 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper catalyzed Heck-like cyclizations of oxime esters†
Adele
Faulkner‡
a,
Nicholas J.
Race‡
a,
James S.
Scott
b and
John F.
Bower
*a
aSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: john.bower@bris.ac.uk
bAstraZeneca, Alderley Park, Macclesfield, Cheshire SK10 4TG, UK
First published on 9th April 2014
Abstract
Copper catalyzed Heck-like cyclizations of oxime esters are described. Mechanistic studies indicate a reaction pathway that proceeds via the generation and cyclization of an intermediate that possesses iminyl radical character. To the best of our knowledge, this work encompasses the first examples of Cu-catalyzed aza-Heck reactions that proceed via oxidative initiation at nitrogen to generate products containing a new alkene. This new protocol is also an effective alternative to Pd-based systems and highlights the value of replacing precious metal catalysts with cheaper and more sustainable variants.
Introduction
The advent of catalysis based upon the oxidative generation and capture of aryl-Pd(II) intermediates has had a profound impact upon the field of organic synthesis. Accordingly, it is estimated that 20% of C–C bond forming reactions employed in the pharmaceutical sector are reliant upon this technology.1 Given the privileged position of nitrogen in drug discovery, it is surprising that related processes involving the oxidative generation and capture of aza-Pd(II) species have been slow to emerge.2 Seminal studies by Narasaka demonstrated that Pd(0)-catalysts undergo oxidative addition into the N–O bond of O-pentafluorobenzoyl oximes 1 to generate imino-Pd(II) intermediates 2 (Scheme 1A).3,4 The reactivity of these species mirrors that of their aryl counterparts and migratory insertion of pendant alkenes provides an aza-variant of the Heck reaction.5 This reactivity manifold is heavily underdeveloped and our studies have focused upon providing efficient catalysis systems that generate synthetically versatile chiral N-heterocyclic scaffolds (e.g.3 rather than 4).6,7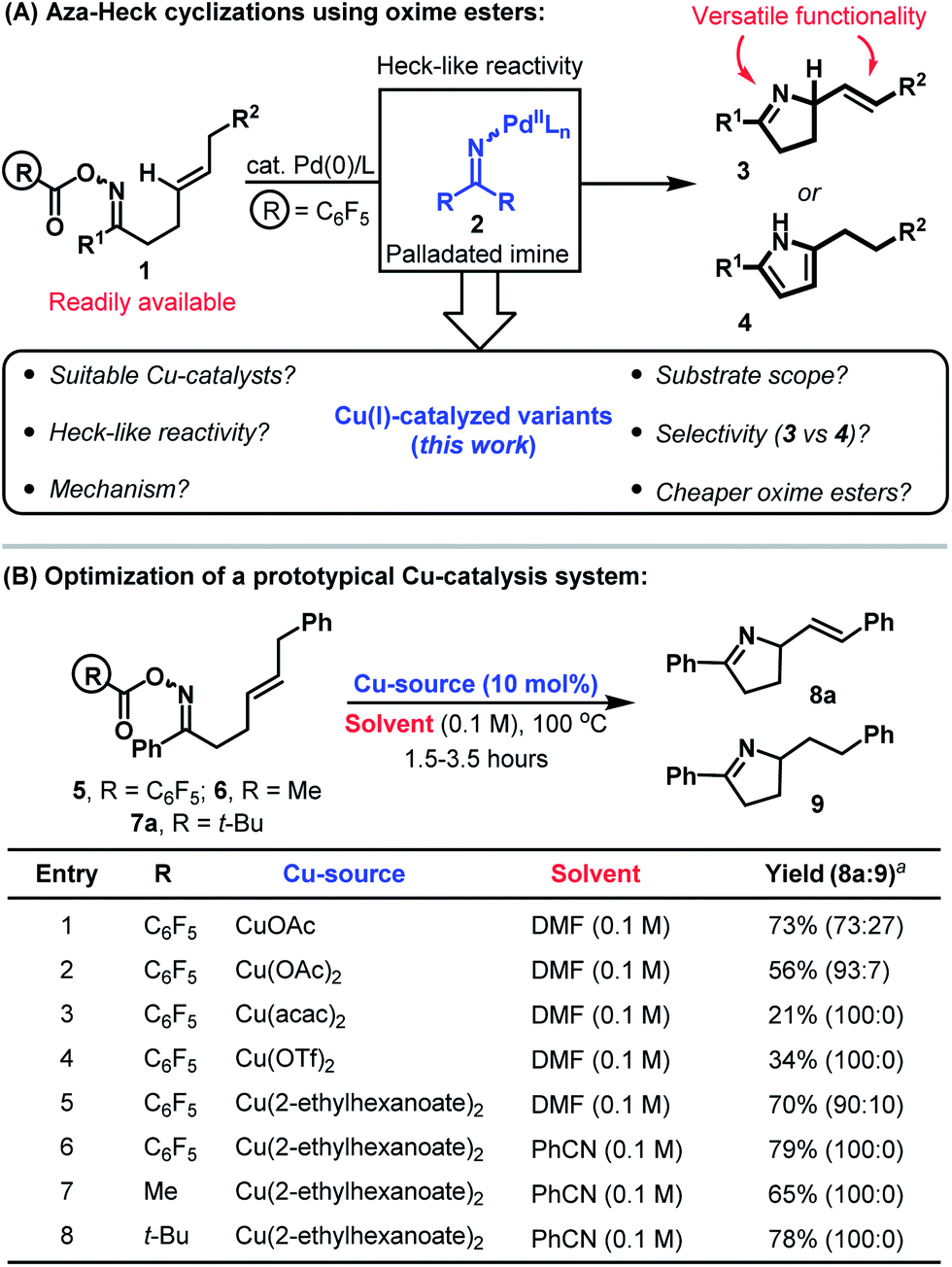 | ||
| Scheme 1 Aza-Heck cyclizations of oxime esters and the development of a Cu-catalyzed protocol. a Isolated yield (product ratios were determined by 1H NMR). | ||
