 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hyperbranched polydendrons: a new controlled macromolecular architecture with self-assembly in water and organic solvents†
Fiona L.
Hatton
a,
Pierre
Chambon
a,
Tom O.
McDonald
a,
Andrew
Owen
b and
Steven P.
Rannard
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, L69 7ZD, UK. E-mail: srannard@liv.ac.uk
bDepartment of Molecular and Clinical Pharmacology, University of Liverpool, Block H, 70 Pembroke Place, Liverpool L69 3GF, UK
First published on 6th February 2014
Abstract
A new macromolecular architecture (hyperbranched polydendrons) is presented. Combining aspects of linear-dendritic hybrids, controlled radical polymerisation and branched vinyl polymerisation, the materials have very high molecular weight (Mw > 1 MDa) and surface functionality. Although dispersities are broad (Đ up to 25) the structures behave with remarkable uniformity upon manipulation of solvent environment. Comparisons of conventional linear-dendritic hybrids and hyperbranched polydendrons are presented, including aspects of their synthesis. Under solvent exchange in organic media, a reversible self-assembly to form monodispersed nanoparticles (PDI as low as 0.013) is observed. Self-assembly and encapsulation is also observed during aqueous nanoprecipitation of the hyperbranched materials, with nanoparticle size (diameters from 60–140 nm) controlled through modification of precipitation conditions and the generation of the ideally branched dendrons at one end of each primary chain. The aqueous nanoparticles are highly stable and offer considerable opportunities for tailored functionality and future advanced applications.
Introduction
The global interest in macromolecular architecture has been driven over the last two and a half decades by advances in controlled polymer synthesis techniques, such as nitroxide-mediated polymerisation,1 atom transfer radical polymerisation (ATRP)2 and reversible addition–fragmentation chain-transfer polymerisation (RAFT),1 allowing the accurate tailoring of chain chemistry, composition and degree of polymerisation. Additionally, the appreciation of branched structures such as hyperbranched polymers3 (Fig. 1A) and dendrimers4 (Fig. 1B) has led to a variety of chemistries, rivalling those available for linear polymers including: polyimines/polyamines,5 polyurethanes,6,7 polyethers,8 polyesters,9 polycarbonates,10 polyamides,11,12 polyamidoamines,13 and polycarbosilanes.14 Structure and function have collectively been iterated to develop potential advanced applications for polymers in areas as diverse as drug delivery,15 biology,16 sensors,17 energy storage17,18 and self-healing coatings.19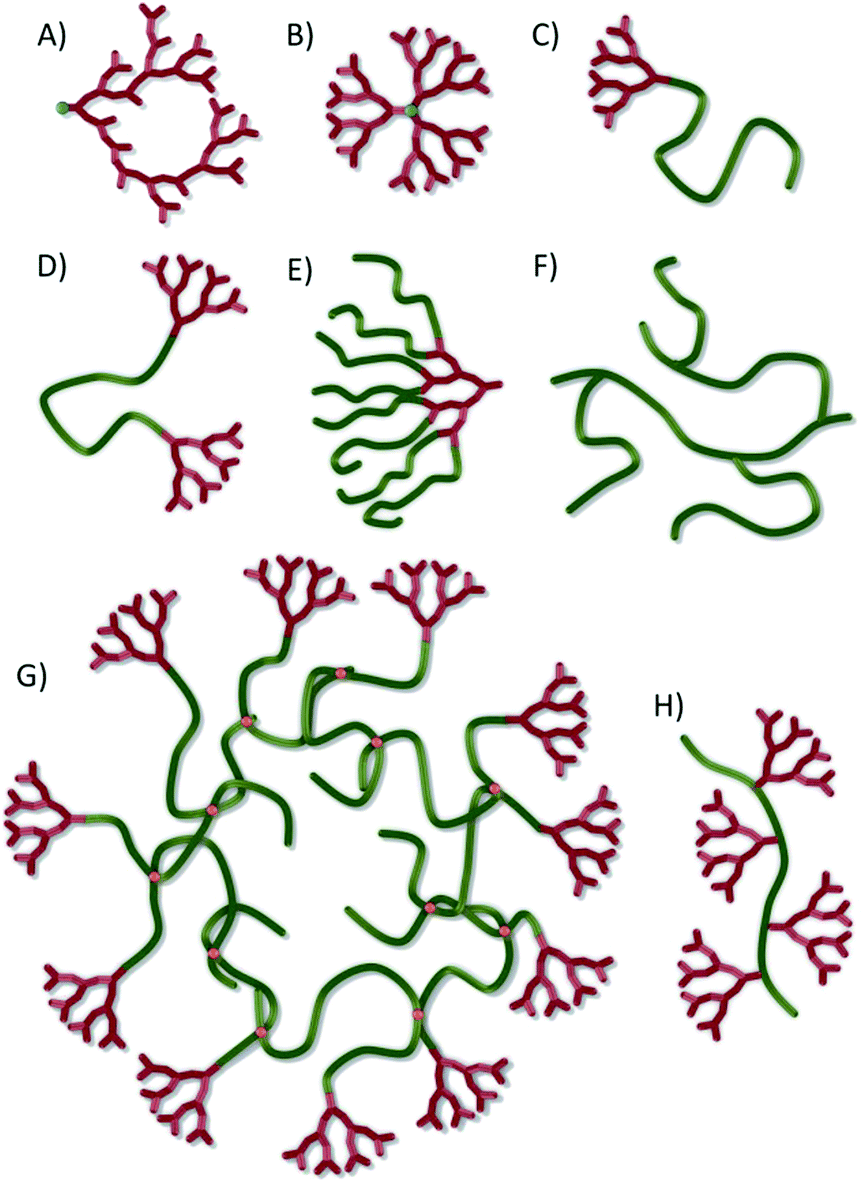 | ||
| Fig. 1 Schematic representation of a range of macromolecular architectures: (A) hyperbranched polymer; (B) ideally branched dendrimer; (C) linear-dendritic (LD) hybrid; (D) DLD hybrid; (E) DLx hybrid; (F) self-condensed or grafted vinyl polymer; (G) hyperbranched-polydendron; (H) dendronised linear polymer. | ||
The control of chemistry and purity have allowed macromolecular science to embrace nanomaterials, establishing various self-assembled nanostructures20 from spherical solid nanoparticles21 to multi-compartment micelles,22,23 core–shell structures,24 vesicles/liposomes,25,26 rods27 and worms.28 Arguably the most diverse organic nanomaterial studies have involved ideally-branched dendrimers.16,29,30 Dendritic polymer application31 is maturing in nanomedicine32 and electronics,33 but, although dendrimers possess uniform molecular weight distributions and well defined shapes, the available size range is typically <15 nm,34 although recent reports have described ideal, generation 13 polytriazine dendrimers with diameters of approximately 30 nm and molecular 8.4 MDa.35 Considerable obstacles continue to exist for cost-effective applications of such materials including the synthetic complexity and molecular weight/physical size limitations.
