 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescent and chemico-fluorescent responsive polymers from dithiomaleimide and dibromomaleimide functional monomers†
Mathew P.
Robin
and
Rachel K.
O'Reilly
*
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: Rachel.OReilly@warwick.ac.uk; Tel: +44 (0)247 652 3236
First published on 16th May 2014
Abstract
A new class of brightly fluorescent and profluorescent methacrylate and acrylate monomers is reported. The fluorescent monomers contain the dithiomaleimide (DTM) fluorophore, which imparts a large Stokes shift (up to 250 nm) and bright emission. Furthermore, the simple and efficient chemistry of the DTM group, as well as its excellent processability (highly soluble, neutral functional group) makes monomer preparation straightforward. Copolymerisation at 10 mol% loading with a range of hydrophobic and hydrophilic monomers is demonstrated by RAFT polymerisation. Reactions proceed to high monomer conversion with excellent control over molecular weight (ĐM < 1.3) under standard polymerisation conditions. Incorporation of these fluorescent DTM-functional monomers has little effect on polymer properties, with PEG (meth)acrylate copolymers retaining their water solubility and thermoresponsive behaviour. A thiol-exchange reaction is also possible, whereby the thiol ligands of the pendent DTM groups can be exchanged by conjugate addition–elimination with an alternative thiol. Monomers containing the dibromomaleimide (DBM) group gave profluorescent copolymers. Reaction of the DBM group with thiols (to form the DTM group) corresponds to a chemico-fluorescent response, leading to an OFF-to-ON switching of fluorescence. This post-polymerisation functionalisation is shown to be fast and highly efficient (>95% conversion in 3 h), and by using thiols of different polarities can be used to progressively tune the LCST cloud point of a thermoresponsive polymer over a range of 11 °C. Therefore, both DTM and DBM functional monomers provide a simple and effective tool for fluorescent labelling of (meth)acrylate polymers.
Introduction
The polymerisation of dye molecules that are functionalised with a vinyl group (fluorescent vinyl monomers) allows incorporation of fluorophore units along a polymer backbone. These fluorescent vinyl monomers are highly versatile, as they are compatible with reversible-deactivation radical polymerisation (RDRP) processes,1 such as nitroxide mediated polymerisation (NMP),2 atom transfer radical polymerisation (ATRP),3 and reversible addition-fragmentation chain transfer (RAFT) polymerisation.4 Copolymers, block copolymers and homopolymers of fluorescent vinyl monomers have found a myriad of applications in organic electronic devices, sensor materials, studying the physical properties of polymers, and for labelling polymer materials (nanoparticles, hydrogels, membranes etc.) for fluorescence detection/imaging in biomedical applications.5 Polymers can also be fluorescently labelled using an end-group modification approach,6 but there are several advantages of using fluorescent vinyl monomers. For example when using these monomers the degree of fluorophore incorporation (and hence its concentration in the final polymer) can be simply varied by altering the monomer feed, as opposed to an end-group labelling approach which is limited to one or two fluorophores per chain. Another advantage is that it doesn't require any modification to either the initiator or the final polymer. Furthermore if a fluorescent vinyl monomer is used, the resultant polymer end-groups remain ‘available’, allowing for further modification of the polymer by end-group functionalisation or conjugation. The importance and utility of fluorescent vinyl monomers is illustrated by the wealth of variations that have been investigated, with a recent review of the literature finding over 200 different examples.5 Popular amongst these include vinyl monomers based on polyaromatic hydrocarbons (such as naphthalene,7 pyrene,8 perylene,9 and anthracene10), fluorescein,11 rhodamine,12 coumarin,13 naphthalimide,14 BODIPY,15 and oxadiazole16 fluorophores. However, the molecular weight and relative dimensions of many of these frequently used fluorophores are significant, with the result that they can significantly alter polymer properties. For example the incorporation of highly hydrophobic aromatic fluorophores can dramatically affect the solubility of hydrophilic polymers in aqueous solution, which has been shown to result in polymer aggregation and increased surface adsorption from solution.17As well as permanently fluorescent labels, it is highly desirable to be able to generate an emissive fluorophore from a non-fluorescent labelling agent (i.e. a latent fluorophore or profluorophore) upon completion of a targeted reaction. As opposed to the attachment of an already emissive species, use of a profluorophore gives a clear indication that fluorescent labelling has been achieved at the desired location, as an OFF-to-ON change in emission will occur. For polymeric systems this has been achieved by using a quenched fluorophore where the target reaction results in the loss of the quenching group. For example, copper-catalysed azide–alkyne cycloaddition (CuAAC) of ‘quenched’ 3-azidocoumarin with an alkyne functional polymer leads to the formation of an emissive triazole-coumarin functional polymer.18 Cleavage of a quenching ‘trimethyl lock’ from a rhodamine functional polymer by intracellular esterases has also been used to generate an OFF-to-ON change in emission,19 as has the nitroxide exchange reaction of an NMP synthesised polymer with an isoindoline conjugated to (and therefore quenched by) a nitroxide.20 Fluorescence emission can also be triggered where the labelling reaction is also the fluorophore forming reaction, i.e. two non-fluorescent groups react to form a fluorophore. These are much rarer, as they require the reaction that generates the fluorophore to be highly efficient, if it is to have utility as a labelling reaction for macromolecules. The tetrazole-alkene/azirine-alkene cycloaddition results in emissive products as demonstrated by Lin and colleagues,21 and this reaction has been used for protein–polymer conjugation (PEGylation),22 and for polymer–polymer conjugation both in solution and on a silicon or cellulose surface.23
We have recently demonstrated that the conjugate-addition of dibromomaleimide (DBM) with thiols results in the formation of a fluorescent dithiomaleimide (DTM) product.24 This fast and highly efficient reaction has been utilised for PEGylation,24,25 disulfide bridging of proteins for bioconjugation,26 glycoprotein synthesis,26a,27 polymer end-group functionalisation,28 polymer–polymer conjugation,29 and the synthesis of cyclic30 and sequence-ordered polymers.31 We have also shown that the DTM fluorophore can be incorporated into a block copolymer micelle at the junction between the core forming poly(lactide) block, and the corona forming poly(PEG acrylate) block. Due to the DTM group's small size and intermediate polarity, it had no detrimental effect on block copolymer self-assembly. In the micellar state the DTM does not self-quench leading to a significant increase in emission, and a concentration-independent emission and anisotropy profile over 3 orders of magnitude concentration range.32 Furthermore, time-domain fluorescence-lifetime imaging (FLIM) was shown to be able to resolve differences in the supramolecular state in vitro, differentiating assembled and dis-assembled micelles. However, one drawback to this approach is the use of a DTM functional dual ROP initiator/RAFT agent for the block copolymer synthesis, limiting the versatility of this approach.
