2|Fc|2)/3. Rw(F2). Rw(F2) refined to 0.174, with R(F) equal to 0.0666 and a goodness of fit, S = 1.13. Definitions used for calculating R(F), Rw(F2) and the goodness of fit, S, are given in ESI†.31 The data were checked for secondary extinction but no correction was necessary. Neutral atom scattering factors and values used to calculate the linear absorption coefficient are from the International Tables for X-ray Crystallography (1992).32 All figures were generated using SHELXTL/PC.33 Tables of positional and thermal parameters, bond lengths and angles and torsion angles are found in the ESI† (Tables S2–S6).
Laser flash photolysis measurements
Femtosecond transient absorption spectroscopy experiments were carried out in PhCN solution. The femtosecond time-resolved transient absorption (fs-TA) setup consisted of Optical Parametric Amplifiers (OPA) (Palitra, Quantronix) pumped by a Ti:sapphire regenerative amplifier system (Integra-C, Quantronix) operating at 1 kHz repetition rate and an optical detection system. The generated OPA pulses had a pulse width of ∼100 fs and an average power of 100 mW in the 280–2700 nm spectral range. These were used as pump pulses. White light continuum (WLC) probe pulses were generated using a sapphire window (3 mm of thickness) by focusing a small portion of the fundamental 800 nm pulses, which were picked off by a quartz plate before entering into the OPA. The time delay between the pump and probe beams was carefully controlled by making the probe beam travel along a variable optical delay (ILS250, Newport). The intensities of the spectrally dispersed WLC probe pulses were monitored by a High Speed spectrometer (Ultrafast Systems). To obtain the time-resolved transient absorption difference signal (DA) at a specific time, the pump pulses were chopped at 500 Hz and the absorption spectra intensities were saved alternately with or without the pump pulse. Typically, 4000 pulses were used to excite the samples and to obtain the fs-TA spectrum at a particular delay time. The polarization angle between the pump and probe beam was set at the magic angle (54.7°) using a Glan-laser polarizer with a half-wave retarder in order to prevent polarization-dependent signals. The cross-correlation full width at half maximum in pump-probe experiments was less than 200 fs and the chirp of the WLC probe pulses was measured to be 800 fs in the 400–800 nm region. To minimize chirp, all reflection optics were used in the probe beam path and a 2 mm path length of quartz cell was employed. After the fs-TA experiments, the absorption spectra of all compounds was carefully checked to detect if there were artifacts due to degradation and photo-oxidation of the samples in question. Three-dimensional data sets of ΔA versus time and wavelength were subjected to singular value decomposition and global fitting to obtain the kinetic time constants and their associated spectra using the Surface Xplorer software (Ultrafast Systems).Nanosecond time-resolved transient absorption measurements were carried out in PhCN solution using a laser system provided by UNISOKU Co., Ltd. Measurements of nanosecond transient absorption spectra were performed according to the following procedure: A deaerated solution was excited by a Panther optical parametric oscillator pumped by a Nd:YAG laser (Continuum, SLII-10, 4–6 ns FWHM) at λ = 430 nm. The photodynamics were monitored by continuous exposure to a xenon lamp (150 W) as a probe light and a photomultiplier tube (Hamamatsu 2949) as a detector.
EPR spectroscopy
The EPR spectra were taken on a JEOL X-band spectrometer (JES-RE1XE) with a quartz ESR tube. The EPR spectrum of the charge-separated state in frozen PhCN was measured under photoirradiation with a high-pressure mercury lamp (USHIO LIGHTING USH–1005D) through a water filter focusing at the sample cell in the EPR cavity using a liquid helium cryostat before making the spectroscopic measurements at 4 K. The g value was calibrated using a Mn2+ marker.Theoretical calculations
Hartree–Fock calculations were performed using the Gaussian 09 program34 suite on a supercomputer (KISTI, IBM). The MO distribution diagram of 1-Zn4−·(2-Zn2+)2 was obtained via the HF method employing LanL2DZ basis set for all atoms.Acknowledgements
Support is acknowledged from the U.S. National Science Foundation (CHE-1057904 to J.L.S.), the Robert A. Welch Foundation (F-1018 to J.L.S.), JSPS (no. 20108010 to S.F, 26620154 and 26288037 to K.O., 26810024 to M.I. and 25·627 to Y.K.), an ALCA fund, JST (to S.F.), the Global Frontier R&D Program on Centre for Multiscale Energy System funded by the National Research Foundation under the Ministry of Science, ICT & Future, Korea (2012-8-2081 to D. K).References
- (a) The Photosynthetic Reaction Centre, ed. J. Deisenhofe and J. R. Norris, Academic Press, San Diego, 1993 Search PubMed; (b) Molecular Level Artificial Photosynthetic Materials, ed. G. J. Meyer, Wiley, New York, 1997 Search PubMed.
