 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective allylboration of imines and indoles under mild conditions. An in situ E/Z isomerization of imines by allylboroxines†
Rauful
Alam
a,
Arindam
Das
a,
Genping
Huang
a,
Lars
Eriksson
b,
Fahmi
Himo
a and
Kálmán J.
Szabó
*a
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: kalman@organ.su.se
bDepartment of Inorganic and Structural Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden
First published on 4th March 2014
Abstract
Direct allylboration of various acyclic and cyclic aldimine, ketimine and indole substrates was performed using allylboronic acids. The reaction proceeds with very high anti-stereoselectivity for both E and Z imines. The allylboroxines formed by dehydration of allylboronic acids have a dual effect: promoting E/Z isomerization of aldimines and triggering the allylation by efficient electron withdrawal from the imine substrate.
Introduction
Reaction of allylboronates with imines is an attractive approach for selective synthesis of functionalized homoallyl amines, which are useful synthetic intermediates in pharmaceutical chemistry and natural product synthesis.1 According to the general view in the synthetic community the allylboration of imines is more difficult than that of carbonyl compounds, due to the lower electrophilicity of the carbon atom in the imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) compared to the carbonyl (CO) group.1a,b,2 Another important issue concerns the stereochemistry of the allylboration. Imines may have E or Z geometry and the isomerization complicates the stereochemical outcome of the process. When E-aldimines and (E)-3-substituted allylboronates react, syn-selectivity is expected on the basis of the Zimmerman–Traxler (Z–T) model (eqn (1)). Yet, in many cases (including also the present study) anti-selectivity has been observed, which is similar to cases involving carbonyl substrates.2a,c
N) compared to the carbonyl (CO) group.1a,b,2 Another important issue concerns the stereochemistry of the allylboration. Imines may have E or Z geometry and the isomerization complicates the stereochemical outcome of the process. When E-aldimines and (E)-3-substituted allylboronates react, syn-selectivity is expected on the basis of the Zimmerman–Traxler (Z–T) model (eqn (1)). Yet, in many cases (including also the present study) anti-selectivity has been observed, which is similar to cases involving carbonyl substrates.2a,c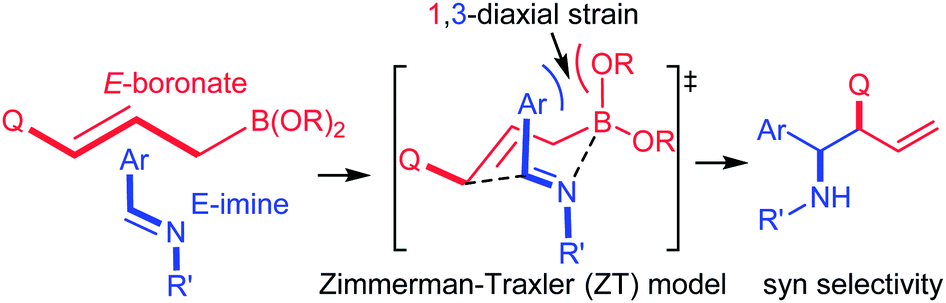 | (1) |