Herein we report the synthesis and RAFT copolymerisation of novel methacrylate and acrylate fluorescent DTM monomers, and chemico-fluorescent responsive DBM monomers. We demonstrate that the fluorescent DTM monomers result in the formation of highly emissive polymers, while the DBM monomers give access to chemico-fluorescent responsive polymers (Fig. 1). We show that these DBM functional polymers undergo a fast and highly efficient conjugation-induced fluorescent labelling reaction with thiols, to form fluorescent products. We believe that these new DTM and DBM monomers allow a much more versatile and efficient route to polymers labelled with the highly desirable DTM fluorophore.
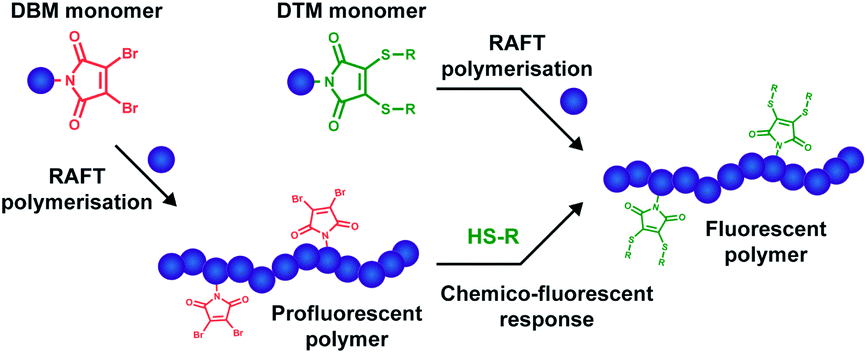 | ||
| Fig. 1 Strategy for the preparation of fluorescent polymers using DTM monomers, and profluorescent polymers that undergo a chemico-fluorescent response using DBM monomers. | ||
Results and discussion
Monomer synthesis
Novel dithiomaleimide (DTM) and dibromomaleimide (DBM) monomers were prepared according to Scheme 1. The methacrylate derivatives dithiomaleimide methylmethacrylate (DTMMA) and dibromomaleimide methylmethacrylate (DBMMA), were synthesised by alkylation of butanethiol-DTM24 (1) or commercially available 2,3-DBM with bromoacetyl methacrylate33 (the latter being prepared in a single step according to literature). The acrylate derivatives dithiomaleimide acrylate (DTMA) and dibromomaleimide acrylate (DBMA) were prepared likewise by alkylation with bromoacetyl acrylate34 (which was also prepared in a single step according to literature). Monomers were purified by flash column chromatography and characterised by 1H and 13C NMR spectroscopy (Fig. S1–S4†) and high resolution mass spectroscopy (see ESI†). HPLC of the fluorescent monomers DTMMA and DTMA demonstrated the presence of a single fluorescent species (Fig. S5†), whose excitation and emission spectra (recorded in CHCl3) were very similar to the butanethiol-DTM precursor (1).24 They had a broad excitation spectra with maxima at ∼260 and ∼420 nm, with the corresponding emission maximum at ∼520 nm (Fig. 2), indicating that the emissive properties of the DTM group had not been compromised by incorporation into (meth)acrylate monomers. In this study a simple n-butyl thiol ligand was chosen for the DTM, however it should be noted that a range of alternative functionality could be incorporated into these DTM monomers by varying the choice of thiol ligand, as demonstrated previously.24 | ||
| Scheme 1 Synthesis of dithiomaleimide (DTM) and dibromomaleimide (DBM) functional methacrylate (MA) and acrylate (A) monomers. (i) K2CO3, tetrabutylammonium iodide (0.1 eq.), acetone, rt; (ii) K2CO3, acetone, rt. | ||
 | ||
| Fig. 2 (a) Excitation and (b) emission spectra for fluorescent DTM containing monomers (DTMMA and DTMA) and polymers (P1–4) as solutions in chloroform. | ||
Polymerisation of fluorescent DTM monomers
Polymers containing the DTM fluorophore could be accessed directly, simply by copolymerisation of the fluorescent DTM monomers with (meth)acrylates. We investigated the RAFT polymerisation of DTMMA and DTMA using commercially available chain transfer agents (CTAs). For methacrylate polymerisations 2-cyano-2-propyl benzodithioate was chosen, and for acrylate polymerisations cyanomethyl dodecyl trithiocarbonate was used, to give optimum control over molecular weight for these monomer classes.4 All polymerisations were performed using typical conditions, namely as a solution in 1,4-dioxane, heating at 65 °C, with the radical initiator AIBN and 10 mol% loading of the DTM monomer; [CTA] :![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) [comonomer] :[DTMMA] :[AIBN] = 1:45:5:0.1 (Scheme 2 and Table 1).