There is a growing interest in replacing Pd(0)-catalysts with more abundant and isoelectronic Cu(I)-variants.2,8 Cu(I)-catalyzed aza-Stille and aza-Suzuki cross-couplings involving oxime esters have been reported by Liebeskind et al. but the corresponding aza-Heck processes have not been developed.9,10 In this report we detail the discovery and mechanism of a Cu-catalyzed protocol for the aza-Heck cyclization of oxime esters. This provides a direct and economic alternative to Pd-based systems, and also addresses selectivity issues that hampered our earlier work (e.g. β-hydride elimination selectivity to 3vs.4, Scheme 1A).6a To the best of our knowledge, the present study also encompasses the first examples of copper-catalyzed aza-Heck reactions that furnish products containing a new alkene by oxidative initiation at nitrogen (i.e. in terms of substrate/product structure, the process is an exact aza-variant of the conventional Heck reaction where the oxime ester takes the place of the aryl halide).10,11
Results and discussion
At the outset of our studies, the prospect of replacing Pd-based systems with Cu(I)-variants was considered tentative. The generation of aza-copper intermediates by oxidative addition into N–O bonds has been invoked in a range of amination processes.9,10,11d,g–j However, reactions involving alkenes provide 1,2-difunctionalization processes and do not afford new alkene containing products.11d,e,i Consequently, the viability of a copper-catalyzed aza-Heck cycle that incorporates the key steps of oxidative initiation and β-hydride elimination was unclear. Our preliminary investigations involved exposing DMF solutions of O-pentafluorobenzoyl oxime 5 to a variety of commercial Cu-salts (Scheme 1B). Gratifyingly, both CuOAc and Cu(OAc)2 provided the desired product 8a with complete selectivity over the alternative pyrrole product (entries 1 and 2; cf.Scheme 1A). However, 8a was accompanied by significant quantities of adduct 9, which contains a saturated side chain. Cu(acac)2 and Cu(OTf)2 both suppressed the formation of this byproduct but provided only modest yields of the target 8a (entries 3 and 4). However, good selectivity and yield was obtained using the more soluble Cu(2-ethylhexanoate)2, which provided adduct 8a in 79% yield and as the only observable product when PhCN was used as solvent (entry 6). Pleasingly, this protocol also tolerates less activated oxime esters and acetyl and pivaloyl variants 6 and 7a cyclized efficiently to provide 8a in 65% and 78% yield respectively (entries 7 and 8). This facet is particularly striking and is in stark contrast to our work with Pd-systems, where O-pentafluorobenzoyl oximes are a requirement for efficient cyclization.6The ability to use acetyl or pivaloyl oxime esters is beneficial from the viewpoint of cost, starting material