Research spanning various chemical disciplines has also generated complex materials such as rotaxanes and catenanes, the mixture of cyclic structures and linear polymer chains, and the growing field of linear-dendritic hybrids where linear polymer chains are decorated either at the chain ends or side-chains with dendrons; fragments of dendrimers. Linear-dendritic polymer hybrids have now been synthesised in varying forms36a to create materials including one dendron at the end of a linear polymer (an LD hybrid; Fig. 1C), dendrons at both ends of a linear chain (a DLD hybrid; Fig. 1D), linear chains conjugated to the periphery of a dendron (a DLx hybrid, Fig. 1E) and dendrons at the centre of complex architectures with one linear chain attached to the focal point and either the same or different polymers synthesised from the peripheral dendron functionality (an LDLx hybrid). Micellar structures of amphiphilic linear-dendritic copolymers (30–70 nm),9 and micelle aggregates (approximately 200 nm),37 have been reported after association of relatively small building blocks.
The introduction of macromolecular complexity is often achieved at the expense of synthetic simplicity. Multiple iterative reactions are required to produce many such materials, with repeated isolation and purification steps and frequently the use of large reagent excesses. This limits access to these architectures to specialist research groups, preventing greater innovation and evaluation. The various reports of hyperbranched step-growth and vinyl polymerisations have attempted to simplify the synthesis of such materials but often structural fidelity or fine control is lost. In a range of cases, simple one-pot hyperbranched vinyl polymerisations38,39 have been shown to introduce nanoparticle formation,40 including shaped nanoparticles,41,42 with controllable stimuli responsive and surface active behaviour,43 even though the control of molecular weight and dispersity is somewhat sacrificed.
A key challenge is the identification of accessible chemistries that generate materials with novel architecture and uniform behaviour. Herein, we describe the first report of a novel approach that combines controlled radical polymerisation, hyperbranched vinyl polymerisation and linear dendritic hybrids to form a new polymer architecture, hyperbranched polydendrons (hyp-polydendrons). We have adopted this term as linear polymers that are decorated with dendrons (Fig. 1H) have been termed ‘polydendrons’ previously.44 Unlike branched vinyl polymers produced from self-condensing vinyl polymerisation45 or grafting techniques46 (Fig. 1F), hyp-polydendrons (Fig. 1G) have two untethered ends on each primary chain of the branched architecture, with one chain end bearing an ideally branched dendron. The materials have unique structure and have been synthesised to very high molecular weights in a one-pot polymerisation strategy; as such these new materials offer novel opportunities. Moreover, we also describe the self-assembly of hyp-polydendrons to controllably form highly monodisperse, stable nanoparticles with control of size and chemistry; a highly surprisingly uniform behaviour from such polydisperse materials.
Results and discussion
Within our group, one-pot branched vinyl copolymerisation techniques have been shown to form nanoparticles in a single synthetic procedure41,42via a concerted propagation and interchain branching of linear polymer chains using monofunctional monomers in the presence of low concentrations of bifunctional monomers. When used in conjunction with controlled radical polymerisation techniques, high molecular weight branched amphiphilic block copolymers have been formed that produce spherical nanoparticles when dialysed using water.41 The introduction of multi-functional initiators allowed dumbbell and clover-leaf nanoparticles to be formed.42 In this current study, we combine aspects of dendrimer and branched vinyl polymer strategies to simultaneously create linear-dendritic hybrid copolymers and introduce interchain branching to produce very high molecular weight complex unimolecular architectures in a one-pot controlled radical polymerisation. The hyp-polydendrons have been extensively characterised and their self-assembly in organic solvents and water is studied under various conditions.Dendron initiator synthesis and linear polymerisation studies
One of the benefits of hyp-polydendrons is the ability to produce high molecular weight materials containing ideally branched dendritic chain ends, without the requirement for lengthy, iterative dendrimer or dendron syntheses. As such, we targeted low generation (G1-Br and G2-Br) dendron ATRP initiators and have compared their efficiency with the simple commercial ethyl α-bromoisobutyrate (EBiBr) initiator.A straightforward synthesis (Scheme 1) was adopted for G1-Br and G2-Br using a symmetrical secondary alcohol 1,3-dibenzyloxy-2-propanol, 1. G1-Br was synthesised in a single reaction with α-bromoisobutyryl bromide; G2-Br required the use of 1,1′-carbonyl diimidazole to form the imidazole carboxylic ester 2 and allow the selective formation of urethane links at the primary amino sites of diethylenetriamine6,47 to form the disubstituted product 3. The ring opening of β-butyrolactone by the unreacted secondary amino functionality of 3 led to the synthesis of 4 containing a secondary hydroxyl which was readily reacted with α-bromoisobutyryl bromide to yield G2-Br. The synthesis of initiators and intermediates was confirmed by 1H and 13C nuclear magnetic resonance spectroscopy (NMR) and electrospray mass spectroscopy (see ESI, Fig. S1–S17†).