The unexpected anti-selectivity was mainly explained by two mechanistic models: (i) either a boat TS (transition state)2a,d instead of a chair TS (eqn (1)) occurs during the course of the reaction or (ii) spontaneous E/Z isomerization of the imines2c takes place prior to the allylation. However, modeling studies for the allylboration of aldehydes have shown that the boat geometry is unlikely in these types of process.3 Besides, the barrier for the thermal E/Z isomerization of aldimines is high; therefore it is unlikely to happen.4
Results and discussion
It is well documented that the reaction of aldehydes and allylboronates proceeds with anti-selectivity in a self-catalyzed process.1a,5 However, the low reactivity of the imines with allylboronates makes it difficult to gain insight into the mechanism of the stereo-selection. Most of the described allylboration methods require external catalysts as the imines have to be activated and/or generated in situ, which complicates the studies of the stereochemistry of self-catalyzed allylboration.1c–h Previously, we have published a convenient method for palladium-catalyzed synthesis of allylboronic acids6 from allyl alcohols and diboronic acids.7 Allylboronic acids proved to be much more reactive with carbonyl compounds than other allylboronates,6 such as allyl-Bpin derivatives. We have now found that allylboronic acids readily react with imines under dry conditions without any external Lewis acid or other additives (eqn (2)). The dry conditions were ensured by adding molecular sieves (MS) (4 Å). Without the addition of a drying agent we observed hydrolysis of the imine substrate to an aldehyde. In fact the tendency of imines to hydrolyse, such as 1a in the presence of allylboronic acids 2 (and absence of molecular sieves), was greater than in the pure form (i.e. without 2).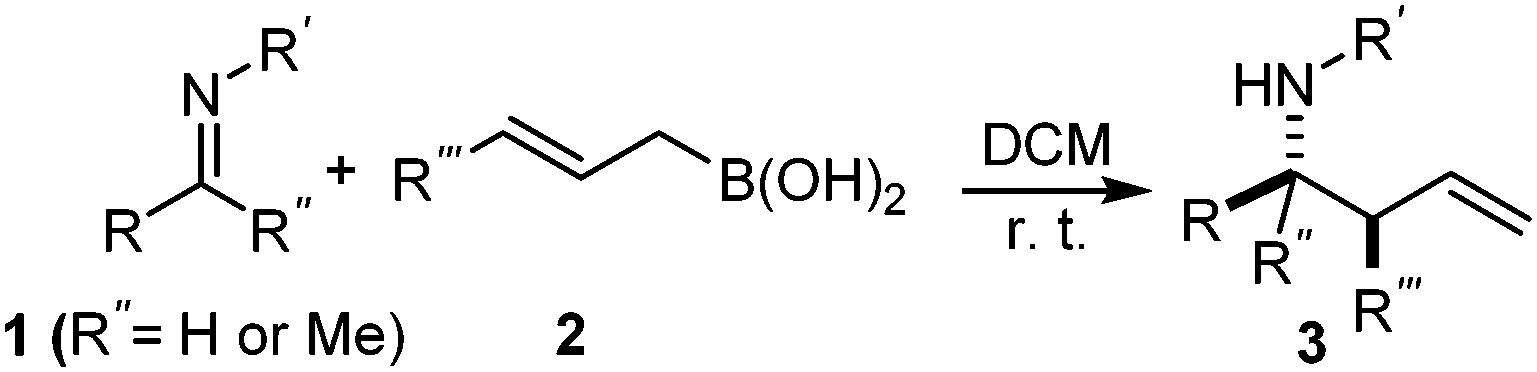 | (2) |
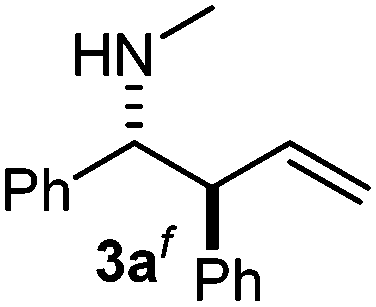
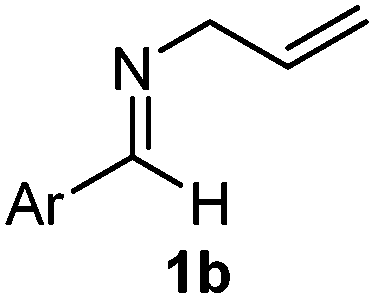
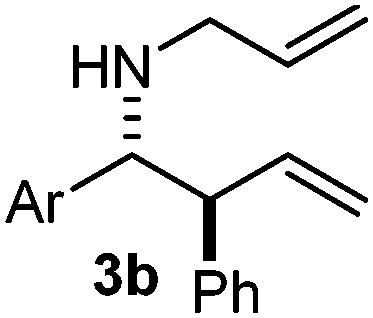
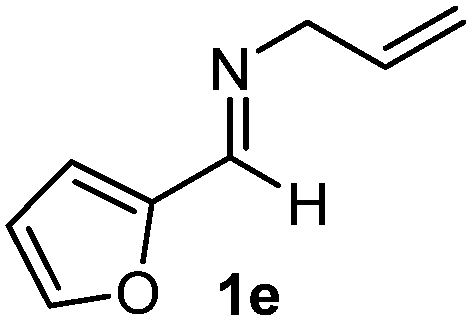
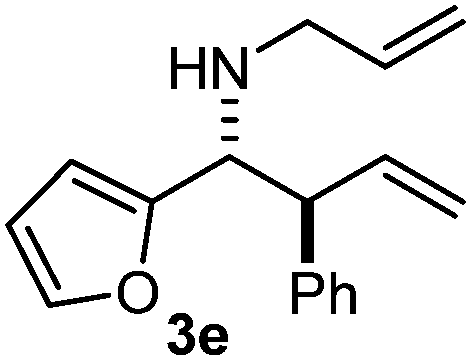
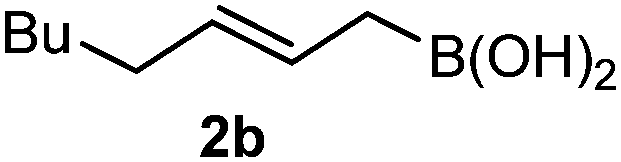
Interestingly, both the E and Z imines gave the same anti-selectivity, which is similar to aldehydes5 and ketones.6 Acyclic aryl and heteroaryl imines (1a–e) with E geometry react readily with cinnamyl and octenyl boronic acids 2a and b in the presence of molecular sieves at room temperature in a couple of hours (Table 1, entries 1–7). The reactions of imines 1a, 1b, 1d and 1e gave single stereoisomers (3a, 3b, 3d and 3e) with anti-selectivity.