stability and atom economy. Consequently, we elected to explore scope using a range of pivaloyl oxime ester substrates 7a–l that possess pendant 1,2-disubstituted alkenes (Table 1). In the majority of cases cyclization proceeded smoothly to generate the target dihydropyrroles 8a–j in good to excellent yield and with complete selectivity over the alternative pyrrole products (cf.Scheme 1A). A range of alkyl and aryl oxime esters can participate in this process and cyclization efficiency is not adversely affected by sterically demanding oximes (e.g.7d). The successful cyclization of 7c, which possesses a potentially problematic Lewis basic pyridyl moiety, is particularly noteworthy. For ease of comparison, and where determined, the results of the cyclization of the analogous O-pentafluorobenzoyl oxime esters with our best Pd-based systems are included.6a Note that in many cases (e.g.8g and 8i) these Pd-catalyzed processes suffered from competing formation of significant quantities of pyrrole products (the ratios of dihydropyrrole to pyrrole products are given in parentheses). Another limitation of Pd-based systems is that aryl bromides are not well tolerated.6c For the copper catalyzed protocol this is not an issue and cyclization of 7h provided 8h in good yield and with Ar–Br bond still intact. This then opens up the option to modify further the initial scaffold using conventional Pd(0)-catalyzed cross-coupling reactions. Certain limitations are evident however, and alkynyl and aldoxime based systems 7k and 7l did not cyclize efficiently. In the former case the issue was the sensitivity of the product 8k to conjugate addition by in situ generated pivalic acid. In the latter case (8l), decomposition of the oxime ester to the corresponding nitrile predominated.12
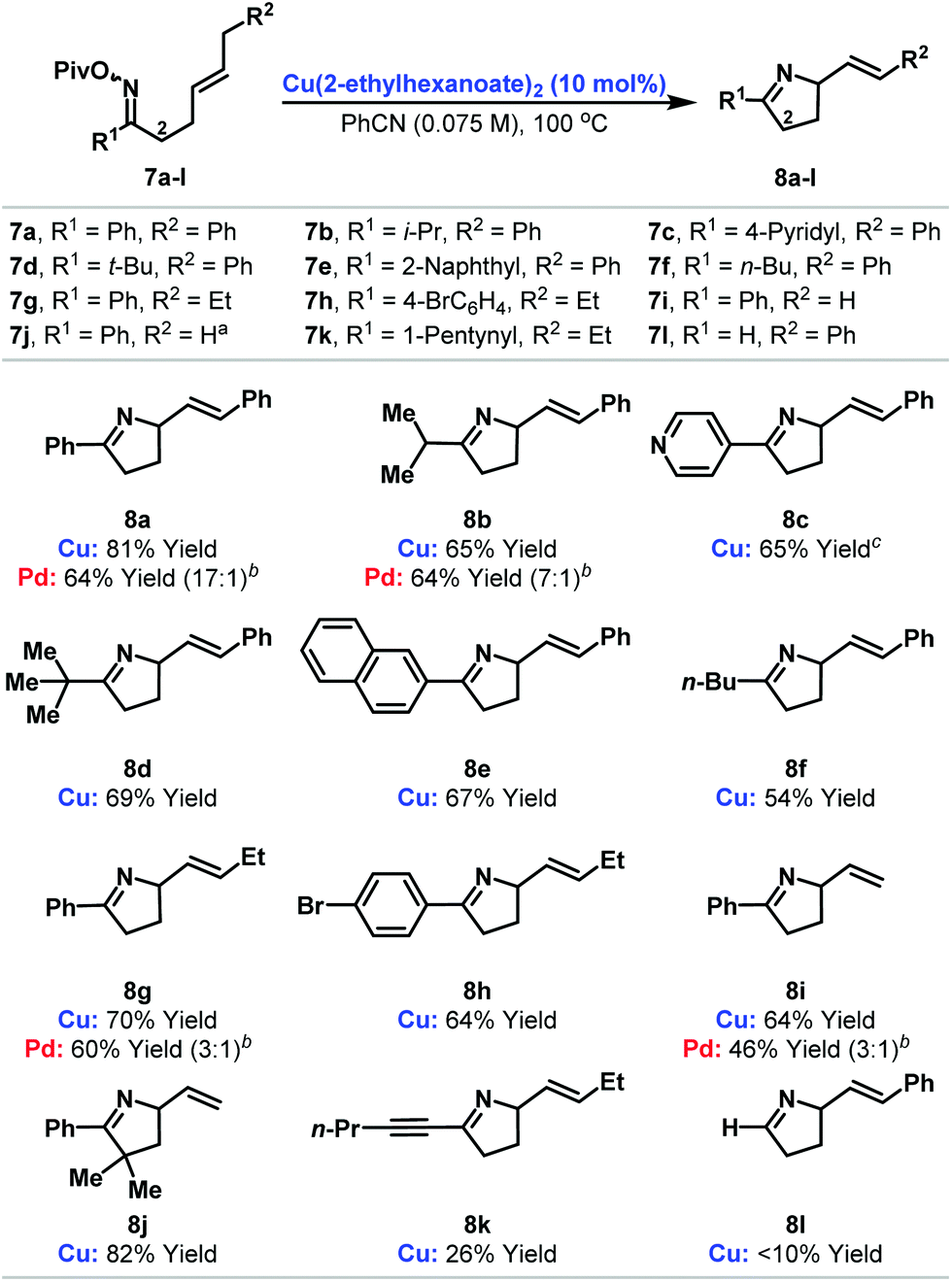
We have also explored cyclizations of more heavily substituted 1,1-disubstituted alkenes 7m–r to provide adducts 8m–r that possess challenging quaternary amino-substituted stereocenters (Table 2). For 7m–q cyclization was efficient independent of the nature of the alkene. For example, cyclization of 7m, which involves an electron deficient acrylate, provided 8m in 76% yield. Notably, under our best palladium catalyzed conditions, the analogous O-pentafluorobenzoyl oxime ester cyclized in only 31% yield.6b Some limitations do exist with respect to the alkene and cyclization of 7r, which generates a benzylic C–N bond, was not efficient. Here, competing formation of the corresponding ketone (the formal hydrolysis product of the oxime ester) was problematic.13