- (a) J. Frey, G. Kodis, S. D.Straight, T. A. Moore, A. L. Moore and D. Gust, J. Phys. Chem. A, 2013, 117, 607 CrossRef CAS PubMed; (b) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 1993, 26, 198 CrossRef CAS; (c) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS PubMed.
- (a) A. B. Ricks, K. E. Brown, M. Wenninger, S. D. Karlen, Y. A. Berlin, D. T. Co and M. R. Wasielewski, J. Am. Chem. Soc., 2012, 134, 4581 CrossRef CAS PubMed; (b) V. L. Gunderson, A. L. Smeigh, C. H. Kim, D. T. Co and M. R. Wasielewski, J. Am. Chem. Soc., 2012, 134, 4363 CrossRef CAS PubMed; (c) F. D. Lewis, R. L. Letsinger and M. R. Wasielewski, Acc. Chem. Res., 2001, 34, 159 CrossRef CAS PubMed; (d) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS PubMed.
- (a) V. Balzani, P. Ceroni, A. Juris, M. Venturi, S. Campagna, F. Puntoriero and S. Serroni, Coord. Chem. Rev., 2001, 219–221, 545 CrossRef CAS; (b) J. A. Faiz, V. Heitz and J.-P. Sauvage, Chem. Soc. Rev., 2009, 38, 422 RSC.
- (a) S. Fukuzumi, Org. Biomol. Chem., 2003, 1, 609 RSC; (b) S. Fukuzumi, Phys. Chem. Chem. Phys., 2008, 10, 2283 RSC; (c) S. Fukuzumi, Bull. Chem. Soc. Jpn., 2006, 79, 177 CrossRef CAS; (d) S. Fukuzumi and T. Kojima, J. Mater. Chem., 2008, 18, 1427 RSC; (e) S. Fukuzumi, T. Honda, K. Ohkubo and T. Kojima, Dalton Trans., 2009, 3880 RSC; (f) K. Ohkubo and S. Fukuzumi, Bull. Chem. Soc. Jpn., 2009, 82, 303 CrossRef CAS; (g) K. Ohkubo and S. Fukuzumi, J. Porphyrins Phthalocyanines, 2008, 12, 993 CrossRef CAS; (h) S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455 CrossRef CAS PubMed.
- (a) D. M. Guldi, Chem. Commun., 2011, 47, 606 RSC; (b) D. M. Guldi, A. Rahman, V. Sgobba and C. Ehli, Chem. Soc. Rev., 2006, 35, 471 RSC; (c) G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768 CrossRef CAS PubMed; (d) N. Martin, L. Sanchez, M. A. Herranz, B. Illescas and D. M. Guldi, Acc. Chem. Res., 2007, 40, 1015 CrossRef CAS PubMed.
- (a) H. Imahori, Y. Sekiguchi, Y. Kashiwagi, T. Sato, Y. Araki, O. Ito, H. Yamada and S. Fukuzumi, Chem.–Eur. J., 2004, 10, 3184 CrossRef CAS PubMed; (b) H. Imahori, D. M. Guldi, K. Tamaki, Y. Yoshida, C. Luo, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 6617 CrossRef CAS PubMed; (c) D. M. Guldi, H. Imahori, K. Tamaki, Y. Kashiwagi, H. Yamada, Y. Sakata and S. Fukuzumi, J. Phys. Chem. A, 2004, 108, 541 CrossRef CAS.
- (a) S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600 CrossRef CAS PubMed; (b) K. Ohkubo, H. Kotani and S. Fukuzumi, Chem. Commun., 2005, 4520 RSC; (c) S. Fukuzumi, H. Kotani and K. Ohkubo, Phys. Chem. Chem. Phys., 2008, 10, 5159 RSC.
- (a) M. Murakami, K. Ohkubo and S. Fukuzumi, Chem.–Eur. J., 2010, 16, 7820 CrossRef CAS PubMed; (b) M. Murakami, K. Ohkubo, T. Nanjo, K. Souma, N. Suzuki and S. Fukuzumi, ChemPhysChem, 2010, 11, 2594 CrossRef CAS PubMed.
- S. Fukuzumi, K. Doi, T. Suenobu, K. Ohkubo, Y. Yamada and K. D. Karlin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15572 CrossRef CAS PubMed.