[comonomer] :[DTMMA] :[AIBN] = 1:45:5:0.1 (Scheme 2 and Table 1).
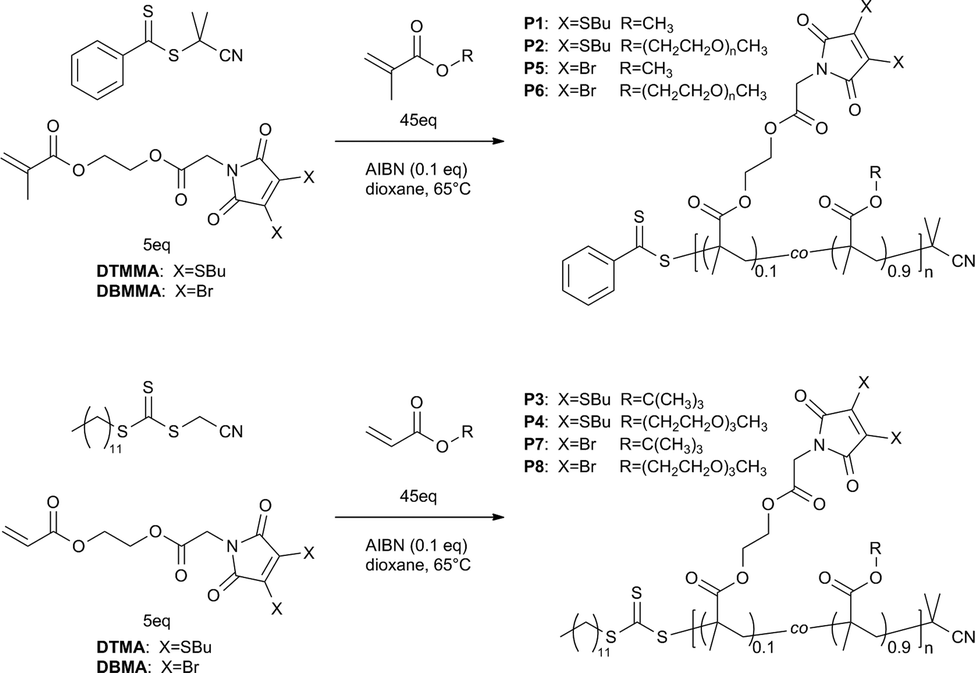 | ||
| Scheme 2 RAFT copolymerisations of DTMMA, DTMA, DBMMA and DBMA. | ||
| Polymer compositiona | M n (kDa) | M n (kDa) | Đ M | |
|---|---|---|---|---|
| a Calculated by NMR end-group analysis. b Measured by SEC (THF or CHCl3 eluent, PS calibration). | ||||
| P1 | P(MMA38.3-co-DTMMA4.3) | 6.0 | 5.3 | 1.13 |
| P2 | P(OEGMA30.3-co-DTMMA3.5) | 10.5 | 7.8 | 1.23 |
| P3 | P(tBA46.4-co-DTMA4.5) | 8.2 | 5.6 | 1.24 |
| P4 | P(TEGA31.6-co-DTMA3.4) | 8.6 | 4.7 | 1.25 |
| P5 | P(MMA37.2-co-DBMMA4.7) | 5.9 | 6.6 | 1.12 |
| P6 | P(OEGMA36.5-co-DBMMA5.3) | 13.1 | 9.3 | 1.24 |
| P7 | P(tBA28.8-co-DBMA3.4) | 5.4 | 4.2 | 1.14 |
| P8 | P(TEGA15.0-co-DBMA1.8) | 4.3 | 3.4 | 1.26 |
| P9 | POEGMA34.7 | 10.1 | 9.6 | 1.20 |
| P10 | PTEGA44.4 | 10.1 | 5.7 | 1.17 |
| P11 | P(OEGMA27.6-co-DBMMA4.7) | 10.3 | 9.3 | 1.24 |
Copolymerisation of DTMMA with the hydrophobic monomer methyl methacrylate (MMA) displayed linear first order consumption of both monomers, with a linear increase of molecular weight with conversion (as measured by SEC), and low dispersities throughout (ĐM < 1.2), indicating a good control over molecular weight during the polymerisation (Fig. S6†). Both MMA and DTMMA were consumed at an approximately equivalent rate, to a final conversion at 9 h of 84% for MMA and 87% for DTMMA. The polymer (P1) was isolated by precipitation into methanol, with 1H NMR spectroscopy of the purified product revealing incorporation of DTMMA at the expected 10 mol% loading (Fig. S7†). SEC analysis of P1 (THF eluent) indicated a narrow molecular weight distribution (ĐM = 1.13), with incorporation of the DTM functional group and retention of the dithiobenzoate RAFT end-group indicated by absorption maxima at 413 nm and 307 nm respectively for the polymer peak in the 3D chromatogram collected using a PDA detector (Fig. S8†). The fact that both MMA and DTMMA were consumed at an approximately equivalent rate, and that the final polymer had both monomers incorporated at their initial feed ratio, as well as having a narrow molecular weight distribution, suggests a random copolymerisation.