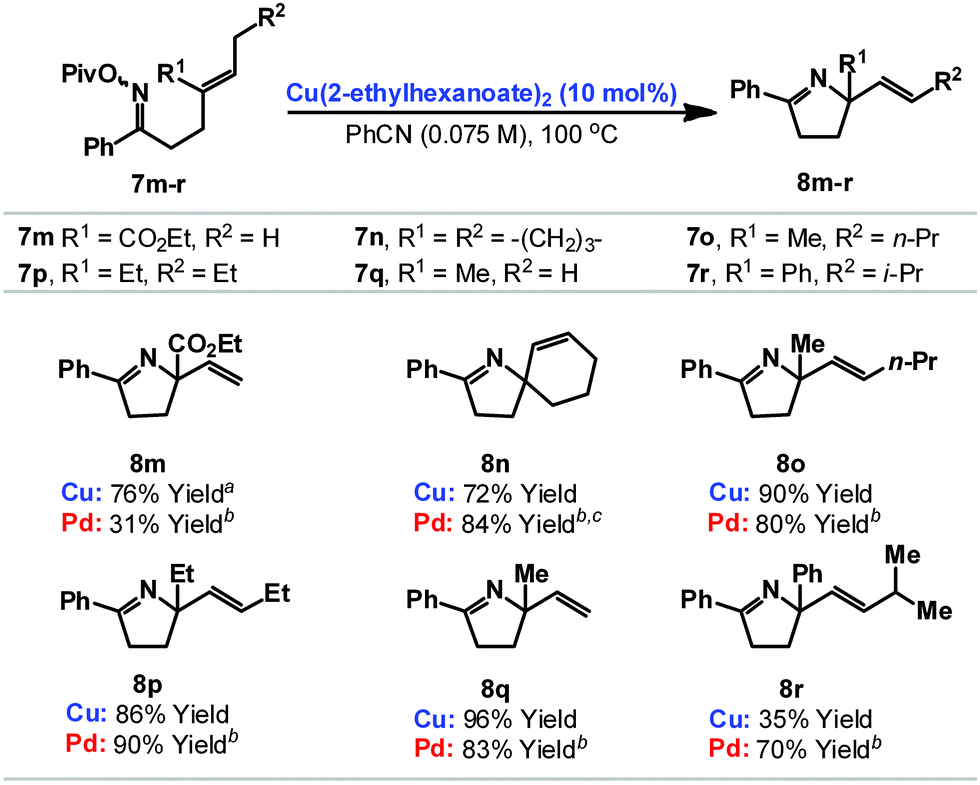
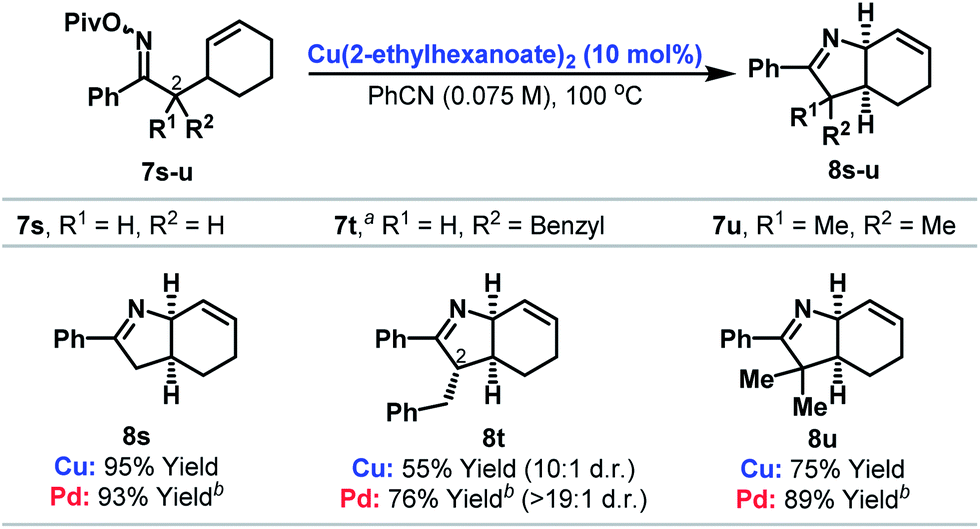
Cyclizations onto pendant cyclohexenes provide a direct entry to cis-configured heterobicycles 8s–u (Table 3). Here, reaction efficiency is comparable to our best Pd-based systems.6c In the case of 7t, cyclization of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers at C-2 provided 8t as a 10:1 mixture of diastereomers at C-2. By analogy with our earlier work,6c we favor epimerization of the C-2 stereocenter under the acidic reaction conditions after cyclization to provide the thermodynamically favored diastereomer 8t.
:1 mixture of diastereomers at C-2 provided 8t as a 10:1 mixture of diastereomers at C-2. By analogy with our earlier work,6c we favor epimerization of the C-2 stereocenter under the acidic reaction conditions after cyclization to provide the thermodynamically favored diastereomer 8t.
Our studies indicate that the copper-catalyzed processes described here are distinct from Pd-catalyzed variants and most likely do not involve migratory insertion of the alkene component into an N–Cu bond. A working mechanistic hypothesis is outlined in Scheme 2A. In situ generation of Cu(I)-carboxylate triggers cyclization to alkyl radical 12. Pathways proceeding via either the generation of iminyl radical 10 or imino-Cu(III) intermediate 11 can be envisaged; in the latter case cyclization occurs by homolytic cleavage of the N–Cu bond.14,15 It is well established that alkyl radicals can undergo oxidative elimination upon exposure to cupric acetate.16 Accordingly, trapping of alkyl radical 12 with Cu(II)-carboxylate17 provides alkyl-Cu(III) intermediate 13. Alkyl-Cu(III) species have significant carbocationic character and can undergo syn-elimination (as depicted) to generate alkene 8i.16 This process is known to favor formation of the less hindered alkene, which accounts for the observed regioselectivities. The minor quantities of saturated product (e.g.9) obtained during optimization are presumably the result of hydrogen atom abstraction by 12 from elsewhere in the reaction system.18 Alkyl-Cu(III) carboxylates (i.e.13) are mechanistically promiscuous and undergo β-hydride elimination or reductive elimination of carboxylate (to generate an alkyl-O(CO)R bond) or solvolysis to a carbocation (which might lead to Ritter-type products).16 It is noteworthy that the current protocol gives high selectivity for alkene 8i over byproducts derived either from these latter two pathways or from alkyl radical 12.