- (a) F. D’Souza and O. Ito, Chem. Commun., 2009, 4913 RSC; (b) S. Fukuzumi, K. Saito, K. Ohkubo, T. Khoury, Y. Kashiwagi, M. A. Absalom, S. Gadde, F. D’Souza, Y. Araki, O. Ito and M. J. Crossley, Chem. Commun., 2011, 47, 7980 RSC; (c) J. L. Sessler, E. Karnas, S. K. Kim, Z. Ou, M. Zhang, K. M. Kadish, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2009, 130, 15366 Search PubMed; (d) T. Kojima, T. Honda, K. Ohkubo, M. Shiro, T. Kushukawa, T. Fukuda, N. Kobayashi and S. Fukuzumi, Angew. Chem., Int. Ed., 2008, 47, 6712 CrossRef CAS PubMed.
- (a) S. Fukuzumi, K. Ohkubo, F. D’Souza and J. L. Sessler, Chem. Commun., 2012, 48, 9801 RSC; (b) S. Fukuzumi and K. Ohkubo, J. Mater. Chem., 2012, 22, 4575 RSC; (c) T. Kamimura, K. Ohkubo, Y. Kawashima, H. Nobukuni, Y. Naruta, F. Tani and S. Fukuzumi, Chem. Sci., 2013, 4, 1451 RSC; (d) K. Ohkubo, Y. Kawashima and S. Fukuzumi, Chem. Commun., 2012, 48, 4314 RSC.
- Y. Yamada, A. Nomura, K. Ohkubo, T. Suenobu and S. Fukuzumi, Chem. Commun., 2013, 49, 5132 RSC.
- N. Mizoshita, K.-i. Yamanaka, T. Shimada, T. Tani and S. Inagaki, Chem. Commun., 2010, 46, 9235 RSC.
- Z. Zakavi, H. Rahiminezhad and R. Alizadeh, Spectrochim. Acta, Part A, 2010, 994 CrossRef PubMed.
- N. L. Bill, M. Ishida, S. Bähhring, J. M. Lim, S. Lee, V. M. Lynch, K. A. Nielsen, J. O. Jeppesen, K. Ohkubo, S. Fukuzumi, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2013, 135, 10852 CrossRef CAS PubMed.
- Meaningful 1H NMR spectroscopic analyses in less polar solvents were precluded by the lack of requisite solubility.
- See the details of the X-ray experiments in ESI.†.
- The excited state dynamics of 2-Zn2+ were also analyzed by fs-TA data; see Fig. S7 in the ESI.†.
- The reference spectrum of radical cation species of 2-Zn2+ generated by thermal electron transfer with Li+-encapsulated fullerene (Li+@C60) was reported in ref. 16; the λmax is at 876 nm.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265 CrossRef CAS.
- The smaller D value reflects a low transition probability for the forbidden transition; this results in the disappearance of the fine structure that might be expected in the EPR spectrum.
- Y. Kawashima, K. Ohkubo, K. Mase and S. Fukuzumi, J. Phys. Chem. C, 2013, 117, 21166 CAS.
- Electrochemical Reactions in Non-aqueous Systems, ed. C. K. Mann and K. K. Barnes, Mercel Dekker, New York, 1970 Search PubMed.
- SAINT V8.27B, Bruker AXS Inc, Madison, Wisconsin, 2012 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. J. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112 CrossRef PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, The Netherlands, 1998 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- P. v. d. Sluis and A. L. Spek, Acta Crystallogr., 1990, A46, 194 CrossRef.
- Rw(F2) = {Σw(|Fo|2 − |Fc|2)2/Σw(|Fo|)4}1/2 where w is the weight given each reflection. R(F) = Σ(|Fo| − |Fc|)/Σ(|Fo|) for reflections with Fo > 4(σ(Fo)). S = [Σw((|Fo|2 −|Fc|2))2/(n – p)]1/2, where n is the number of reflections and p is the number of refined parameters.
- International Tables for X-ray Crystallography, vol. C, Tables 4.2.6.8 and 6.1.1.4, ed. A. J. C. Wilson, Boston, Kluwer Academic Press, 1992 Search PubMed.
- G. M. Sheldrick, SHELXTL/PC (Version 5.03), Siemens Analytical X-ray Instruments, Inc., Madison, Wisconsin, USA, 1994 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting Fig. S1–S7, and Tables S1–S7. CCDC 938473. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc00803k |
| This journal is © The Royal Society of Chemistry 2014 |