 | ||
| Scheme 1 Synthesis of first and second generation atom transfer radical polymerisation initiators used for hyp-polydendron synthesis. | ||
We have previously used EBiBr to form linear and high molecular weight branched vinyl polymers through the ATRP of the commercial isomeric mixture of 2-hydroxypropyl methacrylate (HPMA)48 and similar conditions were employed in this study using Cu(I)Cl and 2,2′-bypyridyl as the catalytic system and methanol as the reaction solvent at a concentration of 50% v/v. The polymerisation of HPMA was targeted to number average degrees of polymerisation (DPn) of 50 monomer units (ESI, Fig. S18 and S19†).
The targeted linear p(HPMA50), and linear-dendritic polymer hybrids G1-p(HPMA50) and G2-p(HPMA50), were readily synthesised at 30 °C (Scheme 2A) and allowed to polymerise to monomer conversions in excess of 99%. Polymers of number average molecular weights (Mn) of <15 kDa and dispersity (Đ) values of <1.45 were generated (Table 1), as determined by triple detection gel permeation chromatography (GPC), and excellent kinetic data was obtained, showing control of radical concentration throughout each polymerisation (Fig. 2).
 | ||
| Scheme 2 Cu-catalysed ATRP Synthesis of (A) linear dendritic hybrids and (B) hyp-polydendrons using HPMA, EGDMA and G2-Br. Scheme shows only two polymer chains conjoined for clarity. Many additional chains are combined into the branched architecture. | ||
| Target polymer | GPCa (THF) | Calc. DPn | |||
|---|---|---|---|---|---|
| M n (Da) | M w (Da) | Đ | GPC | 1H NMRb | |
| a Triple detection gel permeation chromatography. b Calculated from NMR spectra (ESI, Fig. S20). | |||||
| p(HPMA50) | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 250 250 |
13950 |
1.24 | 76.7 | — |
| p(HPMA50-co-EGDMA0.8) | 147100 |
928500 |
6.31 | — | — |
| G1-(HPMA50) | 9800 | 13000 |
1.33 | 65 | 55 |
| G1-(HPMA50-co-EGDMA0.8) | 47250 |
1169000 |
24.74 | — | 57 |
| G2-(HPMA50) | 14400 |
20300 |
1.41 | 93.5 | 49 |
| G2-(HPMA50-co-EGDMA0.8) | 90500 |
1304000 |
14.41 | — | 55 |
 | ||
| Fig. 2 Comparative studies of conversion vs. polymerisation time and the evolution of molecular weight with conversion for the methanolic ATRP of linear-dendritic hybrids and hyp-polydendrons using G2-Br to a targeted DPn = 50 monomer units: G2-(HPMA50) linear dendritic hybrid – (A) conversion vs. time plot (closed blue squares) and corresponding semi-logarithmic plot (closed green triangles), (B) overlaid Mw (closed orange circles) and Mn (closed red triangles) vs. conversion plots and corresponding Đ vs. conversion (closed blue circles); hyp-polydendron G2-(HPMA50-co-EGDMA0.8) – (C) conversion vs. time plot (open blue squares) and corresponding semi-logarithmic plot (open green triangles); (D) overlaid Mw (open orange circles) vs. conversion plot and corresponding Đ vs. conversion (open blue circles). | ||
Variation of the obtained Mn and Đ values suggests differing initiator efficiencies, with G1-Br appearing to be the most efficient, and possible termination by combination at the very high final monomer conversions (>99%).
Synthesis of hyperbranched-polydendrons
The synthesis of a hyp-polydendron involves the polymerisation of a mixture containing a monofunctional monomer and a low concentration of bifunctional branching monomer. The branching monomer concentration must be maintained at a ratio with the initiator below an effective initiator:brancher of 1:1 to avoid gelation. This differs significantly from the reports of arms-first core-crosslinking using linear-dendritic hybrid macroinitiators.36b
EBiBr, G1-Br and G2-Br were therefore utilised (Scheme 2B) to initiate the polymerisation of HPMA in the presence of the branching monomer ethylene glycol dimethacrylate (EGDMA) with target DPn values of 50 monomer units for the primary chains of the hyp-polydendron and an initiator:brancher ratio of 1:0.8 (ESI† for experimental details). We have previously utilised EGDMA in this way to form highly branched p(HPMA) and amphiphilic branched A–B block copolymers of oligoethylene glycol methacrylate and n-butyl methacrylate.41,42
In all cases, the presence of 0.8 equivalents of EGDMA, with respect to the initiator used, led to a considerable increase in the observed Mn and Mw values of the polymerisation and subsequent dispersities of the resulting polymers (Table 1). Mw values as high a 1.17 MDa were seen for the G1-initiated hyp-polydendrons and 1.30 MDa for G2-initiated materials. From a weight average perspective therefore, the recovered G1-initiated polymers comprised structures with approximately 90 primary chains and the G2-initiated polymers comprising approximately 65 primary chains. It was clear from the GPC chromatograms that materials of significantly higher molecular weight were also present (ESI, Fig. S18 and S19†).