Copolymerisation of DTMMA with the hydrophilic monomer oligoethylene glycol methacrylate (OEGMA, Mn = 300 Da) at a 10 mol% loading of DTMMA also proceeded with linear kinetics, and good control over molecular weight (Fig. S9†). Again both monomers were consumed at an equivalent rate, with the purified polymer (P2) showing the expected 10 mol% loading of DTMMA by 1H NMR spectroscopy (Fig. S10†). A narrow molecular weight distribution (ĐM = 1.23), and incorporation of DTM and dithiobenzoate groups was again shown by SEC with a PDA detector (Fig. S11†). Despite the choice of a hydrophobic n-butyl thiol ligand in the DTMMA monomer, the OEGMA copolymer P2 retained its water solubility and thermoresponsive behaviour.35 The LCST cloud point of P2 in water (18.2 MΩ cm) was measured at 10 g l−1 as Tc = 50.2 ± 0.0 °C during the heating cycle, and 50.0 ± 0.1 °C for the cooling cycle (Fig. S12†). An analogous homopolymer of POEGMA prepared by RAFT (P9, Mn = 10.1 kDa) was found to have Tc = 65.8 ± 0.0 °C and 65.7 ± 0.1 °C for heating and cooling respectively, indicating that copolymerisation with DTMMA had caused an increase in hydrophobicity.
Similar success was observed for copolymerisation of DTMA with hydrophobic tert-butyl acrylate (tBA) and hydrophilic triethyleneglycol monomethylether acrylate (TEGA)36 monomers at a 10 mol% loading of DTMA using the same reaction conditions as in the methacrylate polymerisations (Scheme 2). Again, linear first order polymerisation kinetics, a linear increase of molecular weight with conversion, and low dispersities were observed (Fig. S13†). 1H NMR revealed incorporation of DTMA at 9 mol% and 10 mol% for tBA and TEGA copolymerisations respectively (Fig. S14 and S15†), while SEC analysis showed narrow molecular weight distributions and incorporation of the DTM and trithiocarbonate chromophores (Fig. S16†). The water solubility and thermoresponsive behaviour of the P(TEGA-co-DTMA) copolymer was also retained, with an LCST cloud point at 10 g l−1 in water (18.2 MΩ cm) of Tc = 37.3 ± 0.3 °C during the heating cycle, and 36.9 ± 0.2 °C for the cooling cycle (Fig. S17†). The analogous PTEGA homopolymer (P10, Mn = 10.1 kDa) prepared by RAFT had Tc = 65.5 ± 0.0 °C (heating cycle) and 65.2 ± 0.0 °C (cooling cycle) suggesting that the introduction of the hydrophobic DTMA monomer had caused an increase in hydrophobicity, as was observed for DTMMA (P2).
The fluorescence spectra (in CHCl3) of the PDTMMA and PDTMA copolymers (P1–4) are very similar to that of the monomers, retaining the excitation maxima at ∼260 nm and ∼420 nm and emission maximum at ∼520 nm (Fig. 2). The dependence of emission on polymer concentration was studied, using P3 as an example. 2D excitation–emission spectra were collected at concentrations of 5, 1, 0.5 and 0.1 mM (Fig. S18†). At the higher concentrations aggregate/multimer or dimer emission was observed, while at 0.1 mM emission corresponded to that of fluorophore unimers, in accord with the spectrum of the small molecule DTM 1 at the same concentration. This indicates that once the polymer is sufficiently diluted to avoid inter-chain quenching, there is no significant intra-chain quenching caused by neighbouring DTM containing repeat units. Molar emission (integrated emission intensity divided by molar concentration) for P3 in CHCl3 also demonstrates this lack of self-quenching, with a region of concentration independent emission between 0.1 mM and 1 μM (Fig. S19†).
The results presented in this section demonstrate that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the DTM motif was unreactive toward radical polymerisation for DTMMA and DTMA monomers, and that no adverse effects on DTM fluorescence are caused by incorporation into a polymeric structure, in line with previous findings.24,32
C double bond of the DTM motif was unreactive toward radical polymerisation for DTMMA and DTMA monomers, and that no adverse effects on DTM fluorescence are caused by incorporation into a polymeric structure, in line with previous findings.24,32
Polymerisation of chemico-fluorescent responsive DBM monomers
For the chemico-fluorescent responsive methacrylate monomer (DBMMA), RAFT copolymerisations with MMA and OEGMA performed under the same conditions (1,4-dioxane solution, 65 °C, [CTA] :[AIBN] :[MMA or OEGMA] :[DBMMA] = 1:0.1:45:5, Scheme 2) again demonstrated linear first order kinetics, a linear increase of molecular weight with conversion and low dispersity, indicating good control over the polymerisation (Fig. S20†). 1H NMR spectroscopy of the purified polymers (P5 and P6) revealed incorporation of DBMMA at 11 mol% and 13 mol% loading, while 13C NMR of P5 provided additional proof of the incorporation of the DBM monomer as evidenced by the characteristic resonance of the DBM CC at 129.9 ppm, and CO at 166.7 ppm (Fig. S21 and S22†). SEC of P5 and P6 showed narrow molecular weight distributions with ĐM = 1.12 and 1.24 respectively (Fig. S23†).