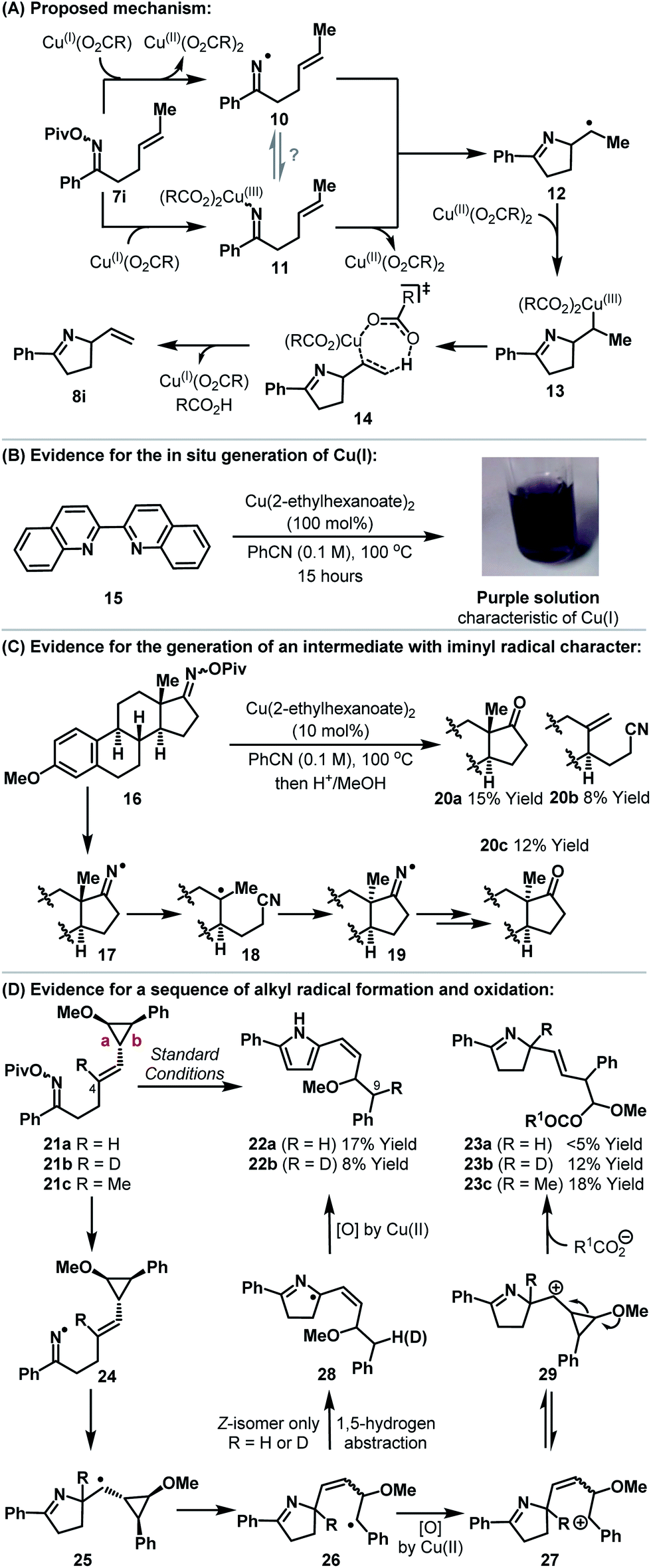 | ||
| Scheme 2 Mechanistic analysis and supporting studies. | ||
A series of experiments underpin the mechanism proposed in Scheme 2A. Heating a PhCN solution of Cu(II)(2-ethylhexanoate)2 in the presence of cuproin 15 resulted in the slow evolution of a deep purple solution (Scheme 2B).19 This is indicative of the formation of a Cu(I)–cuproin complex and is supportive of either reduction or disproportionation of Cu(II)(2-ethylhexanoate)2 under the reaction conditions.20 By way of comparison, exposure of Cu(I)OAc to analogous conditions resulted in the immediate formation of a similar purple solution (see the ESI†). The generation of an intermediate with significant iminyl radical character is evidenced using estrone derived oxime ester 16 (Scheme 2C). Upon exposure to Cu(II)(2-ethylhexanoate)2 and subsequent hydrolysis (MeOH, aq. HCl) the formation of adducts 20a–c was observed. The inversion of the methyl substituted stereocenter in 20c is accounted for by reversible β-scission from iminyl radical 17 (or an imino-Cu(III) species with radical-like character; not depicted), which leads to the thermodynamically favored diastereomer 19.21,22 Multiple mechanistic pathways, including those based upon iminyl radicals, can account for the formation of 20a/b.23
To gain insights into the sequence of events after cyclization we have prepared cyclopropyl substrates 21a–c (Scheme 2D). The substituted cyclopropane moiety is based upon Newcomb's design, which enables differentiation of radical vs. carbocation-like intermediates;24 the latter would be indicative of pathways involving either alkene imino-cupration16 or Lewis acid activation of the oxime ester.10 Because the mechanism proposed in Scheme 2A involves both radical and carbocation-like intermediates, careful analysis of the products arising from cyclization of all three substrates 21a–c was required. Cyclization of 21a resulted in the formation of the unstable cis-configured vinyl pyrrole 22a as the only observable product. This indicates that alkyl radical 25 forms and then rearranges, via cleavage of bond b, to the more stable benzylic radical 26. 1,5-Hydrogen atom abstraction (cis-alkene isomer of 26 only)25 then leads, after in situ oxidation by Cu(II)(carboxylate)2, to pyrrole 22a. Alternatively, benzylic oxidation of 26 followed by 1,5-hydride transfer (not depicted) could also generate 22a. Cyclization of deuterio-variant 21b revealed full deuterium transfer from C-4 of the starting material to C-9 of product 22b. In this case, the formation of adduct 23b, which results from cleavage of bond a, was also observed. For methyl-substituted analogue 21c, only product 23c was observed. Presumably, at the stage of 26, Cu(II)(carboxylate)2 promotes oxidation to benzylic carbocation 2716 which undergoes ring-closure to cyclopropyl stabilized carbocation 29.26 Methoxy-triggered cleavage of bond a generates an oxocarbenium ion which is trapped by carboxylate (R1CO2− = pivalate or 2-ethylhexanoate) to afford adduct 23b,c (R = D or Me).27 Overall, these results support initial cyclization to an alkyl radical and subsequent Cu(II)-promoted oxidation to an alkene. A pathway based upon migratory insertion of the alkene into the N–Cu bond of an imino-Cu(III) intermediate is discounted as this should lead solely to dihydropyrroles 23a–c. An ionic mechanism, involving Lewis acid activation of the oxime ester by Cu(II)(carboxylate)2, is not consistent with the results presented here.