| Entry | Boronic acid | Imine | Time (h) | Product | Yieldb |
|---|---|---|---|---|---|
a Unless otherwise specified 2 (0.28 mmol) and the MS (4 Å) were stirred in DCM (0.6 mL) then 1 (0.20 mmol) was added. The mixture was stirred at rt for the indicated times and isolated as a single diastereomer.
b Isolated yield.
c dr = 91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9.
d dr > 95:5.
e Boronic acid solution in CDCl3 (0.3 M) was used.
f The structure determination is based on X-ray. Ar = p-bromophenyl. PMP = p-methoxyphenyl. :9.
d dr > 95:5.
e Boronic acid solution in CDCl3 (0.3 M) was used.
f The structure determination is based on X-ray. Ar = p-bromophenyl. PMP = p-methoxyphenyl.
|
|||||
| 1 |
|
|
1 |
|
73 |
| 2 | 2a |
|
3 |
|
84d |
| 3 | 2a |
|
1 |
|
72c |
| 4 | 2a |
|
1 |
|
78 |
| 5 | 2a |
|
3 |
|
92 |
| 6 |
|
1d | 1 |
|
80d |
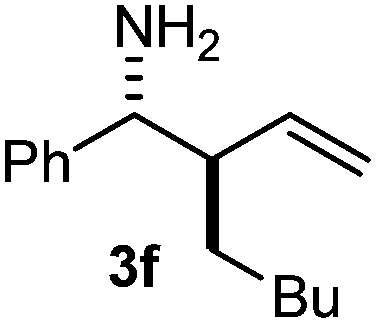
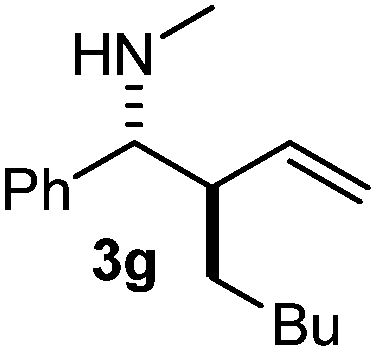
| 7 | 2b | 1a | 3 |
|
74d |
| 8 |
|
1d | 1 |
|
66d,e |
| 9 | 2a |
|
1 |
|
93 |
| 10 | 2a |
|
24 |
|
65 |
| 11 | 2a |
|
3 |
|
72 |
| 12 | 2a |
|
1 |
|
71c |
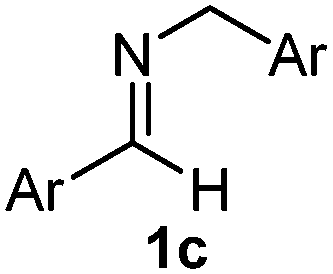
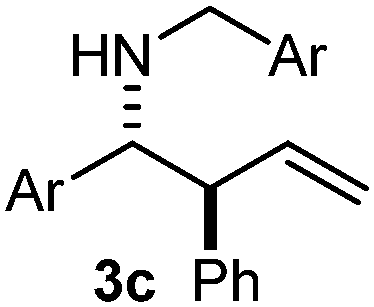
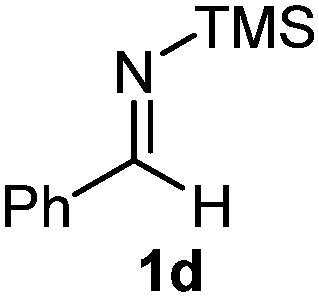
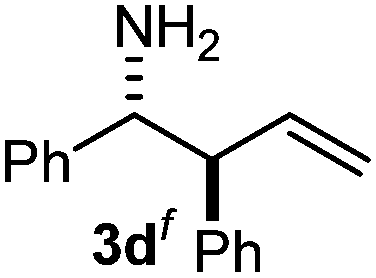
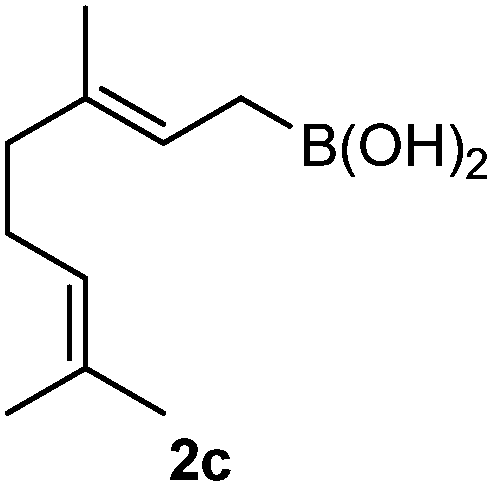
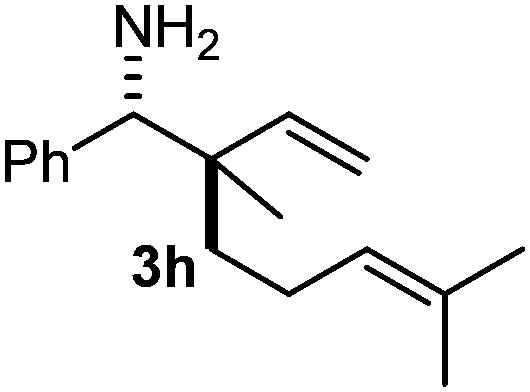
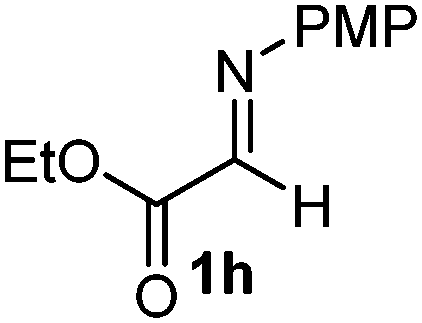
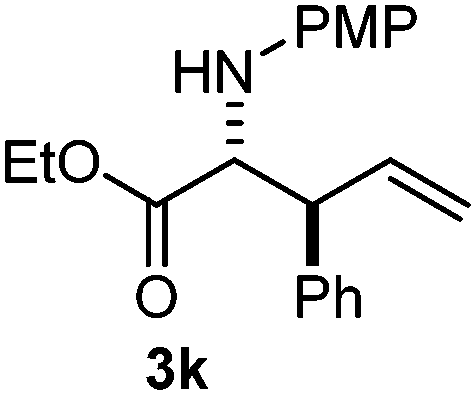
The assignment of the stereochemistry for 3a and 3d is based on X-ray diffraction. Imine 1d underwent desilylation during the reaction and thus it gave the homoallyl amine product 3d (entry 4). Benzyl imine 1c also reacted with very high stereo-selectivity but in this case two diastereomers were formed in a 91:9 ratio. The reaction of geranylboronic acid 2c with imine 1d was surprisingly fast (only one hour) and resulted in 3h (entry 8) with adjacent quaternary and tertiary stereocenters, with a diastereomeric ratio (dr) of 95:5.
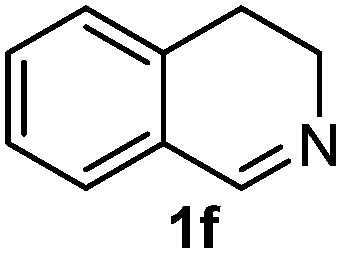
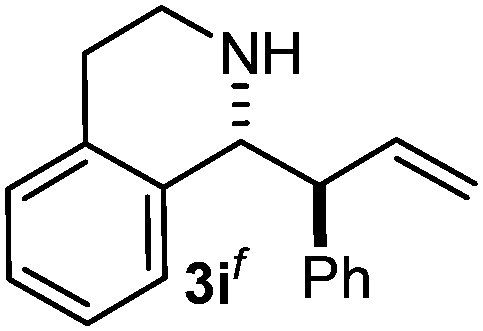
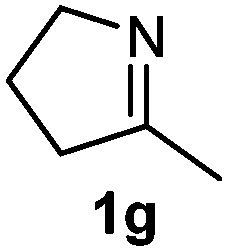
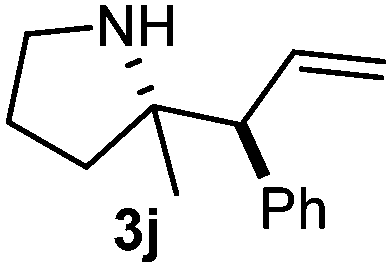
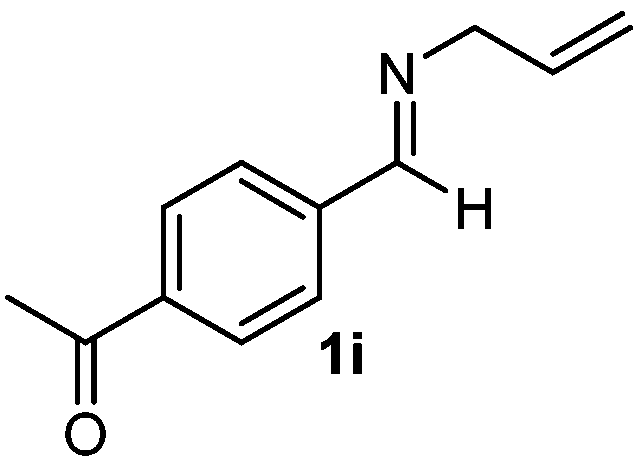
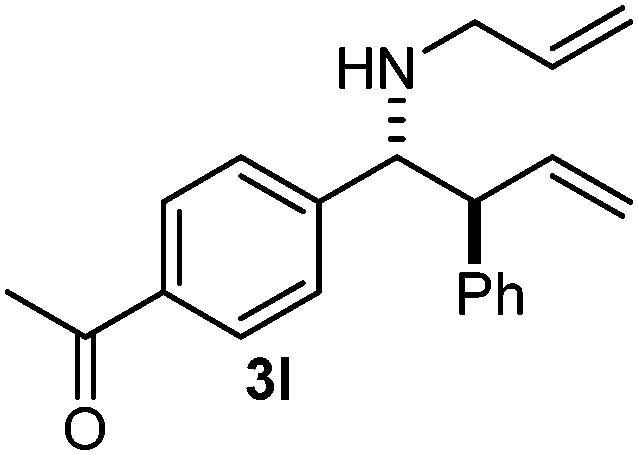
Cyclic imine1h1f has a Z geometry, yet the stereochemistry of the sole product 3i also has anti-geometry (entry 9), which was confirmed by X-ray diffraction. Thus 1a with a stable E-geometry4b and its closely related analog 1f with Z-geometry gave the same product, the anti-stereoisomer (cf. entries 1 and 9) at room temperature in DCM/1 h without an external catalyst. Moreover, the stereochemistry