Copolymerisations of DBMA (1,4-dioxane solution, 65 °C, [CTA] :[AIBN] :[tBA or TEGA] :[DBMA] = 1:0.1:45:5) initially proceeded with linear first order consumption of monomer, however once a certain total monomer conversion had been reached (∼60% and ∼35% for tBA and TEGA respectively) a complete retardation of polymerisation was observed (Fig. S24†). No loss of control over the evolution of molecular weight was observed (ĐM ≤ 1.2 throughout the polymerisations), indicating that chain transfer to DBM or branching via DBM CC double bond polymerisation were not the cause of the retardation. To demonstrate that this retardation is caused by the DBM group we conducted a series of RAFT polymerisations of tBA in the presence of 2,3-dibromo-N-methyl-maleimide (DBMM) at a range of DBMM loadings. By measuring initial rates of monomer consumption the order of reaction was found to be −0.68 for DBMM (Fig. S25†), using the method of Bell et al.37 In comparison, RAFT polymerisations of MMA in the presence of DBMM gave an order of reaction = −0.14 for DBMM, explaining why no significant retardation of polymerisation was observed for copolymerisations of DBMMA. This suggests that interaction of the propagating radical with the DBM group is the cause of the retardation, with either the greater stability of the methacrylate radical over the acrylate radical, or its greater steric bulk decreasing this effect. The external order of butanethiol-dithio-N-methyl-maleimide (DTMM32) in tBA and MMA RAFT polymerisations was found to be 0.08 and −0.01 respectively, indicating that the DTM group doesn't interfere in the polymerisations. This is in line with copolymerisations of DTMMA and DTMA (above), and with previous reports.24,32,38
1H NMR spectra of P7 and P8 showed incorporation of DBMA at 11 mol% in both cases (Fig. S26 and 27†), with no obvious deviation from the expected product. The 13C NMR spectrum of P7 clearly showed peaks attributed to the DBM group's CC (129.8 ppm) and CO (166.6 ppm) resonances. SEC again revealed good control over molecular weight, with ĐM = 1.14 and 1.24 for P7 and P8 respectively (Fig. S28†). In line with previous results for polymerisations with a DBM functional RAFT agent,28 this data suggests that the polymerisation retardation does not lead to loss of the DBM group.
Post polymerisation functionalisation of chemico-fluorescent responsive P(OEGMA-co-DBMMA)
Post-polymerisation functionalisation of polymers derived from the DBM monomers gives an alternative route to fluorescent DTM containing polymers (Fig. 1). The pendent DBM units undergo a highly efficient conjugation reaction with two equivalents of a thiol, allowing introduction of further functionality along the polymer backbone, while simultaneously inducing a chemico-fluorescent response resulting in OFF-to-ON switching of fluorescence emission. The efficient conjugation of pendent reactive groups along a thermoresponsive polymer backbone has been shown to allow subtle tuning of poly(N-isopropylacrylamide) and POEGMA LCST cloud points.39 We therefore anticipated that the reaction of P(OEGMA-co-DBMMA) with thiols would also allow for LCST cloud point modification.To demonstrate this approach we performed the reaction of P(OEGMA27.6-co-DBMMA4.7) (P11) with a range of thiols (HS-R) bearing different functional groups (R) according to Scheme 3 and Table 2. P11 was first dissolved in pH 6 buffer (100 mM sodium phosphate, 150 mM NaCl) at 10 g l−1, before addition of a small excess of thiol (12 eq. relative to the polymer which corresponds to 2.6 eq. per DBM group) as a 1 M solution in DMF.
 | ||
| Scheme 3 Post-polymerisation functionalisation of chemico-fluorescent responsive P(OEGMA27.6-co-DBMMA4.7) (P11) with thiols (HS-R). | ||
| Thiol | Conversiona (%) | Cloud pointb (°C) | M n (kDa) | Đ M | |
|---|---|---|---|---|---|
| a Measured by 1H NMR spectroscopy. b Measured by temperature-dependent light transmission at 10 g l−1 in water (18.2 MΩ cm). c Measured by SEC (CHCl3 eluent, PS calibration). d Polymer interacted with SEC column due to acid groups. e DTM CH2 and OH resonances overlapped with POEGMA resonances. | |||||
| P11 | — | — | 46.3 ± 0.3 | 9.3 | 1.24 |
| P12 | HS(CH2)2CO2H | 95 | 55.2 ± 0.7 | —d | —d |
| P13 | HS(CH2)2OH | —e | 54.9 ± 0.6 | 7.0 | 1.38 |
| P14 | HSCH(CH3)2 | 96 | 48.1 ± 0.8 | 7.7 | 1.39 |
| P15 | HSBn | 97 | 44.3 ± 0.3 | 8.9 | 1.34 |
The reaction was found to be very fast, with the immediate formation of the yellow/green DTM fluorophore observed. After purification by dialysis 1H NMR spectroscopy analysis revealed new resonances attributed to the successfully added thiols. For those products where the new resonances didn't overlap with major POEGMA resonances, conversion was calculated as ≥95% for double thiol substitution (Fig. S29†). SEC revealed slight changes in polymer Mn as a result of the substitution of –Br with –SR. There was some low molecular weight tailing for P13 due to interactions between the column and the pendent diol groups, while the acid functional P12 failed to elute entirely due to column interactions. The use of a UV-vis SEC detector confirmed transformation of the pendent groups to DTMs, as the polymers absorbed at the DTM absorption maximum (λmax = 420 nm in the CHCl3 SEC solvent) as shown in Fig. S30.† Fluorescence spectra recorded in CHCl3 solution showed the presence of the DTM fluorophore, with excitation maxima at ∼420 nm and corresponding emission maxima of ∼520 nm, therefore indicating that the P11 had successfully undergone an OFF-to-ON switching of fluorescence emission (Fig. S31†). LCST cloud point measurements (at 10 g l−1 in 18.2 MΩ cm water) of the thiol substituted polymers P12–15 revealed that the transition temperature could be subtly tuned either above or below that of the initial polymer through R group choice, with a 11 °C range in cloud points obtained (Fig. 3 and Table 2). The trend in LCST cloud points was found to follow the relative polarities of the thiols used, suggesting that water solubility of the pendent DTM groups was the determining factor in cloud point temperature.