As further support for the mechanism outlined in Scheme 2A, it is pertinent to consider the results of an attempted 6-ring cyclization (Scheme 3). Exposure of oxime ester 30 (the homologue of 7s) to optimized conditions did not result in the formation of Heck-type product 31. Instead, adducts 8n and 34 were generated in 46% and 31% yield respectively. The formation of these products can be accounted for by copper-catalyzed generation of iminyl radical 32 (or an imino-Cu(III) species with radical like character). 1,5-Hydrogen atom abstraction then generates an allylic radical which undergoes copper-catalyzed oxidation to the corresponding cation 33. This is trapped by either the imine moiety or pivalate to provide 8n or 34. These processes represent interesting approaches to allylic C–H amination or oxidation. The generation of 8n can be viewed as a copper-catalyzed variant of the Hofmann–Löffler–Freytag reaction and further investigations into the scope of this process are ongoing.28–30
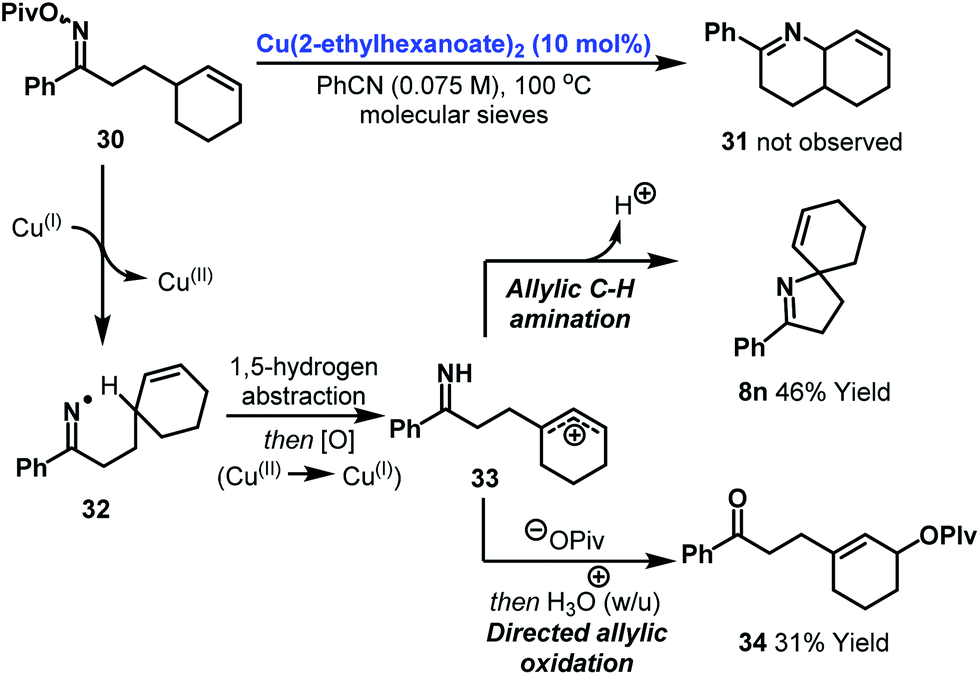 | ||
| Scheme 3 Attempted 6-ring cyclization and mechanistic pathways to allylic C–H functionalization products. | ||
Conclusions
In summary, we demonstrate that simple copper salts can replace phosphine ligated palladium catalysts for aza-Heck cyclizations of oxime esters. The Cu-catalyzed protocol proceeds via a mechanistically distinct pathway involving radical-based C–N bond formation and does not involve migratory insertion of the alkene into the N–Cu bond of an imino-Cu(III) intermediate. The net result is an easy catalytic entry to a range of synthetically flexible pyrrolidine derivatives that seem well suited to applications in medicinal chemistry. Key synthetic benefits of the current work include (a) the replacement of expensive Pd-based systems with more economical Cu-variants, (b) the use of cheap pivaloyl oxime esters instead of O-pentafluorobenzoyl variants, (c) complete selectivity for chiral products over the corresponding pyrroles for processes involving 1,2-disubstituted alkenes and (d) a catalyst system that tolerates aryl bromides. In a broader context, these studies also provide unique examples of Cu-catalyzed aza-Heck reactions that proceed via oxidative initiation at nitrogen to generate new alkene containing products. Replacing precious metal catalysts with cheaper and more sustainable variants is an important goal and this study highlights a case where this can be achieved in a particularly effective manner.Acknowledgements
A.F. thanks AstraZeneca and the University of Bristol for a Ph.D. studentship. N.J.R. thanks the Bristol Chemical Synthesis Doctoral Training Centre, funded by the EPSRC (EP/G036764/1), for the provision of a Ph.D. studentship and the SCI for a postgraduate scholarship. EPSRC (EP/J007455/1) are thanked for support. J.F.B. is indebted to the Royal Society for a University Research Fellowship.Notes and references
- (a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC; (b) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- Review: I. P. Beletskaya and A. V. Cheprakov, Organometallics, 2012, 31, 7753 CrossRef CAS . We refer specifically to processes that rely upon an internal oxidant (cf. oxidative addition of Pd(0)-catalysts into aryl-halide bonds).