of the allylboration (using 2a) of 1a and its aldehyde analog (benzaldehyde) are identical.8 Most of the ketimines, such as the methyl analogs of 1a and 1b resisted allylboration under the applied uncatalyzed conditions. However, ketimine 1g reacted with excellent stereoselectivity but much slower (in 24 h) than the aldimines. This indicates that allylboronic acids are able to react with ketimines as well but the reaction is sensitive to steric factors. Thus bulkier ketimines than 1g could be useful substrates for asymmetric allylation. For example, chiral Lewis acids1c,d,9 or chiral auxiliaries10 on the ketimine can be employed to increase the reactivity of the reactants. Glyoxylate imine 1h also reacted readily with allylboronic acids, opening a new synthetic route8,11 for allyl boronate based stereoselective synthesis of amino acid derivatives. In previous studies6 we have shown that allylboronic acids react readily with ketones. Compound 1i has both keto and aldimine functionalities (entry 12) but only the imine functionality was transformed when 2a was added. The high chemoselectivity indicates that aldimines react faster with allylboronic acids than ketones. Cyclic ketimine 1g was the only aliphatic imine that we could employ, as acyclic aliphatic imines underwent rapid hydrolysis even in the presence of molecular sieves. Our efforts to remove minute trace amounts of water proved to be fruitless.
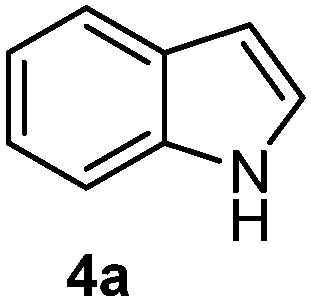
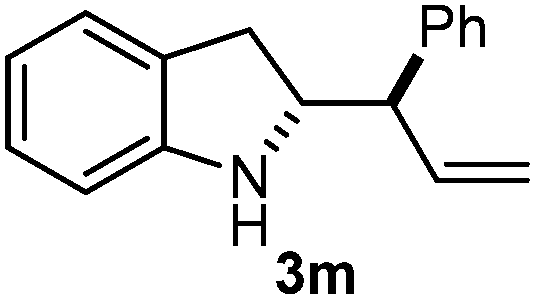
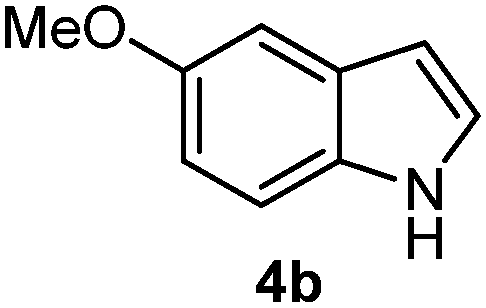
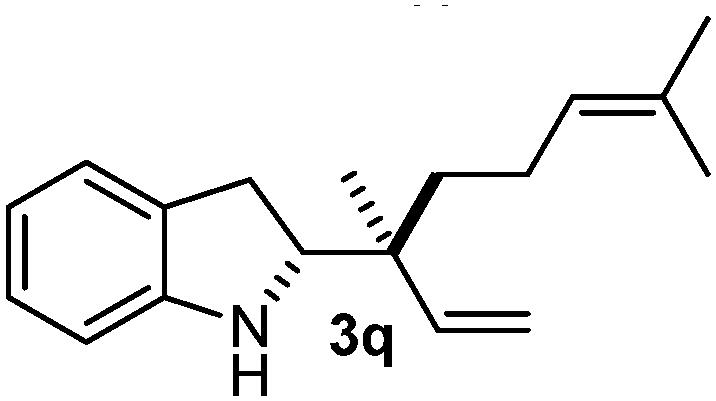
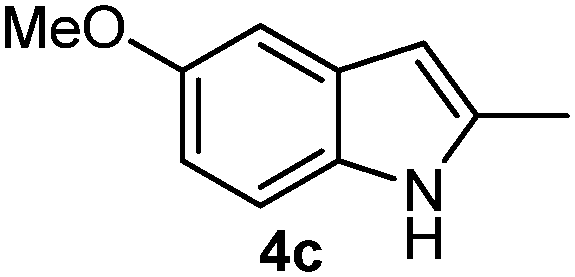
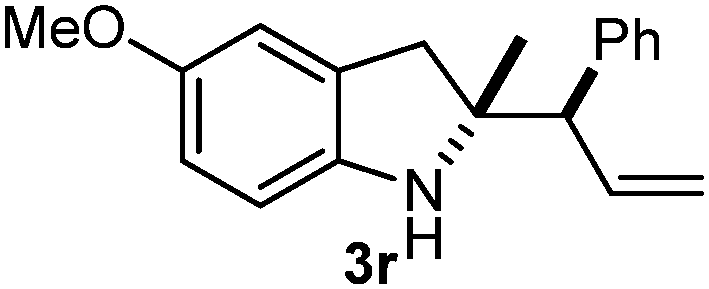
Batey and co-workers12 have recently shown that indoles react with allyl-BF3K derivatives in the presence of BF3via in situ formation of allyl-BF2 species. We have found that allylboronic acids react readily with indoles 4a–c without any additives (Table 2). The allylation proceeded with very high stereoselectivity, affording a single product. The reaction was complete in a couple of hours using 2a or 2b. Geranylboronic acid 2c reacted with 4a with high selectivity creating adjacent quaternary and tertiary stereocenters (3q) in 24 hours (entry 5). Methyl indole derivative 4c was also reacted at 60 °C with 2a to selectively give 3r with adjacent quaternary and tertiary stereocenters (entry 6). The longer reaction times and higher temperatures (entries 5 and 6) required for completion of these two latter processes indicate that the reaction is slower in the presence of bulky groups.