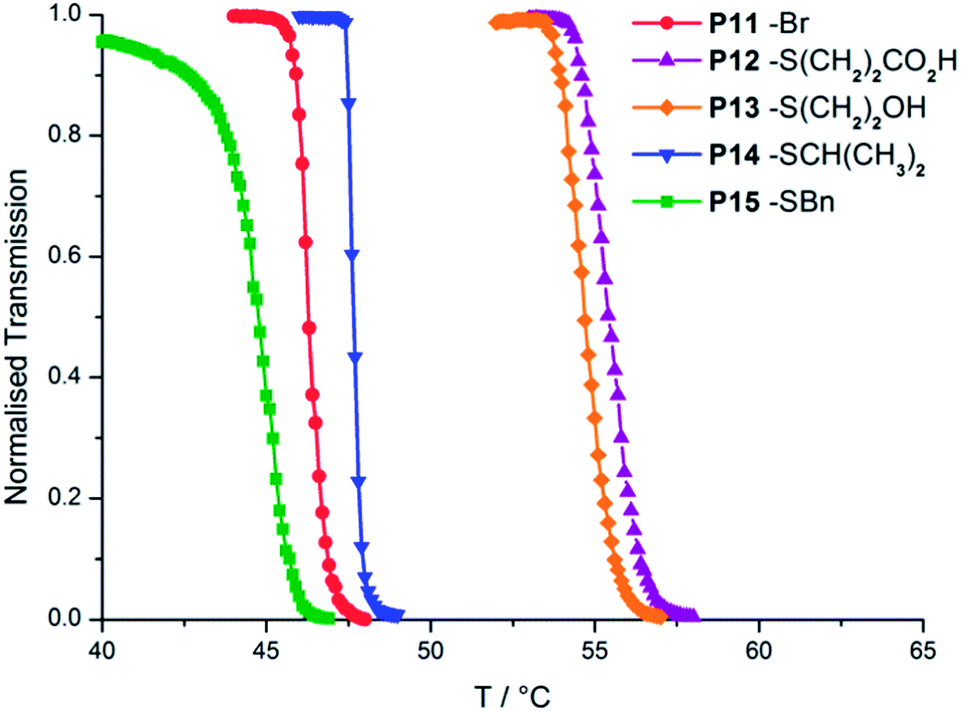 | ||
| Fig. 3 LCST cloud point determination by temperature-dependent light transmission for solutions of P11–15 at 10 g l−1 in water. | ||
In order to monitor the rate of site-group modification the absorbance due to the DTM fluorophore was measured in situ during a post-polymerisation functionalisation reaction. The UV-vis spectrum of a solution of P12 in pH 6 buffer was measured. A concentration of 1 g l−1 was required to obtain absorbance <1 at the λmax of 409 nm, and this absorbance was taken to correspond to 95% conversion for the post-polymerisation functionalisation reaction (Table 2). Then, to a 1 g l−1 solution of P11 in buffer was added 12 eq. of HS(CH2)2CO2H as a 1 M solution in DMF, and the absorbance at 409 nm monitored as a function of time. Even at this 10× dilution from the optimum conditions, the reaction was found to be very fast, reaching 50% conversion within 10 min, and >95% conversion after 3 h (Fig. 4).
 | ||
| Fig. 4 Conversion vs. time as measured by UV-vis spectroscopy (λabs = 409 nm) for the post-polymerisation functionalisation of P(OEGMA27.6-co-DBMMA4.7) (P11) with HS(CH2)2CO2H in pH 6 buffer at 1 g l−1. Inset: photograph of reaction at 180 min observed under natural light (left) and 365 nm handheld lamp (right). | ||
Reversible chemico-fluorescent response by thiol-exchange
The work of Baker and Caddick has shown that it is possible to perform a thiol-exchange reaction on DTM, resulting in the elimination of the original thiol ligands and addition of two new thiol ligands.26a This is due to retention of the maleimide CC double bond in the DTM group, which therefore allows further conjugate addition–elimination reactions to occur. Furthermore, we have previously shown that the DTM which contains thiophenol (–SPh) ligands has a drastically decreased emission, due to conjugation of the phenyl groups to the DTM ring.24 Therefore, by performing a thiol-exchange reaction on an emissive DTM-functional polymer with thiophenol, it should be possible to achieve an ON-to-OFF switching of fluorescence.
To illustrate this possibility a thiol-exchange reaction was performed using the emissive P(TEGA31.6-co-DTMA3.4) copolymer (P4). In order to achieve complete conversion an excess of thiophenol was used, corresponding to 10 eq. per –SBu ligand in P4 (Scheme 4). P4 was dissolved in pH 6 buffer at 10 g l−1, before addition of thiophenol as a 2.5 M solution in DMF. Emission (λex = 435 nm) was monitored during the reaction, with a drastic reduction in emission observed within 1 hour (Fig. S32†). After purification by dialysis, 1H NMR spectroscopy analysis of the product (P16) revealed new resonances attributed to the successfully added –SPh groups, with loss of resonances corresponding to the –SBu groups (Fig. S33†).