- (a) H. Tsutsui and K. Narasaka, Chem. Lett., 1999, 45 CrossRef CAS; (b) H. Tsutsui, M. Kitamura and K. Narasaka, Bull. Chem. Soc. Jpn., 2002, 75, 1451 CrossRef CAS, For reviews, see: (c) M. Kitamura and K. Narasaka, Chem. Rec., 2002, 2, 268 CrossRef CAS PubMed; (d) K. Narasaka and M. Kitamura, Eur. J. Org. Chem., 2005, 4505 CrossRef CAS . Pentafluorobenzoyl oxime esters are usually employed for these processes because they are stable to Beckmann rearrangement.
- Imino-Pd(II) intermediates have been characterized and exploited in catalytic C–H amination: (a) Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676 CrossRef CAS PubMed; see also: (b) W. P. Hong, A. V. Iosub and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 13664 CrossRef CAS PubMed.
- Previous studies are consistent the direct insertion of the alkene component into the N–Pd(II) bond in a manner that is analogous to the conventional Heck reaction (see ref. 6b and c).
- (a) N. J. Race and J. F. Bower, Org. Lett., 2013, 15, 4616 CrossRef CAS PubMed; (b) A. Faulkner, J. S. Scott and J. F. Bower, Chem. Commun., 2013, 49, 1521 RSC; (c) A. Faulkner and J. F. Bower, Angew. Chem., Int. Ed., 2012, 51, 1675 CrossRef CAS PubMed.
- There is a pressing demand for the development of efficient methodologies that target low molecular weight (200–350 Da), 3D (sp3-rich) scaffolds: A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114 CrossRef CAS PubMed.
- Selected examples of copper catalyzed Heck-like couplings: (a) R. J. Phipps, L. McMurray, S. Ritter, H. A. Duong and M. J. Gaunt, J. Am. Chem. Soc., 2012, 134, 10773 CrossRef CAS PubMed; (b) V. Declerck, J. Martinez and F. Lamaty, Synlett, 2006, 3029 CAS; (c) Y. Peng, J. Chen, J. Ding, M. Liu, W. Gao and H. Wu, Synthesis, 2011, 213 Search PubMed; (d) V. Calò, A. Nacci, A. Monopoli, E. Ieva and N. Cioffi, Org. Lett., 2005, 7, 617 CrossRef PubMed; (e) J.-H. Li, D.-P. Wang and Y.-X. Xie, Tetrahedron Lett., 2005, 46, 4941 CrossRef CAS.
- (a) S. Liu, Y. Yu and L. S. Liebeskind, Org. Lett., 2007, 9, 1947 CrossRef CAS PubMed; (b) S. Liu and L. S. Liebeskind, J. Am. Chem. Soc., 2008, 130, 6918 CrossRef CAS PubMed. These processes are proposed to involve oxidative addition of Cu(I) into the N–O bond. For mechanistically similar processes that involve O-acyl hydroxylamine derivatives, see: (c) Z. Zhang, Y. Yu and L. S. Liebeskind, Org. Lett., 2008, 10, 3005 CrossRef CAS PubMed. For examples of mechanistically similar processes that involve N–Cl bonds, see: (d) C. He, C. Chen, J. Cheng, C. Liu, W. Liu, Q. Li and A. Lei, Angew. Chem., Int. Ed., 2008, 47, 6414 CrossRef CAS PubMed and references cited therein. Copper can catalyze SN2 substitutions of N–O bonds. For example, see: (e) M. J. Campbell and J. S. Johnson, Org. Lett., 2007, 9, 1521 CrossRef CAS PubMed.
- Zard has reported recently two examples of cyclizations of oxime esters that are stoichiometric in Cu(OAc)2 and provide aza-Heck-type products by an ionic mechanism that involves Lewis acid activation of the oxime ester: M. Bingham, C. Moutrille and S. Z. Zard, Heterocycles, 2014, 88, 953 CrossRef CAS.
- Intramolecular copper-catalyzed alkene difunctionalization reactions that use external oxidants: (a) T. W. Liwosz and S. R. Chemler, J. Am. Chem. Soc., 2012, 134, 2020 CrossRef CAS PubMed; (b) P. H. Fuller, J. W. Kim and S. R. Chemler, J. Am. Chem. Soc., 2008, 130, 17638 CrossRef CAS PubMed; (c) M. C. Paderas, J. B. Keister and S. R. Chemler, J. Org. Chem., 2013, 78, 506 CrossRef PubMed, Intramolecular copper-catalyzed alkene difunctionalization reactions that use internal oxidants: (d) alkene amino-hydroxylation: M. Noack and R. Göttlich, Chem. Commun., 2002, 536 RSC; (e) alkene amino-chlorination: G. Heuger, S. Kalsow and R. Göttlich, Eur. J. Org. Chem., 2002, 1848 CrossRef CAS. Intermolecular oxidative aza-Heck reactions that employ an external oxidant and do not rely on oxidative initiation at nitrogen: (f) T. W. Liwosz and S. R. Chemler, Chem.–Eur. J., 2013, 19, 12771 CrossRef CAS PubMed. The activation of oxime ester N–O bonds with catalytic Cu(I) to form new C–N bonds has been employed in various contexts. For leading references, see: (g) aza-copper enolate generation: Y. Wei and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 3756 CrossRef CAS PubMed; (h) aryl-C–H amination: K. Tanaka, M. Kitamura and K. Narasaka, Bull. Chem. Soc. Jpn., 2005, 78, 1659 CrossRef CAS; (i) alkene imino-bromination: Y. Koganemaru, M. Kitamura and K. Narasaka, Chem. Lett., 2002, 784 CrossRef CAS; For the activation of oxime ester N–O bonds with catalytic Cu(I) to form new N–N bonds, see: (j) M.-N. Zhao, H. Liang, Z.-H. Ren and Z.-H. Guan, Synthesis, 2012, 44, 1501 CrossRef CAS.