| Entry | Boronic acid | Indole | Time (h) | Product | Yieldb |
|---|---|---|---|---|---|
| a Unless otherwise stated, allylboronic acid 2a–c (0.15 mmol) was reacted with indoles 4a–c (0.1 mmol) at rt in DCM (0.4 mL). b Isolated yield as a single diastereomer. c Reaction scale up to 0.5 mmol of indole. d Reaction performed at 60 °C. | |||||
| 1 | 2a |
|
3 |
|
90 |
| 2 | 2a |
|
1 |
|
96/97c |
| 3 | 2b | 4a | 3 |
|
95 |
| 4 | 2b | 4b | 1 |
|
85 |
| 5 | 2c | 4a | 24 |
|
74 |
| 6d | 2a |
|
12 |
|
75 |
The most intriguing mechanistic aspect of the above allylboration of E and Z imines is the very fast anti-selective allylation. Since the stereochemistry is the same for the allylboration of aldehydes and ketones, we hypothesized that the reaction with imines also takes place according to the Z–T model13via a chair-type TS. However, according to this model a Z-geometry is required for the imines (such as in 1f) to predict anti-selectivity via a chair TS (cf.eqn (1)). Thus, the acyclic E-aldimines 1a–d and 1h–i should undergo rapid isomerization to the corresponding Z-form prior to the allylboration. The thermal isomerization of aldimines has a high activation energy.4b For example, according to the 1H NMR spectrum 1a exists as a stable E isomer in CDCl3 even at elevated temperatures (50 °C). Application of organoboronic acids as organocatalysts has attracted great interest in the synthetic community.14 Moreover, Piers and co-workers15 have shown that boron-based Lewis acids, such as B(C6F5)3 are able to catalyze the isomerization of aldimines. Accordingly, we assumed that allylboronic acid or its boroxine may catalyze the isomerization of E- to Z-aldimines prior to the allylboration process. We have observed several indications of possible interactions of allylboronates and imines prior to the allylation. As mentioned above, the hydrolysis of aldimines to aldehydes is much faster in the presence, rather than in the absence, of allylboronic acids. Without the use of molecular sieves we observed partial hydrolysis of imines 1a–d and 1h–i leading to the formation of homoallyl alcohols by the allylboration of the hydrolyzed products. The application of molecular sieves solved this problem but also gave rise to the dehydration of allylboronic acids. This leads to the formation of allyl boroxines, such as 2ab from 2a, which are detectable by 1H NMR.6a Since allylboronic acid 2a allylates Z-aldimines (such as 1f) rapidly, we studied the E/Z isomerization of 1a in the presence of aryl boroxine 5 (Fig. 1), which is obviously not able to allylate imines. Boroxine 5 was prepared from the corresponding arylboronic acid by stirring with molecular sieves. Before the isomerization experiment the molecular sieves were removed by filtration in a glove box. It was found that 1a rapidly isomerized to 6 in the presence of boroxine 5. The process was monitored by 1H NMR, indicating the formation of a 1:1 mixture of 1a and 6. In 6 the phenyl and N-methyl groups are in the Z-geometry, which was ensured by detection of the dNOE effect between the N-methyl and ortho-phenyl protons (Fig. 1). In 1a a dNOE effect was observed between the N-methyl group and the imine C–H, which shows that in isolated 1a the phenyl and N-methyl groups are in the E-geometry.
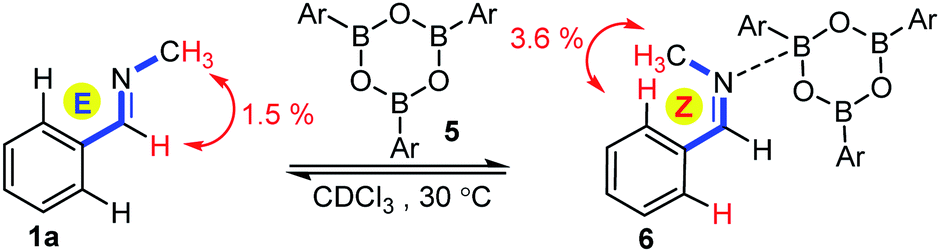 | ||
| Fig. 1 E/Z isomerization of 1a in the presence of aryl boroxine (Ar = 4-fluorophenyl). The major 1H dNOE is indicated for the two observed isomeric forms. | ||
Although, the reaction mixture (Fig. 1) contained 100% boroxine 5 based on the 1H-NMR spectrum, we also considered the possibility that traces of water could generate arylboronic acid by the hydrolysis of 5. Hall and co-workers14d reported that molecular sieves may act as reservoirs of water and, thus traces of active boronic acid may be available by the hydrolysis of boroxine. When small amounts of water were added to boroxine solution 5, the appearance of the 1H-NMR shift of the corresponding boronic acid was observed. Under these conditions we did not observe any E/Z isomerization of 1a. Thus, we conclude that boroxine under dry conditions is required for the efficient isomerization of E-imines (such as 1a) to Z-imines.
We employed molecular sieves (4 Å) to remove residual water completely from the reaction mixture. However, molecular sieves may act as (weak) acid catalysts in certain processes.16 To check this possibility we performed the allylation of 1a with 2a under standard conditions (entry 1) in the presence of NaHCO3 to buffer the acidity of the employed molecular sieves. We did not observe any effect by NaHCO3 on the outcome of the reaction, and thus we conclude that molecular sieves do not act as acid catalysts for the presented allylation process.
The Z relationship of the N-methyl and phenyl groups in 6 may satisfactorily explain the anti-selectivity of the allylboration via a chair TS in line with the Z–T model. To prove this assumption we performed a computational DFT study using the B3LYP functional17 (for computational details see ESI†). The results show (Fig. 2) that the formation of imine–boroxine complex 7a from 1a and allyl boroxine 2ab is an exergonic process (by −4.1 kcal mol−1). This assumes that facile E/Z isomerization of the imine takes place, as established above for 1a (Fig. 1). It is interesting to note that 7a, in which the N-methyl and phenyl groups are in a Z-geometry (like in 6), is more stable by 6.2 kcal mol−1 than 7b, which has an E-geometry.
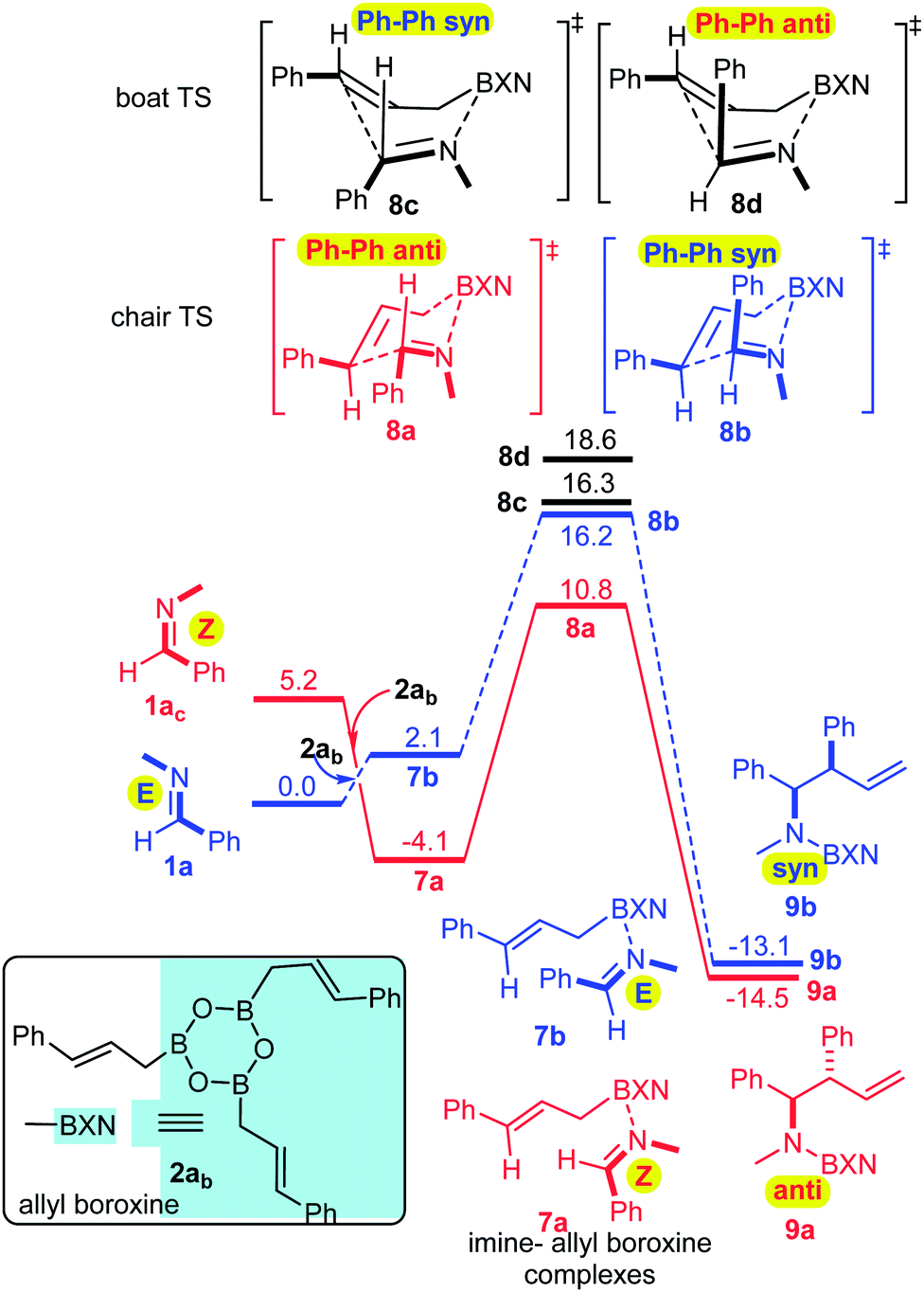 | ||
| Fig. 2 Reaction profile for the allylboration of 1a in the presence of allylboroxine 2ab. The ΔG values are given in kcal mol−1. | ||
This trend is reversed compared to the free imines, 1avs.1ac. From 7a, the allylboration proceeds via chair TS 8a with a low activation barrier (14.9 kcal mol−1) affording 9a with anti-selectivity. This is in agreement with the Z–T model. The chair-shape of TS structure 8a and the TS geometry for the allylboration of aldehydes3 are very similar, which is in line with the identical stereochemistry observed for the two processes. Allylation of the other imine–allyl boroxine complex (7b) or 1a, in which the N-methyl and phenyl are in an E geometry, requires 5.4 kcal mol−1 higher activation barriers to give the syn product 9b. The high barrier is apparently because of the axial position of the phenyl group in 8b, which is sterically unfavorable in line with the Z–T model (see eqn (1)). We have also calculated the activation barriers via boat TSs2a,b (8c and 8b). However, formation of the anti-product 9avia boat TS 8d involves a much higher barrier than via chair TS 8a (by 7.8 kcal mol−1). The high energy of the boat forms 8c and 8d compared to the chair forms 8a and 8b is not surprising, as the unfavorable eclipsing strains and 1,4-diaxial strain in the boat form are well known by analysis of the conformational energy surface of cyclohexane.18 Due to the relatively short B–C (2 Å) and B–N (1.5 Å) distances, the steric strains in TS structures 8a–d (Fig. 4) and the corresponding stationary points in the potential energy surface of the “ideal” cyclohexane structure are surprisingly similar. In fact, one of the main reasons for the remarkably high stereoselectivity of the allylboration of carbonyls and imines is due to the short B–C, B–O/B–N, and C–C distances in the TSs.
Due to this geometry feature the bulky substituents are brought into close proximity, which allows very efficient stereo-differentiation. A good example is the strong 1,3-diaxial strain between the axial phenyl and the boroxine groups in 8b (Fig. 4), which leads to the less favorable formation of the syn product 9b over the anti product 9a (Fig. 2).
We have also performed modeling studies for allylation with allylboronic acid 2a instead of its boroxine 2ab (Fig. 3). The corresponding reaction profiles show the same mechanistic features as the above processes with boroxine (Fig. 2). Thus, the lowest energy path involves isomerization of E-imine 1a to Z-imine via the formation of an imine–boronic acid complex, which eventually gives the anti-diastereomer. However, there are also notable differences between the reaction profiles for the allylation with boroxine 2ab (Fig. 2) and boronic acid 2a (Fig. 3). Formation of the boroxine–imine complex 7a is exergonic, while formation of the boronic acid–imine complex 10a is endergonic. Furthermore, the activation barrier involving allyl boroxine 2abvia the 1a → 7a → 8a → 9a path (Fig. 2) is substantially lower (by 5.7 kcal mol−1) than the corresponding activation barrier involving allylboronic acid 2a.
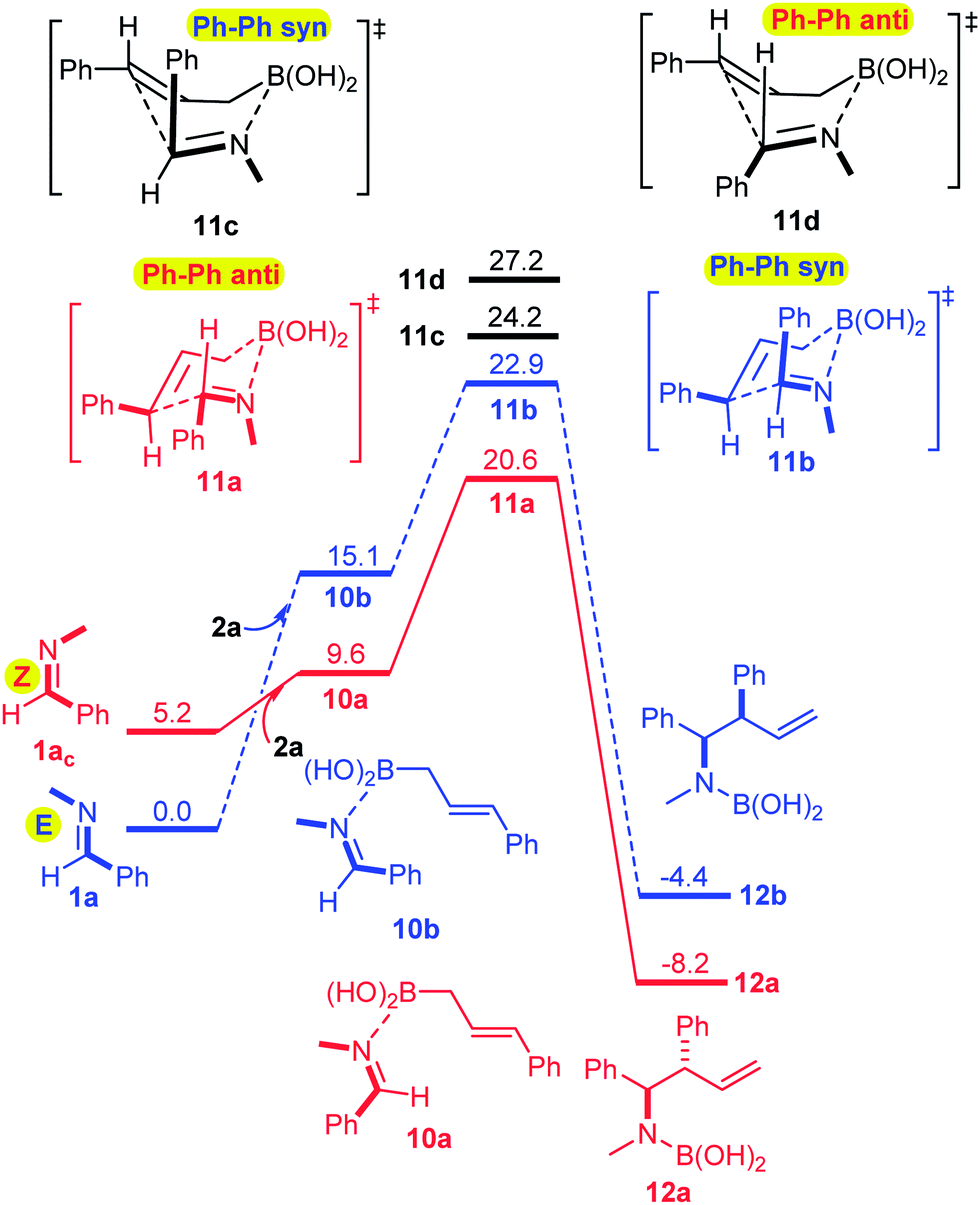 | ||
| Fig. 3 Allylboration of 1a with cinnamyl boronic acid 2a. The ΔG values are given in kcal mol−1. | ||
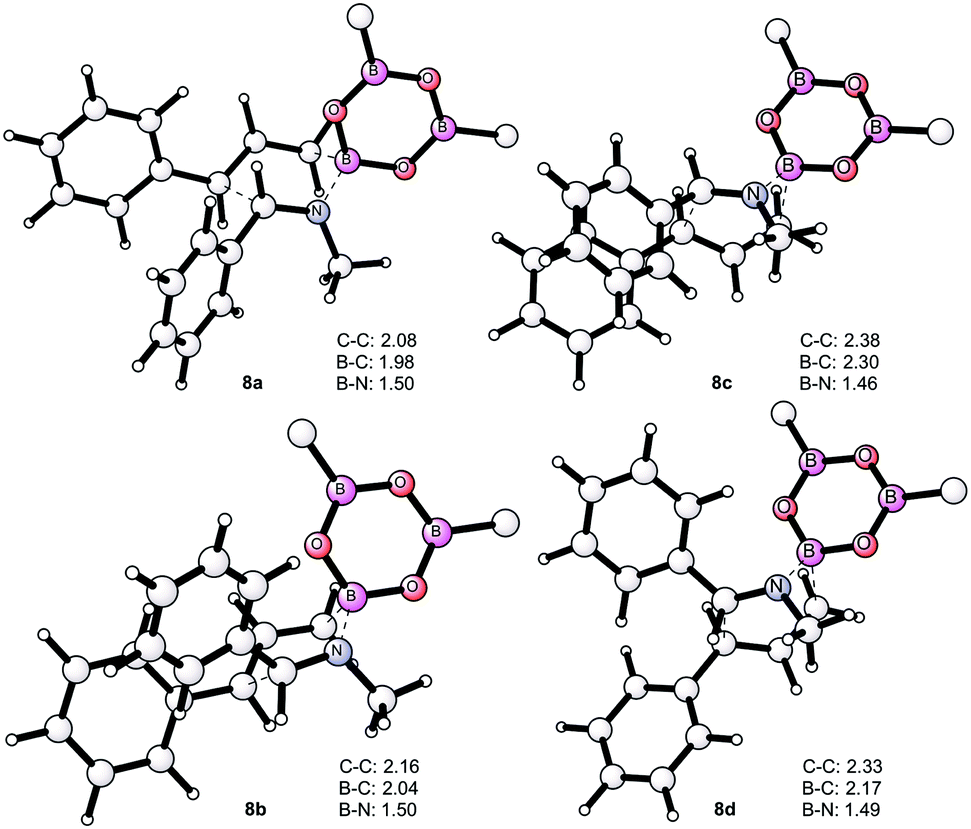 | ||
| Fig. 4 Optimized geometries of the TS structures 8a–d. Two of the allyl moieties of the boroxines have been removed for clarity. The distances are in Å. | ||
The higher efficiency of 2abvs.2a for the allylation of 1a can be explained by the higher B/O ratio in boroxine (1:1) than in allylboronic acid (1:2). Accordingly, less electron density is transferred from the oxygen O(nπ) lone-pair to the empty B(pπ) orbital of boron in boroxine 2ab than in allylboronic acid 2a. This leads to a much higher electrophilicity (Lewis acidity) of the boron B(pπ) in boroxine than in allylboronic acid. The high electrophilicity of boron in boroxine is favorable for both the E/Z isomerization of the aldimines (such as 1a) and the allylation of the imine. A possible failure of direct allylboration of imines, such as 1a–d, with allyl-Bpin and analogs may arise from the fact that the boron atom of the Bpin functionality is not sufficiently electrophilic for the E/Z isomerization of acyclic aldimines and/or triggering the allylation (by interacting with the N-lone-pair of the imine substrate).
To our knowledge, until now allylboroxine mediated E/Z isomerization of imines has not been suggested for the anti-selective allylation of imines. However, Leighton and co-workers19 have reported E/Z isomerization of 2-aminophenol derived imines during cinnamylation of imines with cinnamyl chlorosilanes (Cl-silane analog of 2a). The proposed isomerization is based on the chelation of the hydroxyl unit of 2-aminophenol imine with the silyl group of cinnamyl chlorosilane. An interesting analogy between the allylboronic acid and allyl chlorosilane based cinnamylation reactions is that in both cases in situ E/Z isomerization of the imine may occur by the allylation reagent leading to excellent anti-selectivity.
Conclusions
We have demonstrated that allylboronic acids may readily react with imines. The reaction proceeds under mild conditions with E-aldimine, cyclic aldimine, ketimine and indole substrates with very high anti-stereoselectivity. The process is chemoselective, as aldimines can be allylated in the presence of a keto group. The experimental and DFT mechanistic studies show that boroxines (formed by dehydration of allylboronic acids) have a dual activating effect in this reaction: promoting E/Z isomerization of aldimines, and as efficient electron acceptors/Lewis acids triggering the allylation process. Allylboration is a widely used methodology in natural product synthesis and in advanced organic chemistry.1c–h,20 Based on the above results the scope of allylboration can be further extended for synthesis of complex stereodefined amine structures. In addition, new insights into the stereochemistry of allylboration and into the validity of the Z–T model are helpful for the design of new selective transformations.Conflict of interest
The authors declare no competing financial interests.Acknowledgements
The authors thank the financial support of the Swedish Research Council (VR) and the Knut och Alice Wallenbergs Foundation. The authors also thank Dr Carolina Fontana for helping with some of the NMR experiments. GH thanks the Carl Tryggers Foundation for a postdoctoral fellowship.Notes and references
-
(a)
D. G. Hall, Boronic Acids, Wiley, Weinheim, 2011 Search PubMed
; (b) T. R. Ramadhar and R. A. Batey, Synthesis, 2011, 1321 CAS
-
(a) R. W. Hoffmann and A. Endesfelder, Liebigs Ann. Chem., 1983, 2000 CrossRef CAS
-
(a) Y. Li and K. N. Houk, J. Am. Chem. Soc., 1989, 111, 1236 CrossRef CAS
-
(a) J. E. Johnson, N. M. Morales, A. M. Gorczyca, D. D. Dolliver and M. A. McAllister, J. Org. Chem., 2001, 66, 7979 CrossRef CAS PubMed
-
D. Hall and H. Lachance, Allylboration of Carbonyl Compounds, Wiley, Hoboken, New Jersey, 2012 Search PubMed
-
(a) M. Raducan, R. Alam and K. J. Szabó, Angew. Chem., Int. Ed., 2012, 51, 13050 CrossRef CAS PubMed
-
(a) G. A. Molander, S. L. J. Trice, S. M. Kennedy, S. D. Dreher and M. T. Tudge, J. Am. Chem. Soc., 2012, 134, 11667 CrossRef CAS PubMed
- N. Selander, A. Kipke, S. Sebelius and K. J. Szabó, J. Am. Chem. Soc., 2007, 129, 13723 CrossRef CAS PubMed
- D. L. Silverio, S. Torker, T. Pilyugina, E. M. Vieira, M. L. Snapper, F. Haeffner and A. H. Hoveyda, Nature, 2013, 494, 216 CrossRef CAS PubMed
-
(a) M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600 CrossRef CAS PubMed
-
N. Selander and K. J. Szabó, in Current Frontiers in Asymmetric Synthesis and Application of alpha-Amino Acids, ed. V. A. Soloshonok and K. Izawa, ACS Symposium Series, Oxford University Press, Oxford, 2009 Search PubMed
- F. Nowrouzi and R. A. Batey, Angew. Chem., Int. Ed., 2013, 52, 892 CrossRef CAS PubMed
- R. W. Hoffmann, Angew. Chem., Int. Ed., 1982, 21, 555 CrossRef
-
(a) E. Dimitrijević and M. S. Taylor, ACS Catal., 2013, 3, 945 CrossRef
- J. M. Blackwell, W. E. Piers, M. Parvez and R. McDonald, Organometallics, 2002, 21, 1400 CrossRef CAS
-
(a) N. Fontes, J. Partridge, P. J. Halling and S. Barreiros, Biotechnol. Bioeng., 2002, 77, 296 CrossRef CAS PubMed
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed
-
(a)
D. Cremer and K. J. Szabó, in Conformational Behavior of Six-Membered Rings; Analysis, Dynamics, and Stereoelectronic Effects, VCH, 1995, p. 59 Search PubMed
- J. D. Huber and J. L. Leighton, J. Am. Chem. Soc., 2007, 129, 14552 CrossRef CAS PubMed
-
(a) J. Y. Ding and D. G. Hall, Angew. Chem., Int. Ed., 2013, 52, 8069 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 985818–985820. See DOI: 10.1039/c4sc00415a |
| This journal is © The Royal Society of Chemistry 2014 |