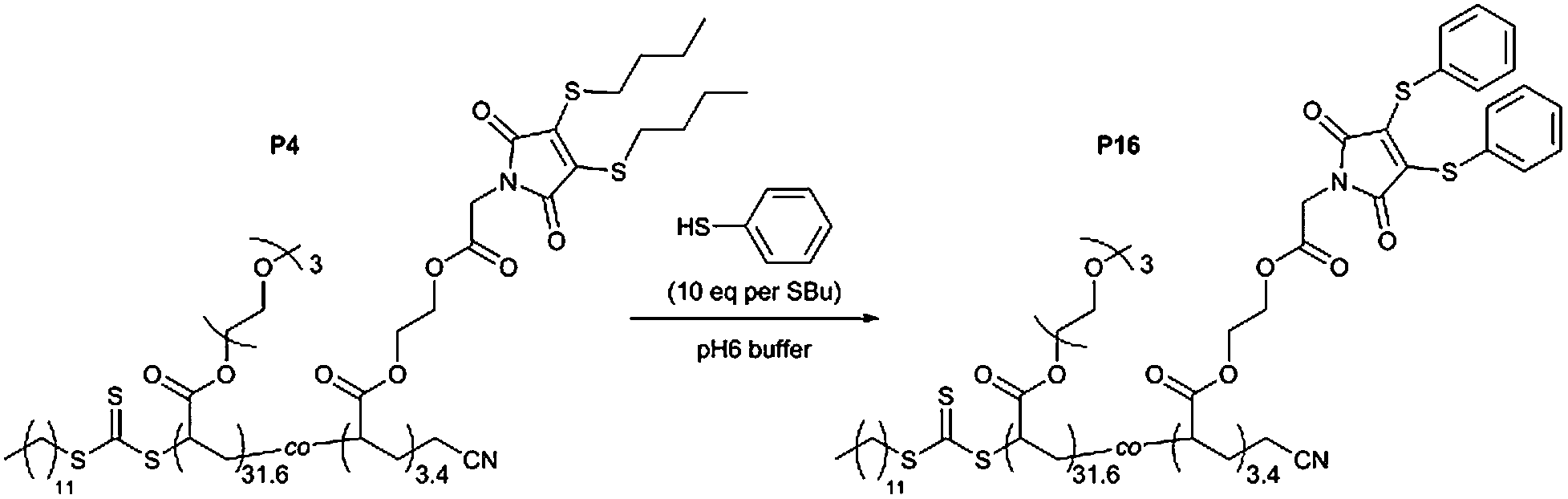 | ||
| Scheme 4 Thiol-exchange reaction of emissive P(TEGA31.6-co-DTMA3.4) (P4) with thiophenol. | ||
Conclusions
Methacrylate and acrylate monomers bearing dithiomaleimide (DTM) or dibromomaleimide (DBM) functional groups have been synthesised, and successfully polymerised by RAFT polymerisation. The fluorescent DTM monomers were found to give fluorescent copolymers with MMA, OEGMA, tBA, and TEGA with good control over molecular weight distribution. Copolymers with OEGMA and TEGA retained their thermoresponsive properties, with the 10 mol% loading of DTM monomers causing a decrease in LCST cloud point.The DBM monomers gave chemico-fluorescent responsive copolymers with MMA, OEGMA, tBA and TEGA, again with good control over molecular weight distribution. Polymerisations with DBM acrylate were found to reach a limiting conversion, due to retardation by the DBM group, however no loss of DBM functionality or molecular weight control was observed. The DBM copolymer with OEGMA was shown to undergo a highly efficient chemico-fluorescent responsive conjugation with thiols in aqueous media, leading to an OFF-to-ON switching of fluorescence emission. These substituted OEGMA copolymers retained an LCST cloud point, which could be progressively tuned by judicious choice of thiol in the conjugation-induced fluorescent labelling reaction.
It was also possible to exchange the thiol ligands on the pendent DTM groups of a DTM-functional copolymer. This thiol-exchange reaction was demonstrated using thiophenol, as the resultant dithiophenol maleimide group has drastically reduced emission. Therefore this thiol-exchange reaction results in an ON-to-OFF switching of fluorescence emission, demonstrating that the chemico-fluorescent response of these polymers can be considered to be reversible.
These new monomers present a straightforward and versatile approach for the labelling of polymeric materials with the DTM fluorophore. The effectiveness of DTM labelled polymer nanoparticles for in vitro fluorescence-lifetime imaging microscopy (FLIM) has previously been demonstrated,32 and we anticipate that these new (meth)acrylate monomers will present a convenient route to a wide range of new DTM labelled fluorescent materials.
Acknowledgements
The authors would like to thank Dr Andrew J. Clark and Dr Jeffery E. Raymond for useful discussions, and the EPSRC and IAS early career fellowship (University of Warwick) for funding. Equipment used in this research was funded in part through Advantage West Midlands (AWM) Science City Initiative and in part by the ERDF.Notes and references
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- A. M. Breul, M. D. Hager and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 5366–5407 RSC.
- M. Beija, M.-T. Charreyre and J. M. G. Martinho, Prog. Polym. Sci., 2011, 36, 568–602 CrossRef CAS PubMed.
- J. Xue, L. Chen, H. L. Wang, Z. B. Zhang, X. L. Zhu, E. T. Kang and K. G. Neoh, Langmuir, 2008, 24, 14151–14158 CrossRef CAS PubMed.
- P. B. Zetterlund, M. N. Alam and M. Okubo, Polym. J., 2008, 40, 298–299 CrossRef.
- M. Sommer, S. Hüttner, U. Steiner and M. Thelakkat, Appl. Phys. Lett., 2009, 95, 183308 CrossRef PubMed.
- M. Demetriou and T. Krasia-Christoforou, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 52–60 CrossRef CAS.
- A. Nese, N. V. Lebedeva, G. Sherwood, S. Averick, Y. Li, H. Gao, L. Peteanu, S. S. Sheiko and K. Matyjaszewski, Macromolecules, 2011, 44, 5905–5910 CrossRef CAS.
- J. Madsen, N. J. Warren, S. P. Armes and A. L. Lewis, Biomacromolecules, 2011, 12, 2225–2234 CrossRef CAS PubMed.
- M. Spiniello, A. Blencowe and G. G. Qiao, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2422–2432 CrossRef CAS.
- P. Zhao, J. Jiang, B. Leng and H. Tian, Macromol. Rapid Commun., 2009, 30, 1715–1718 CrossRef CAS PubMed.
- C. Grazon, J. Rieger, R. Méallet-Renault, B. Charleux and G. Clavier, Macromolecules, 2013, 46, 5167–5176 CrossRef CAS.
- C. Li, Y. Zhang, J. Hu, J. Cheng and S. Liu, Angew. Chem., Int. Ed., 2010, 49, 5120–5124 CrossRef CAS PubMed.
- (a) J. Du, H. Willcock, J. P. Patterson, I. Portman and R. K. O'Reilly, Small, 2011, 7, 2070–2080 CrossRef CAS PubMed; (b) M. Long, D. W. Thornthwaite, S. H. Rogers, F. R. Livens and S. P. Rannard, Polym. Chem., 2012, 3, 154–161 RSC.
- (a) R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, Chem.–Eur. J., 2006, 12, 6776–6786 CrossRef CAS PubMed; (b) A. J. Dirks, J. J. L. M. Cornelissen and R. J. M. Nolte, Bioconjugate Chem., 2009, 20, 1129–1138 CrossRef CAS PubMed; (c) C. F. Hansell and R. K. O'Reilly, ACS Macro Lett., 2012, 1, 896–901 CrossRef CAS.
- S. L. Mangold, R. T. Carpenter and L. L. Kiessling, Org. Lett., 2008, 10, 2997–3000 CrossRef CAS PubMed.
- J. P. Blinco, K. E. Fairfull-Smith, A. S. Micallef and S. E. Bottle, Polym. Chem., 2010, 1, 1009–1012 RSC.
- (a) W. Song, Y. Wang, J. Qu, M. M. Madden and Q. Lin, Angew. Chem., Int. Ed., 2008, 47, 2832–2835 CrossRef CAS PubMed; (b) R. K. V. Lim and Q. Lin, Acc. Chem. Res., 2011, 44, 828–839 CrossRef CAS PubMed.
- R. K. V. Lim and Q. Lin, Chem. Commun., 2010, 46, 7993–7995 RSC.
- M. Dietrich, G. Delaittre, J. P. Blinco, A. J. Inglis, M. Bruns and C. Barner-Kowollik, Adv. Funct. Mater., 2012, 22, 304–312 CrossRef CAS.
- M. P. Robin, P. Wilson, A. B. Mabire, J. K. Kiviaho, J. E. Raymond, D. M. Haddleton and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 2875–2878 CrossRef CAS PubMed.
- (a) F. F. Schumacher, M. Nobles, C. P. Ryan, M. E. B. Smith, A. Tinker, S. Caddick and J. R. Baker, Bioconjugate Chem., 2011, 22, 132–136 CrossRef CAS PubMed; (b) M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 1847–1852 CrossRef CAS PubMed.
- (a) M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed; (b) F. F. Schumacher, V. A. Sanchania, B. Tolner, Z. V. F. Wright, C. P. Ryan, M. E. B. Smith, J. M. Ward, S. Caddick, C. W. M. Kay, G. Aeppli, K. A. Chester and J. R. Baker, Sci. Rep., 2013, 3, 1525 Search PubMed.
- C. P. Ryan, M. E. B. Smith, F. F. Schumacher, D. Grohmann, D. Papaioannou, G. Waksman, F. Werner, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 5452–5454 RSC.
- M. P. Robin, M. W. Jones, D. M. Haddleton and R. K. O'Reilly, ACS Macro Lett., 2012, 1, 222–226 CrossRef CAS.
- Y. Cui, Y. Yan, Y. Chen and Z. Wang, Macromol. Chem. Phys., 2013, 214, 470–477 CrossRef CAS.
- S. Long, Q. Tang, Y. Wu, L. Wang, K. Zhang and Y. Chen, React. Funct. Polym., 2013 DOI:10.1016/j.reactfunctpolym.2013.11.006.
- J.-J. Yan, D. Wang, D.-C. Wu and Y.-Z. You, Chem. Commun., 2013, 49, 6057–6059 RSC.
- M. P. Robin, A. B. Mabire, J. C. Damborsky, E. S. Thom, U. H. Winzer-Serhan, J. E. Raymond and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 9518–9524 CrossRef CAS PubMed.
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, ACS Macro Lett., 2013, 2, 581–586 CrossRef CAS.
- B. Mu, M. Zhao and P. Liu, J. Nanopart. Res., 2008, 10, 831–838 CrossRef CAS.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- F. Hua, X. Jiang, D. Li and B. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2454–2467 CrossRef CAS.
- C. A. Bell, Q. Sun, H. Zhang, S. C. Smith, P. V. Bernhardt and M. J. Monteiro, Polym. Chem., 2010, 1, 207–212 RSC.
- M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, Chem. Commun., 2012, 48, 4064–4066 RSC.
- (a) K. Rahimian-Bajgiran, N. Chan, Q. Zhang, S. M. Noh, H.-i. Lee and J. K. Oh, Chem. Commun., 2013, 49, 807–809 RSC; (b) S. Reinicke, P. Espeel, M. M. Stamenović and F. E. Du Prez, ACS Macro Lett., 2013, 2, 539–543 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterisation data, and supplementary Fig. S1–S33. See DOI: 10.1039/c4sc00753k |
| This journal is © The Royal Society of Chemistry 2014 |