Analysis of the kinetics of the ATRP reactions showed almost identical conversion vs. time and semi-logarithmic plots in the presence and absence of brancher, however, the development of Mn and Mw with conversion was noticeably different for the branched ATRP and non-linear at monomer conversions above 80% (Fig. 2). 1H NMR analysis of the polymers after purification allowed a determination of the DPn of the primary chains of the hyp-polydendron due to the clear resonance of the aromatic protons forming the peripheral groups of the dendron end-group (ESI, Fig. S20†). Good correlation between the targeted number average chain length and that determined by 1H NMR was observed.
Solvent-driven hyperbranched-polydendron assembly
The G1-Br and G2-Br initiated linear-dendritic hybrids underwent similar behaviour on addition of hexane, with initial measurements of particles observed at Φhexane = 0.333 and 0.286 respectively. Neither of the linear-dendritic hybrids led to a plateau of Dz values with increasing hexane fraction and particles with Dz up to 1.75 μm (G2-(HPMA50); Φhexane = 0.750) and PDI values as high as 0.44 (G2-(HPMA50); Φhexane = 0.333) were observed. Measurements using DLS are unreliable above 1 μm and, interestingly, the G2-(HPMA50) material precipitated at Φhexane > 0.750; the only polymer in this series to do so at Φhexane values up to 0.800.
The hyperbranched EBiBr-initiated polymer, p(HPMA50-co-EGDMA0.8), and the hyp-polydendrons within Table 1 were subjected to the same addition of non-solvent (Fig. 3 and ESI Table S1†) as discussed above. Measurable Dz values were observed within the initial acetone solutions at values varying from 42 nm (p(HPMA50-co-EGDMA0.8)) to 46 nm (G1-(HPMA50-co-EGDMA0.8)) and 37 nm (G2-(HPMA50-co-EGDMA0.8)) and PDI values from 0.4 (p(HPMA50-co-EGDMA0.8)) to 0.38 (G2-(HPMA50-co-EGDMA0.8)) and 0.37 (G1-(HPMA50-co-EGDMA0.8)) (Fig. 3A).
 | ||
| Fig. 3 Self-assembly of branched polymers on addition of hexane (bad solvent) to acetone (good solvent) solutions: (A) impact of hexane solvent fraction on z-average diameter for p(HPMA50-co-EGDMA0.8) (open red circles), G1-(HPMA50-co-EGDMA0.8) (open blue triangles) and G2-(HPMA50-co-EGDMA0.8) (open green diamonds); (B) impact of initial solution concentration (CI) on self-assembly of G1-(HPMA50-co-EGDMA0.8); (C) reversible assembly of G1-(HPMA50-co-EGDMA0.8) (arrows indicate direction of solvent addition); and (D) robustness of G1-(HPMA50-co-EGDMA0.8) nanoparticles to dilution (Φhexane = 0.800). | ||
Progressive increases in Φhexane led to corresponding decreases in the hyp-polydendron Dz values with comparably broad dispersities. At a Φhexane = 0.167, the EBiBr-initiated hyperbranched polymer underwent a significant increase in size and decrease in PDI (Dz = 181 nm; PDI = 0.055) and a similar increase in size and decrease in dispersity was seen for the hyp-polydendrons at Φhexane = 0.231 (G1-(HPMA50-co-EGDMA0.8), Dz = 162 nm, PDI = 0.021; G2-(HPMA50-co-EGDMA0.8), Dz = 178 nm, PDI = 0.018) (Fig. 3A). Further addition of non-solvent for the p(HPMA) core architectures, up to Φhexane = 0.667, generated a steady but small decrease in Dz for all hyperbranched materials studied, with the hyp-polydendron bearing the G2 dendron being the largest under all solvent conditions and the EBiBr initiated hyperbranched polymer consistently forming the smallest nanoparticles; in all cases the PDI values remained <0.075. At Φhexane between 0.667 and 0.800, the nanoparticles from each material started again to increase in size whilst maintaining their order of size and low PDI values. The largest nanoparticle formed using this process had a Dz = 230 nm and PDI = 0.097 (G2-(HPMA50-co-EGDMA0.8), Φhexane = 0.800).
The impact of the concentration of polymer in the initial acetone solution, prior to hexane addition, was studied using 0.5 mg mL−1, 5 mg mL−1 and 20 mg mL−1 acetone solutions of the G1-(HPMA50-co-EGDMA0.8) hyp-polydendron (Fig. 3B). The observed large increases in Dz at a Φhexane = 0.231 were generally followed by a subsequent decrease until Φhexane = 0.500. At values above this solvent ratio, the particles derived from the 20 mg mL−1 starting concentration rapidly increased in size and subsequently precipitated. The lower polymer concentrations proceeded to Φhexane = 0.667 with an increase observed on addition of further hexane up to Φhexane = 0.800. Across the samples after Φhexane = 0.231, the low initial concentration of hyp-polydendron of 0.5 mg mL−1 gave consistently the smallest nanoparticles (Dz as low as 84 nm) and the highest initial concentration of 20 mg mL−1 gave the largest nanoparticles (Dz as high as 396 nm). After Φhexane = 0.286, PDI values varied from <0.175 across the sample starting at 20 mg mL−1, to <0.075 for materials generated from an initial concentration of 5 mg mL−1 and <0.145 for particles from the 0.5 mg mL−1 acetone solution.
The reversibility of the assembly into nanoparticles was also studied by preparing nanoparticles of the G1-(HPMA50-co-EGDMA0.8) hyp-polydendron (initial concentration (acetone) = 5 mg mL−1) directly at a Φhexane = 0.667 (Dz = 157 nm, PDI = 0.064) and slowly adding more acetone to decrease the hexane volume fraction (Fig. 3C). With the steady decrease in Φhexane a small increase in size was initially seen, but Dz values only varied between 161 nm and 151 nm over a range of Φhexane from 0.667 to 0.286. Addition of further acetone led to a considerable decrease to 125 nm (Φhexane = 0.231) and then a sharp decrease to 44 nm at Φhexane = 0.167. The addition of a good solvent, therefore, led to the disassembly of the nanoparticles at the same ratio of acetone–hexane required to initiate assembly. To establish whether the reversible phenomenon was driven by dilution, nanoparticles of the G1-(HPMA50-co-EGDMA0.8) hyp-polydendron (generated directly at a concentration of 1 mg mL−1 and a Φhexane = 0.800; 5 mg mL−1 initial acetone concentration) were diluted with various volumes of an acetone–hexane mixture of identical composition (Fig. 3D). Across a 16-fold dilution, the nanoparticles were particularly robust and maintained both size and dispersity.
A direct comparison of the DLS analysis of the linear-dendritic hybrids and the branched polymers are shown in Table 2 at a fixed Φhexane = 0.800 and generated from an initial concentration in acetone of 5 mg mL−1. As can be seen, the branched materials produce much smaller nanoparticles with considerably lower PDI values in all cases.
To investigate the aqueous nanoprecipitation of hyp-polydendrons, a similar study was undertaken using a rapid addition of varying volumes of polymer–THF solutions (10 mg mL−1 or 5 mg mL−1) into 5 or 10 mL of water (30 °C), using the linear-dendritic hybrids and the branched polymers (ESI† for experimental details). Each nanoparticle dispersion formed was stirred overnight at ambient temperature to remove the THF and generate aqueous nanoparticle dispersions ranging from 2 mg mL−1 to 0.05 mg mL−1. As seen previously with linear p(HPMA50), the linear-dendritic hybrids underwent a self-assembly to form nanoparticles, however, the dispersions were not stable, showing increases in Dz and PDI over time with visible precipitate over a small number of days. The branched p(HPMA50-co-EGDMA0.8) generated nanoparticles with Dz values between 60 and 85 nm and PDI values ranging from 0.111 to 0.216 however the hyp-polydendrons displayed a wider range of sizes (60–135 nm) with lower PDI values (as low as 0.064). In all cases, the nanoparticles formed from G2-(HPMA50-co-EGDMA0.8) were larger than those derived from G1-(HPMA50-co-EGDMA0.8) under identical conditions, and showed much lower dispersities (Table 3). Those formed from p(HPMA50-co-EGDMA0.8) were similar in size or smaller than nanoparticles formed from G1-(HPMA50-co-EGDMA0.8), displaying the same overall trend in nanoparticle sizes that were seen when hexane was added to acetone solutions: ethyl < G1 dendron < G2 dendron (Fig. 3A).
| Polymer | Concentration (mg mL−1) | Dilution factor (CF/CI) | D z (nm) | PDIc | |
|---|---|---|---|---|---|
| Initiala (CI) | Finalb (CF) | ||||
| a Polymer concentration in THF. b Nanoparticle concentration in water. c Measured by dynamic light scattering. | |||||
| p(HPMA50-co-EGDMA0.8) | 10 | 2 | 0.2 | 83 | 0.111 |
| 10 | 0.1 | 0.01 | 76 | 0.114 | |
| 5 | 1 | 0.2 | 66 | 0.190 | |
| 5 | 0.05 | 0.01 | 62 | 0.216 | |
| G1-(HPMA50-co-EGDMA0.8) | 10 | 2 | 0.2 | 86 | 0.110 |
| 10 | 0.1 | 0.01 | 91 | 0.101 | |
| 5 | 1 | 0.2 | 64 | 0.130 | |
| 5 | 0.05 | 0.01 | 67 | 0.160 | |
| G2-(HPMA50-co-EGDMA0.8) | 10 | 2 | 0.2 | 106 | 0.083 |
| 10 | 0.1 | 0.01 | 134 | 0.064 | |
| 5 | 1 | 0.2 | 81 | 0.083 | |
| 5 | 0.05 | 0.01 | 93 | 0.071 | |
The aqueous nanoparticles generated from all branched polymers were extremely stable; those from hyp-polydendrons still showed monomodal size distributions (Dz < 100 nm) with little measureable difference over two years storage at ambient temperature (ESI, Fig. S22†). The effect of varying the initial polymer concentration in THF can be seen when comparing the nanoparticles generated at identical dilution factors (CF/CI). Higher CI generally formed larger materials at each dilution factor and higher dilution factors generally formed larger nanoparticles (Table 3).
The controlled addition of hexane allowed samples to be taken at varying volume fractions to observe the nanoparticle formation. An example of this study is shown in Fig. 4A–D for the self-assembly of G2-(HPMA50-co-EGDMA0.8). Images of the dried acetone–hexane mixtures at a Φhexane = 0.090 (Fig. 4A) showed very little structured material and particles resembling uncontrolled aggregates. At an increased Φhexane = 0.286 a higher quantity of poorly-shaped material was observed (Fig. 4B), however, at Φhexane = 0.500 very distinct spherical nanoparticles were readily observed (Fig. 4C) which are essentially unchanged at Φhexane = 0.800 (Fig. 4D). The nanoparticles were generally seen to be separate structures although some aggregated nanoparticles were observed (Fig. 4C), possibly as an artefact of sample preparation.
 | ||
| Fig. 4 Scanning electron microscopy of self-assembled nanoparticles from G2-(HPMA50-co-EGDMA0.8). Hexane addition to acetone solutions with solvent fractions of: (A) Φhexane = 0.090, (B) Φhexane = 0.286, (C) Φhexane = 0.500, and (D) Φhexane = 0.800. (E) Nanoparticles generated by aqueous nanoprecipitation from a THF solution (CI = 5 mg mL−1) an aqueous concentration of CF = 1 mg mL−1. | ||
The aqueous nanoprecipitation of p(HPMA50-co-EGDMA0.8) has been shown previously to be observable using SEM,48 with clearly distinguishable nanoparticles within highly concentrated areas, presumably formed during the drying of the aqueous continuous phase of the nanodispersion. An example of a typical SEM image of G2-(HPMA50-co-EGDMA0.8) is shown in Fig. 4E, generated from addition of a THF solution (CI = 5 mg mL−1) to water to form a CF = 1 mg mL−1 (dilution factor = 0.2) after THF removal. The highly concentrated areas of nanoparticles were observed in all cases and dilution of the sample prior to drying did not significantly affect the ability to generate images of distinct and separate nanoparticles.
Nanomedicines formed by careful control of linear polymer structure, and nanoprecipitation within the 100–200 nm size range,55,56 have shown high drug loadings and targeting aided by aptamer functionality (the so-called Accurins™). Positive phase I evaluation of Accurins have recently led to progression to phase II human studies with the ability to target delivery to sites of prostate and lung cancer.56,57
The new architectures generated during this current study also appear to self-assemble through a nucleation/aggregation mechanism, although further work is required to confirm this. We speculate that the studies of hexane addition to acetone solutions of the branched polymers (Fig. 3A) initially show a high molecular weight expanded coils in solution and the decrease in diameter observed for all study materials demonstrates a steady decrease in the branched core p(HPMA) solvation, forming an increasingly dense structure (shown schematically in Fig. 5Ai–iii). The nuclei that form under these conditions are stable until a critical density or size, leading to a sudden increase in the observed diameter (Fig. 3A) as they aggregate to form larger structures (Fig. 5Aiii–v). These larger structures appear to be somewhat swollen within the solvent environment (Fig. 5v) and steadily decrease in size to form more dense nanoparticles in the progressively poorer solvent environment (Fig. 5v–vi) before aggregating to precipitate.
 | ||
| Fig. 5 Proposed mechanism for hyp-polydendron self-assembly and nanoprecipitation: (A) progressive collapse of solvated hyp-polydendrons to form nuclei (i–iii) and subsequent assembly to form swollen assemblies (iii–v) which finally desolvate and decrease slightly in hydrodynamic diameter (vi). (B) Dynamic light scattering showing the impact of Φhexane on the observed DLS size distributions; (C) DLS measurements of aqueous hyp-polydendron nanoprecipitates formed from varying THF concentrations and different dilutions. | ||
This behaviour is quite clearly seen in the DLS measurements taken at different ratios of acetone and hexane (Fig. 5B). The initially swollen and solvated hyp-polydendrons show appreciable scattering when studied before hexane addition and a broad size distribution is seen with two distinct populations. Addition of a small volume of hexane leads to a narrowing of this distribution and enhanced scattering, with the complete disappearance of this material in later DLS measurements at higher hexane fractions and sharp monomodal peaks for the assembled structures (Fig. 5B). The effect of nanoprecipitation into water using varying CI values, is also readily seen when overlaying DLS traces (Fig. 5C). It is clear that higher CI values lead to larger nanoparticle assemblies and higher dilutions (CF/CI) produce larger nanoparticles when using hyp-polydendrons. This is counter to our previous nanoprecipitation reports of branched p(HPMAx) (using EBiBr and ATRP) where, consistently and across different primary chain lengths, higher dilutions produced smaller nanoparticles – also seen in this study (Table 3). The dendritic end-group of the primary chain appears, therefore, to influence the nucleation/aggregation mechanism and further work will study this in more detail.
The impact of the generation of the dendron chain ends of the hyp-polydendrons can also be observed within the nanoparticle diameters that are formed during this process. The larger chain ends lead to increased nanoparticle diameters, providing both a new strategy for controlling nanoprecipitation (previously unexplored) and an option for placing considerable functionality at the surface of the nanoparticles that are formed. The branched architectures also provide opportunities that are clearly not available from the linear polymer ‘equivalent’ structures, although it is important to note that the branched polymers have considerably higher molecular weights and therefore the formation of nuclei is expected to be a more efficient process as the desolvation of a single molecule of a branched polymer will form a larger nucleus than single chain collapse; the weight average structures of the branched polymers comprise >60 primary polymer chains.
The acetone–hexane studies have indicated a nucleation/aggregation/desolvation mechanism which also is intuitively correct for the aqueous nanoprecipitation although the process of THF solution addition to water was conducted extremely quickly and no intermediate structures were studied. The rapid diffusion of the THF into the surrounding water would lead to a desolvation of the expanded structures at the rapidly retreating good-solvent/anti-solvent interface with collapse and aggregation of nuclei to form the large nanoprecipitated aggregates. The influence of the chain end is also apparent, again offering additional options for functionalisation in aqueous systems and size control. The stability of branched nanoprecipitates has previously been attributed to a combination of negative surface charge and branched architecture48 and the nanoprecipitates formed in this study also exhibit a negative zeta potential ranging from −57 to −63 mV for linear polymers and −31 to −65 mV for branched polymer nanoprecipitates (values relate to specific nanoprecipitation conditions: CI = 5 mg mL−1; CF = 1 mg mL−1).
Pyrene and Nile Red were simply dissolved within THF along with the G2-(HPMA50-co-EGDMA0.8) polymer and subsequently nanoprecipitated following the identical process as described previously for a 5 mg mL−1 to 1 mg mL−1 dilution (see ESI†); in these explorative studies, a low level of each guest molecule (0.1 w/w%) was utilised. Very small decreases in Dz were observed when the guest molecules were present, with the empty nanoprecipitates showing diameters of 81 nm and Nile Red and pyrene encapsulation leading to Dz = 76 and 69 nm respectively (Fig. 6A). Narrow dispersities were maintained (PDIblank = 0.083; PDINile Red = 0.109; PDIpyrene = 0.061) across the samples.
 | ||
| Fig. 6 Encapsulation of guest molecules during G2-(HPMA50-co-EGDMA0.8) hyp-polydendron nanoprecipitation into water: (A) DLS measurements of empty nanoprecipitates (black solid line), Nile Red containing nanoprecipitates (red long dashed line) and pyrene (blue short dashed line). (B) Fluorescence spectroscopy of Nile Red loaded nanoprecipitates (red dashed line) vs. unloaded nanoprecipitates. (C) Fluorescence spectroscopy of pyrene loaded nanoprecipitates (blue dashed line) vs. unloaded nanoprecipitates. | ||
Fluorimetric analysis of the aqueous nanoparticles in the absence and presence of the guest molecules was conducted. The blank hyp-polydendron nanoprecipitates showed no measureable fluorescence when excited at either 552 nm or 335 nm; however, a clear fluorescence was observed from both samples contained the hydrophobic probes when using the appropriate excitation conditions (Fig. 6B and C). The fluorescence observed from Nile Red (630 nm) clearly indicates that the dye guest molecule is present within a hydrophobic environment and has been encapsulated during nanoprecipitation.58 The fine structure of the fluorescence from the encapsulated pyrene allows an indication of the polarity of the immediate environment within the nanoparticle. The ratio of the intensities of the first and third vibrational bands59 (I1/I3) were measured to be 1.420, which lies between values associated in the literature with pyrene fluorescence when dissolved in dichloromethane (I1/I3 = 1.370) or ethyl acetate (I1/I3 = 1.449).60
Conclusions
Collectively, we report the first syntheses of a new polymer architecture (hyperbranched polydendrons), a comparison of the behaviour of the linear-dendritic hybrids that form the primary chains of the new architecture, and the antisolvent-driven self-assembly and encapsulation of the branched polymers to form nanoparticles. The nanoparticles display remarkable size control, stability and uniformity of behaviour despite their combined very high molecular weight and dispersity. The ability to modify the self-assembly through end-group dendron generation, and formation of materials with diameters >100 nm, opens avenues of research for dendron-functional materials at much higher particle sizes and increased synthetic ease than previously described through conventional dendrimer or hyperbranched materials. This report therefore offers a straight-forward strategy for exploring the effects of the architectural components of hyp-polydendrons and their application within fields that have conventionally evaluated dendrimers and nanoprecipitates. Further reports will extend this platform including the optimisation of guest molecule loading, studies of biological interactions with cells and structural component modification to allow increased behavioural control.Acknowledgements
The authors wish to thank the Engineering and Physical Sciences Research Council for a PhD studentship (FL) and grant funding (EP/I038721/1) that underpinned the research. The University of Liverpool and the Centre for Materials Discovery at Liverpool is also gratefully acknowledged for access to scanning electron microscopy.Notes and references
- E. Rizzardo and D. H. Solomon, Aust. J. Chem., 2012, 65, 945 CrossRef CAS
.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015 CrossRef CAS
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924 CrossRef CAS PubMed
- D. G. van der Poll, H. M. Kieler-Ferguson, W. C. Floyd, S. J. Guillaudeu, K. Jerger, F. C. Szoka and J. M. Fréchet, Bioconjugate Chem., 2010, 21, 764 CrossRef CAS PubMed
- T. M. Hermans, M. A. C. Broeren, N. Gomopoulos, P. van der Schoot, M. H. P. van Genderen, N. A. J. M. Sommerdijk, G. Fytas and E. W. Meijer, Nat. Nanotechnol., 2009, 4, 721 CrossRef CAS PubMed
- A. Stoddart, W. J. Feast and S. P. Rannard, Soft Matter, 2012, 8, 1096 RSC
- S. P. Rannard, N. J. Davis and I. Herbert, Macromolecules, 2004, 37, 9418 CrossRef CAS
- S. Grayson and J. M. J. Fréchet, Org. Lett., 2002, 4, 3171 CrossRef CAS PubMed
- A. Carlmark, E. Malmström and M. Malkoch, Chem. Soc. Rev., 2013, 42, 5858 RSC
- S. P. Rannard and N. J. Davis, J. Am. Chem. Soc., 2000, 122, 11729 CrossRef CAS
- S. Rannard, N. Davis and H. McFarland, Polym. Int., 2000, 49, 1002 CrossRef CAS
- F. Aulenta, M. G. B. Drew, A. Foster, W. Hayes, S. Rannard, D. W. Thornthwaite, D. R. Worrall and T. G. A. Youngs, J. Org. Chem., 2005, 70, 63 CrossRef CAS PubMed
- M. S. Diallo, L. Balogh, A. Shafagati, J. H. Johnson, W. A. Goddard and D. A. Tomalia, Environ. Sci. Technol., 1999, 33, 820 CrossRef CAS
- P. Ortega, M. J. Serramia, M. A. Munoz-Fernandez, F. J. de la Mata and R. Gomez, Tetrahedron, 2010, 66, 3326 CrossRef CAS PubMed
- N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840 CrossRef CAS PubMed
- C. C. Lee, J. A. MacKay, J. M. J. Fréchet and F. Szoka, Nat. Biotechnol., 2005, 23, 1517 CrossRef CAS PubMed
- C.-L. Liu, C.-H. Lin, C.-C. Kuo, S.-T. Lin and W.-C. Chen, Prog. Polym. Sci., 2011, 36, 603 CrossRef CAS PubMed
- C. P. Rhodes, J. W. Long, K. A. Pettigrew, R. M. Stroud and D. R. Rolison, Nanoscale, 2011, 3, 1731 RSC
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446 RSC
- Y. Chen, Macromolecules, 2012, 45, 2619 CrossRef CAS
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887 CrossRef CAS PubMed
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2012, 45, 2 CrossRef CAS
- L. N. Pilon, S. P. Armes, P. Findlay and S. P. Rannard, Eur. Polym. J., 2006, 42, 1487 CrossRef CAS PubMed
- H. Wang, L. Chen, Y. Feng and H. Chen, Acc. Chem. Res., 2013, 46, 1636 CrossRef CAS PubMed
- R. B. Grubbs and Z. Sun, Chem. Soc. Rev., 2013, 42, 7436 RSC
- Z.-Y. Qiao, R. Ji, X.-N. Huang, F.-S. Du, R. Zhang, D.-H. Liang and Z.-C. Li, Biomacromolecules, 2013, 14, 1555 CrossRef CAS PubMed
- W. Li, Y. Kim and M. Lee, Nanoscale, 2013, 5, 7711 RSC
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081 RSC
- S. M. Grayson and J. M. J. Fréchet, Chem. Rev., 2001, 101, 3819 CrossRef CAS PubMed
- L. Röglin, E. H. M. Lempens and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 102 CrossRef PubMed
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857 CrossRef CAS PubMed
-
(a) J. M. Oliveira, A. J. Salgado, N. Sousa, J. F. Mano and R. L. Reis, Prog. Polym. Sci., 2010, 35, 1163 CrossRef CAS PubMed
- L. M. Bronstein and Z. B. Shifrina, Chem. Rev., 2011, 111, 5301 CrossRef CAS PubMed
- A. R. Menjoge, R. M. Kannan and D. A. Tomalia, Drug Discovery Today, 2010, 15, 171 CrossRef CAS PubMed
- J. Lim, M. Kostiainen, J. Maly, V. C. P. da Costa, O. Annunziata, G. M. Pavan and E. E. Simanek, J. Am. Chem. Soc., 2013, 135, 4660 CrossRef CAS PubMed
-
(a) M. V. Walter, P. Lundberg, A. Hult and M. Malkoch, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2990 CrossRef CAS
- Y. Chang and C. Kim, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 918 CrossRef CAS
- N. O'Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027 CrossRef CAS
- S. Graham, S. P. Rannard, P. A. G. Cormack and D. C. Sherrington, J. Mater. Chem., 2007, 17, 545 RSC
- J. Weaver, R. Williams, P. Findlay, B. Royles, A. I. Cooper and S. P. Rannard, Soft Matter, 2008, 4, 985 RSC
- T. He, D. J. Adams, M. F. Butler, A. I. Cooper and S. P. Rannard, J. Am. Chem. Soc., 2009, 131, 1495 CrossRef CAS PubMed
- T. He, D. J. Adams, M. F. Butler, C. T. Yeoh, A. I. Cooper and S. P. Rannard, Angew. Chem., Int. Ed., 2007, 46, 9243 CrossRef CAS PubMed
- J. V. M. Weaver, S. P. Rannard and A. I. Cooper, Angew. Chem., Int. Ed., 2009, 48, 2131 CrossRef CAS PubMed
- T. Kaneko, T. Horie, M. Asano, T. Aoki and E. Oikawa, Macromolecules, 1997, 30, 3118 CrossRef CAS
- J. M. J. Fréchet, M. Henmi, I. Gitsov, S. Aoshima, M. R. Leduc and R. B. Grubbs, Science, 1995, 269, 1080 CrossRef PubMed
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS
- S. P. Rannard and N. J. Davis, Org. Lett., 2000, 2, 2117 CrossRef CAS PubMed
-
(a) R. A. Slater, T. O. McDonald, D. J. Adams, E. R. Draper, J. V. M. Weaver and S. P. Rannard, Soft Matter, 2012, 8, 9816 RSC
- O. Thioune, H. Fessi, J. P. Devissaguet and F. Puisieux, Int. J. Pharm., 1997, 146, 233 CrossRef CAS
- S. Schubert, J. T. Delaney and U. S. Schubert, Soft Matter, 2011, 7, 1581 RSC
- V. K. Lamer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847 CrossRef CAS
- C. Zhang, V. J. Pansare, R. K. Prud'homme and R. D. Priestly, Soft Matter, 2012, 8, 86 RSC
- R. Karnik, F. Gu, P. Basto, C. Cannizzaro, L. Dean, W. Kyei-Manu, R. Langer and O. C. Farokhzad, Nano Lett., 2008, 8, 2906 CrossRef CAS PubMed
- I. Y. Perevyazko, J. T. Delaney, A. Vollrath, G. M. Pavlov, S. Schubert and U. S. Schubert, Soft Matter, 2011, 7, 5030 RSC
- P. M. Valencia, P. A. Basto, L. Zhang, M. Rhee, R. Langer, O. C. Farokhzad and R. Karnik, ACS Nano, 2010, 4, 1671 CrossRef CAS PubMed
- J. Cheng, B. A. Teply, I. Sherifi, J. Sung, G. Luther, F. X. Gu, E. Levy-Nissenbaum, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Biomaterials, 2007, 28, 869–876 CrossRef CAS PubMed
- J. Shi, Z. Xiao, N. Kamaly and O. C. Farokhzad, Acc. Chem. Res., 2011, 44, 1123 CrossRef CAS PubMed
- D. L. Sackett and J. Wolff, Anal. Biochem., 1987, 167, 228 CrossRef CAS
- D. López-Díaz and M. M. Velázquez, Chem. Educ., 2007, 12, 327 Search PubMed
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Materials, full experimental details and characterisation. See DOI: 10.1039/c4sc00360h |
| This journal is © The Royal Society of Chemistry 2014 |