- The mechanism for nitrile formation is not clear. Possible pathways include (but are not limited to) Lewis acid promoted Beckmann type-II rearrangement of the oxime ester or β-hydride elimination from an imino-Cu(III) intermediate (vide infra). See also ref. 9a.
- Addition of molecular sieves to the reaction mixture did not suppress the formation of this byproduct. Consequently, we favor a pathway involving decomposition of the oxime ester to the corresponding NH-imine and hydrolysis to the ketone during work-up or chromatography. The NH-imine may form via either an imino-Cu(III) intermediate or an iminyl radical (vide infra).
- For a review on the one electron reduction of oxime derivatives, see: K. Narasaka and M. Kitamura, ARKIVOC, 2006, vii, 245 Search PubMed.
- Iminyl radical and imino-Cu(III) intermediates may exist in equilibrium as depicted in Scheme 2A. Imino-Cu(III) intermediates have been proposed previously (for example, see ref. 9a and b). An alternative possibility is that a radical anion of the oxime ester is generated which then cyclizes, with concomitant loss of pivalate, to generate an alkyl radical (see ref. 11i).
- (a) J. K. Kochi, J. Am. Chem. Soc., 1962, 84, 3271 CrossRef CAS; (b) J. K. Kochi, A. Bemis and C. L. Jenkins, J. Am. Chem. Soc., 1968, 90, 4616 CrossRef CAS; (c) J. K. Kochi and C. L. Jenkins, J. Am. Chem. Soc., 1972, 94, 843 CrossRef; (d) J. K. Kochi and C. L. Jenkins, J. Am. Chem. Soc., 1972, 94, 856 CrossRef.
- “Carboxylate” refers to either pivalate or 2-ethylhexanoate.
- Even though we propose a Cu(I)-initiated process, Cu(II)-salts are preferred. Higher concentrations of Cu(II) may increase the efficiency of oxidative elimination from secondary alkyl radical 12.
- J. Hoste, Anal. Chim. Acta, 1950, 4, 23 CrossRef CAS . For an application of this method as a mechanistic probe in Cu-catalysis, see: T. P. Lockhart, J. Am. Chem. Soc., 1983, 105, 1940 CrossRef.
- Based upon the considerations outlined in ref. 18, we favor partial disproportionation to provide small quantities of Cu(I). For the reduction potentials of copper ions in benzonitrile, see: R. C. Larson and R. T. Iwamoto, J. Am. Chem. Soc., 1960, 82, 3239 CrossRef CAS.
- J. Boivin, A. M. Schiano and S. Z. Zard, Tetrahedron Lett., 1992, 33, 7849 CrossRef CAS.
- It is unclear whether the processes described here proceed via an imino-Cu(III) species or the direct formation of an iminyl radical or, indeed, a radical anion of the oxime ester. To date, all attempts to isolate an imino-Cu(III) intermediate have been unsuccessful.
- 20a,b are formally hydrolysis and Beckmann rearrangement products of 16: C. Wang, X. Jiang, H. Shi, J. Lu, Y. Hu and H. Hu, J. Org. Chem., 2003, 68, 4579 CrossRef CAS PubMed . Both products may arise also via an iminyl radical or imino-Cu(III) intermediate.
- (a) M. Newcomb and D. L. Chestney, J. Am. Chem. Soc., 1994, 116, 9753 CrossRef CAS; (b) M. H. Le Tadic-Biadatti and M. Newcomb, J. Chem. Soc., Perkin Trans. 2, 1996, 1467 RSC . For an application of this type of probe to a copper catalyzed Heck reaction, see ref. 8a.
- The fate of trans-26 (R = H) is unknown. For examples of copper mediated additions of imines to alkenes that generate alkyl radicals, see: S. Sanjaya, S. H. Chua and S. Chiba, Synlett, 2012, 23, 1657 CrossRef CAS.
- For the relative stabilities of benzyl and cyclopropyl stabilized carbocations, see: J. P. Pezacki, D. Shukla, J. Lusztyk and J. Warkentin, J. Am. Chem. Soc., 1999, 121, 6589 CrossRef CAS.
- A mixture of diastereomeric products arising from trapping of the oxocarbenium ion with pivalate or 2-ethylhexanoate was observed (see the ESI†).
- (a) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1883, 16, 558 CrossRef; (b) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1885, 18, 5 CrossRef; (c) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1885, 18, 109 CrossRef; (d) M. E. Wolff, Chem. Rev., 1963, 63, 55 CrossRef CAS.
- For a mechanistically similar process that employs an amidoxime ester, see: H. Chen and S. Chiba, Org. Biomol. Chem., 2014, 12, 42 CAS.
- Exposure of the analogous O-pentafluorobenzoyl oxime ester to our optimized Pd-based systems (see ref. 6) did not result in the formation of 8n or products related to 34 and only formal hydrolysis to the corresponding ketone was observed. See ref. 6a for a discussion on mechanistic pathways to the ketone.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for all compounds are provided. See DOI: 10.1039/c4sc00652f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |