 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Blue transition metal complexes of a natural bilin-type chlorophyll catabolite†
Chengjie
Li
a,
Markus
Ulrich
a,
Xiujun
Liu
a,
Klaus
Wurst
b,
Thomas
Müller
a and
Bernhard
Kräutler
*a
aInstitute of Organic Chemistry & Centre of Molecular Biosciences, University of Innsbruck, Innrain 80/82, A-6020 Innsbruck, Austria. E-mail: bernhard.kraeutler@uibk.ac.at
bInstitute of General, Inorganic & Theoretical Chemistry, University of Innsbruck, Innrain 80/82, A-6020 Innsbruck, Austria
First published on 2nd June 2014
Abstract
“Non-fluorescent” chlorophyll catabolites (NCCs) are ubiquitous, colourless bilane-type natural products, first identified about 20 years ago. In various senescent leaves NCCs are oxidized, in part, to yellow chlorophyll catabolites (YCCs), which undergo further oxidation to unique pink chlorophyll catabolites (PiCCs). The present work presents the crystal structure of a PiCC, the first of a natural chlorophyll catabolite from a higher plant. The PiCC binds (divalent) zinc-, cadmium-, copper- and nickel-ions with high affinity. Binding of these metal ions to the PiCC is rapid at room temperature. The resulting deep blue complexes represent the first transition metal complexes of a bilin-type chlorophyll catabolite. The structure of the metal complexes has been deduced from spectroscopic analyses, which has revealed an effective tridentate nature of the tetrapyrrolic PiCC ligand. The zinc and cadmium complexes show bright red luminescence, the nickel and copper complexes are non-luminescent. Binding of Zn- and Cd-ions to the PiCC ‘lights-up’ the intensive red fluorescence of the metal-complexes, which is detectable at nM levels of these closed shell metal ions. Formation of transition metal complexes with PiCCs, and related chlorophyll catabolites, may thus also occur in the tissues of plants, notably of ‘heavy metal (hyper)-accumulating’ plants.
Introduction
Chlorophyll breakdown is commonly associated with the appearance of the fall colours.1–3 Thus, chlorophyll catabolites from higher plants were primarily expected to be coloured compounds.1 However, when they were first identified, they were revealed to be colourless linear, bilane-type tetrapyrroles.4,5 In senescent leaves the typical major products of chlorophyll breakdown were shown to belong either to the so called “nonfluorescent” chlorophyll catabolites (NCCs),5–7 or to the related linear tetrapyrroles that were classified as colourless dioxobilane-type NCCs (DNCCs).5,8–10 Recently, also natural yellow chlorophyll catabolites (YCCs) and pink-red catabolites (PiCCs) were detected in some senescent leaves.11–13 The same (types of) compounds could also be prepared by chemical oxidation of an NCC (Fig. 1).11 Here, we report on the blue complexes of the bilin-type pink chlorophyll catabolite (PiCC) 1 with four biologically relevant transition metals, as well as on the crystal structure of 1, the first of a chlorophyll-derived ‘phyllobilin’ from a higher plant.5 | ||
| Fig. 1 Chlorophyll breakdown products in higher plants: nonfluorescent chlorophyll catabolites (NCCs), such as to the colorless NCC 3,6,14,15 accumulate in various senescent leaves as rapidly formed products of the degradation of the chlorophylls a and b. In some leaves, the NCC 3 was oxidized to the yellow catabolite (YCC) 2,13 followed by further oxidation to the pink catabolite (PiCC) 1.11 | ||
Results
Pink PiCC 1 is a racemic linear tetrapyrrole found in extracts of senescent leaves of the deciduous Katsura tree (Cercidiphyllum japonicum) as oxidation product of the yellow YCC 2.11 PiCC 1 was also obtained from the ubiquitous NCC 3 by oxidation with dicyano-dichlorobenzoquinone (DDQ),11 or by oxidation of YCC 2 (see below). The UV/Vis-spectrum of 1 in MeOH exhibited strong absorbance bands at 313 and 523 nm (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε = 4.56) (Fig. 2).
ε = 4.56) (Fig. 2).
 | ||
| Fig. 2 UV/Vis-spectra of the PiCC 1 (6.97 μM) and of its zinc-complex Zn-1 (9.67 μM) in MeOH, normalized to 1.00 at the absorption maxima of both spectra. | ||
Detailed NMR-analyses of the non-fluorescent linear tetra-pyrrole 1 revealed its extended structure and an E-configuration at the double bond (between C10 and C11) near the molecular centre.11 Crystals of the PiCC 1 were obtained by vapor diffusion of dichloromethane into a methanolic solution of 1. A single crystal of the potassium salt K-1 of 1 was subjected to X-ray analysis (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2)), confirming the basic features of the NMR-derived structure and revealing further details of the covalent bonding within the PiCC molecules. Thus, the heavy atoms constituting the main chromophore were spanning a nearly planar array, in which the individual bond distances fitted a structure with conjugated alternating single and double bonds, as is, indeed, represented by the conventional formula shown (Fig. 1 and 3).
(no. 2)), confirming the basic features of the NMR-derived structure and revealing further details of the covalent bonding within the PiCC molecules. Thus, the heavy atoms constituting the main chromophore were spanning a nearly planar array, in which the individual bond distances fitted a structure with conjugated alternating single and double bonds, as is, indeed, represented by the conventional formula shown (Fig. 1 and 3).
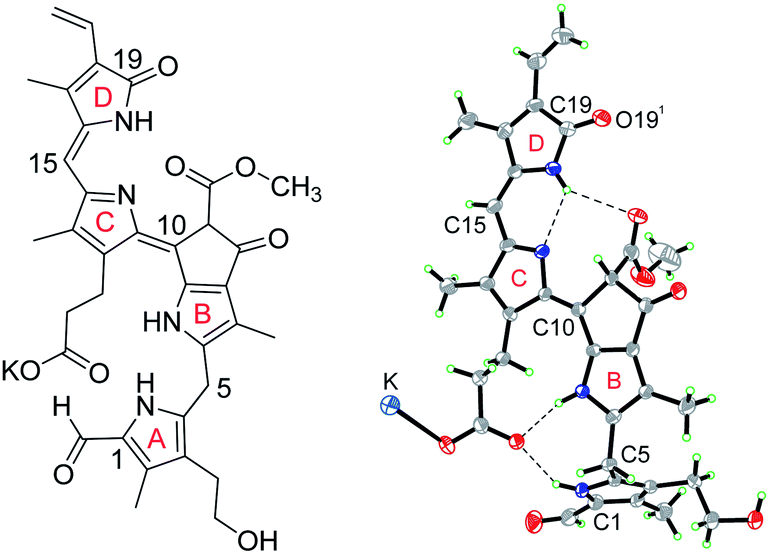 | ||
| Fig. 3 Chemical formula of the potassium salt (K-1) of the pink phyllobiladiene-b,c (PiCC) 1 and ORTEP-model based on the crystal structure of K-1 (atoms are coloured black (C), red (O), blue (N), green (H), and cyan (K)). | ||
The structure of 1 featured a short C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (1.362(8) Å) in E-configuration between C10 and C11, as well as a short CC bond (1.345(8) Å) in Z-configuration between C15 and C16. It also revealed the formation of H-bonded and potassium-bridged pair of enantiomers of K-1 in the crystal. The pair of enantiomers of K-1 showed nearly parallel planes of the π-system extending over rings B to D, and, at a distance of ca. 3.55 Å, indicating tight packing with van der Waals' contact of the conjugated main chromophores. The de-conjugated ring A stands out of the plane of the main chromophore and is positioned by an intra-molecular H-bond between pyrrole-N and a carboxylate O-atom (see Fig. 3 and ESI, Fig. S1†).
C bond (1.362(8) Å) in E-configuration between C10 and C11, as well as a short CC bond (1.345(8) Å) in Z-configuration between C15 and C16. It also revealed the formation of H-bonded and potassium-bridged pair of enantiomers of K-1 in the crystal. The pair of enantiomers of K-1 showed nearly parallel planes of the π-system extending over rings B to D, and, at a distance of ca. 3.55 Å, indicating tight packing with van der Waals' contact of the conjugated main chromophores. The de-conjugated ring A stands out of the plane of the main chromophore and is positioned by an intra-molecular H-bond between pyrrole-N and a carboxylate O-atom (see Fig. 3 and ESI, Fig. S1†).
In contrast to the more saturated phyllobilanes, such as the ubiquitous NCCs, pink-red PiCC 1 proved to be an excellent ligand for divalent transition metal ions (Fig. 4). The synthesis of Zn-1, the complex of 1 with a Zn(II)-ion, proceeded cleanly in Ar-saturated MeOH at room temperature, and could be followed by a rapid colour change of the reaction mixture from pink-red to intense blue (see below). From experiments with Zn(OAc)2, Zn-1 could be isolated with a yield of about 74%. However, work-up was more efficient when Zn(acac)2 was used, furnishing Zn-1 in a yield of 95%. The complex Zn-1 showed intense and red shifted absorbance bands, with maxima at 578 nm (logε = 4.23) and at 620 nm (logε = 4.43, in MeOH, see Fig. 2). A solution of Zn-1 in MeOH exhibited bright red luminescence with an emission maximum at 650 nm (Fig. 5). The molecular formula of Zn-1 was confirmed in a positive ion ESI mass spectrum, in which a base peak at m/z = 725 of [M + Na]+ was recorded. The structure of Zn-1 was largely established by 2-dimensional NMR spectroscopy (Fig. 6, Exptl part and ESI, Table S3 and Fig. S2†). These analyses indicated Zn-coordination of the three N-atoms that belong to the conjugated chromophore at rings B–D, whereas metal-coordination of ring A was shown to be unimportant by a consistent set of similar 1H and 13C-chemical shift data for PiCC (1) and for Zn-1, as well as by the presence of a broad signal of the ring A-NH at 11.9 ppm. 1H1H-NOE-corelations and 13C-chemical shift values indicated Z-configuration at the C10–C11 and C15–C16 double bonds, as expected for a mononuclear assembly of Zn-1 in solution with three-fold N-coordination by the ligand 1. However, polar solvent molecules are likely to be (one) additional ligand(s) (L) at the Zn-ion of Zn-1 (Fig. 4 and 6). In pyridine, a significant red shift of the UV/Vis-spectra indicated coordination of pyridine. Indeed, from such solutions of Zn-1 a dark blue-green powder was isolated, suggested by its 1H-NMR spectrum in CD3OD to be a 1:1 pyridine complex (Zn-1-py).
 | ||
| Fig. 4 Incorporation of divalent transition metal ions into the PiCC 1 gives the complexes M-1 (e.g. with M = Zn, Cd, Cu, Ni). | ||
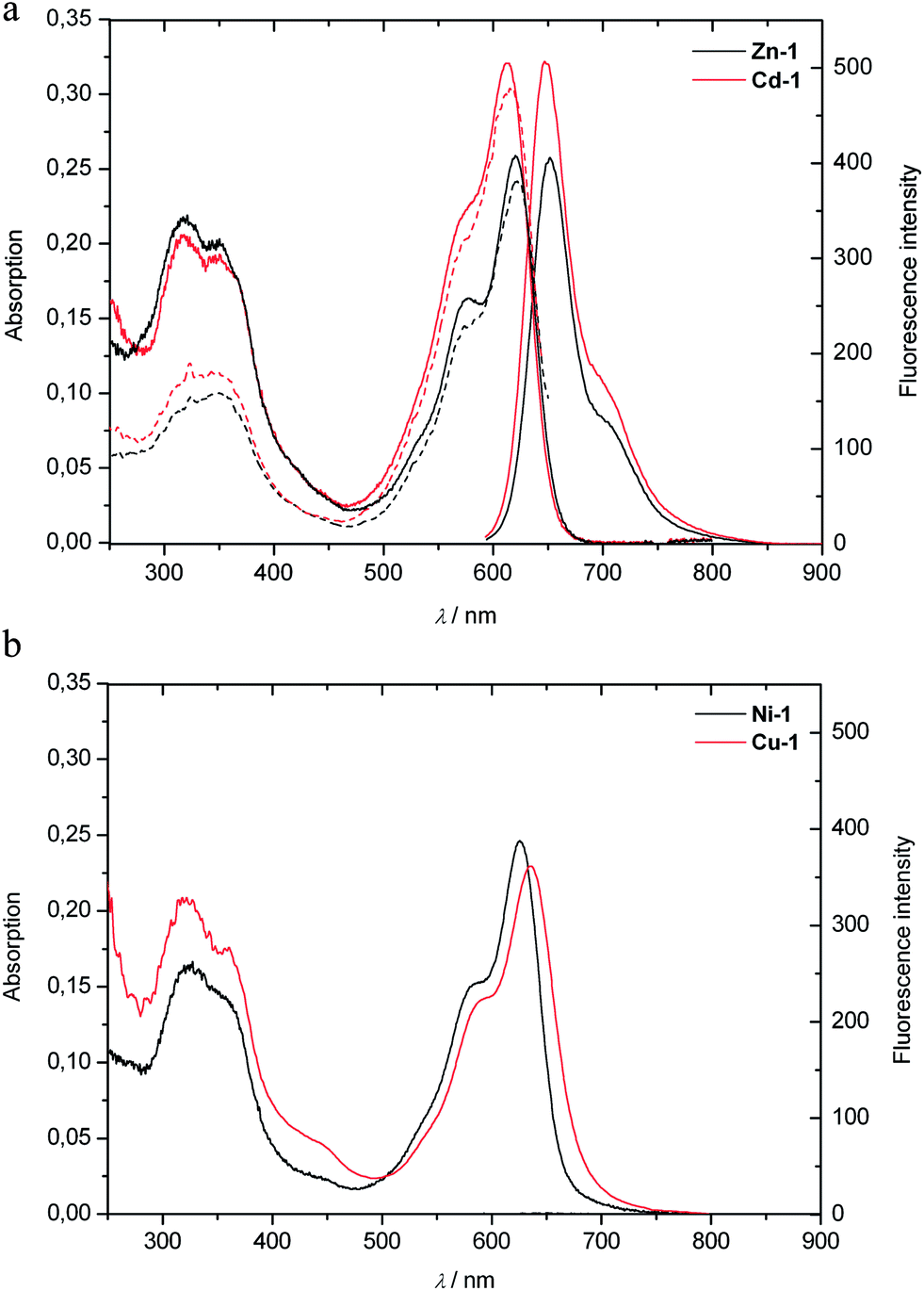 | ||
| Fig. 5 UV/Vis-spectra and fluorescence emission spectra (full lines), as well as fluorescence excitation spectra (dashed lines) of solutions of M-1 in MeOH. Top: spectra of Zn-1 and Cd-1 (9.67 μM). Bottom: spectra of Ni-1 and Cu-1 (9.67 μM). | ||
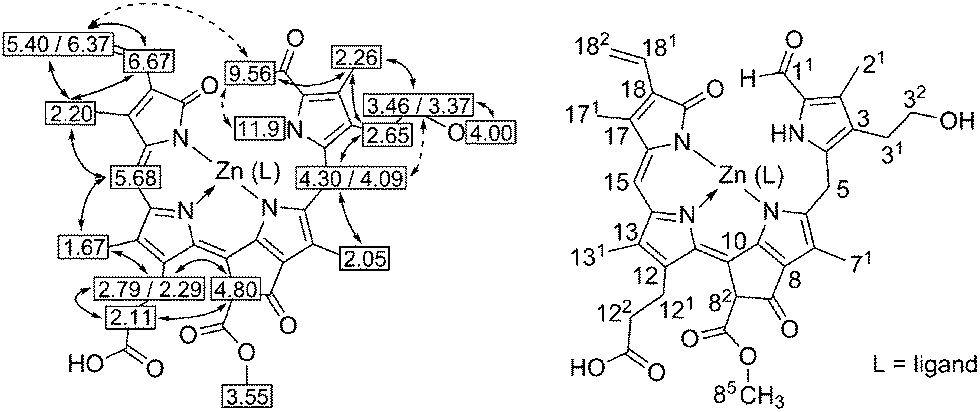 | ||
| Fig. 6 Left: graphical representation of 1H-chemical shift data from a 500 MHz 1H-NMR spectrum of Zn-1 in CD3CN/d6-DMSO (10:1) with correlations from H,H-ROESY-spectra (solid and dashed arrows). Right: deduced constitutional formula of Zn-1 (atom numbering used follows a convention for linear tetrapyrroles16 and is exemplified here). | ||
In an alternative procedure, the zinc complex Zn-1 was obtained from treatment of an air-saturated yellow solution of YCC 2 in DMF with Zn(OAc)2, and storage overnight at room temperature, giving a dark blue solution of Zn-1. Thus, from air oxidation, Zn-1 could be prepared directly from YCC 2 in a yield of about 94%. Via this path, a further method for the partial synthesis of the PiCC 1 from 2 was developed, as the Zn(II)-ion was readily removed from the blue Zn-complex by exposure of Zn-1 to an aqueous phosphate solution. By this procedure, PiCC (1) was obtained from 2 in an overall yield of 91% (see Fig. 7).
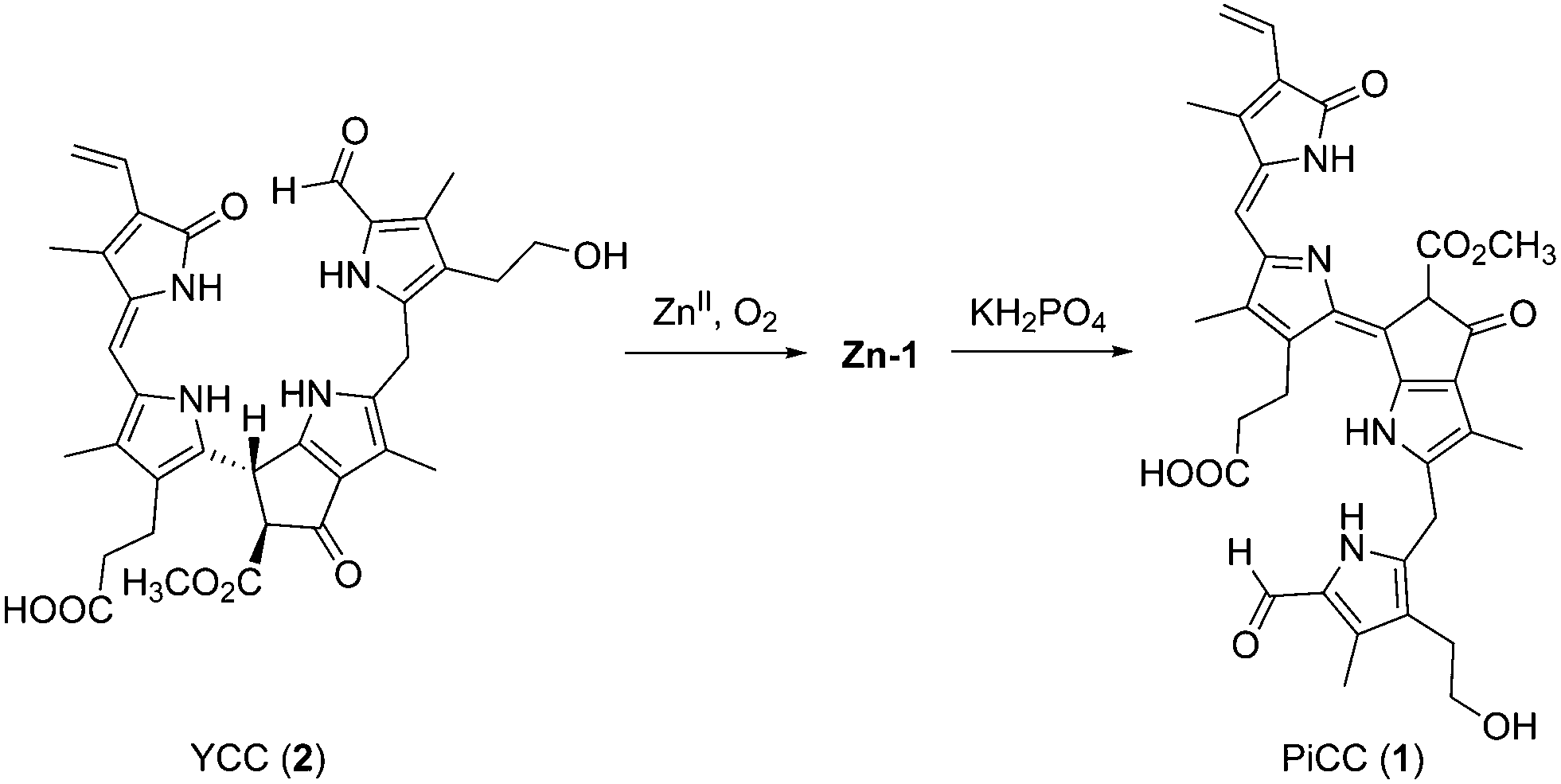 | ||
| Fig. 7 Oxidation of the YCC 2 in aerated solution in the presence of Zn(II)-ions, and yields Zn-1, from which the PiCC 1 can be prepared by decomplexation with phosphate. | ||
The preparation of the Cd(II)-complex Cd-1 was carried out analogous to that of Zn-1: treatment of 1 with Cd(acac)2 in MeOH at room temperature for 2 hours, gave Cd-1, isolated in 95% yield as a dark blue powder. The absorbance spectrum of a solution of Cd-1 in MeOH showed maxima at 570 nm (shoulder) and at 613 nm (logε = 4.52), and it emitted red light with emission maxima at 648 and 706 nm (Fig. 5). The structure of Cd-1 was clarified by ESI mass (base peak at m/z = 775 of [M + Na]+) and 1H-NMR spectra. NMR-analyses provided a set of similar chemical shift values and correlations, as observed for the Zn-complex Zn-1 indicating a closely similar structure (see Exptl. part and ESI, Fig. S3†).
The non-luminescent Ni(II)- and Cu(II)-complexes (Ni-1 and Cu-1) were prepared similarly from 1 in 88% and 90% yield, respectively, by treatment of 1 with Ni(acac)2 or Cu(acac)2 in Ar-saturated MeOH at room temperature. Slower formation of the Ni-complex required a reaction time of about 20 hours. The molecular formulas of Ni-1 and Cu-1 were confirmed by their ESI mass spectra, in which the base peaks were recorded at m/z = 697 [M + H]+ for Ni-1, and at m/z = 740 [M + K]+ for Cu-1. The absorbance spectra of solutions of Ni-1 or Cu-1 in MeOH were similar to those of Zn-1 and Cd-1, and showed long wavelength maxima at 626 nm and at 635 nm, respectively (see Fig. 5). The paramagnetic Cu(II)-complex Cu-1 and the Ni(II)-complex Ni-1 did not emit (visible) light, when photo-excited near 620 nm.
The nickel-complex Ni-1 was diamagnetic in d6-DMSO (or in d3-acetonitrile). In both solvents, 1H-NMR-spectra indicated the presence of two structurally similar compounds (possibly diastereo-isomers), as shown by two sets of closely spaced signals with similar coupling patterns. This signal pattern may be caused by (very slowly equilibrating) coordination isomers from solvent coordination or from intra-molecular coordination of the 32-OH. Interestingly, a well-resolved tripletoid signal near 4.5 ppm could be assigned to the (exchange labile) H-atom of the primary 32-OH group at the side chain extending from ring A (see ESI, Fig. S4†). In contrast, a solution of Ni-1 in deuteromethanol did not show any relevant 1H-NMR-signals, suggesting a paramagnetic nature of Ni-1 in this solvent.
The rates of formation of complexes of the PiCC 1 with four biologically important divalent transition metal ions were studied qualitatively (see Table 1 and ESI, Fig. S5†): in a solution of 1 and of an excess of Zn(OAc)2 in MeOH Zn-1 formed quantitatively within a few minutes at room temperature with pseudo-first order kinetics (kobs (22 °C) = 6.1 × 102 M−1 s−1, see ESI, Fig. S5†). Likewise, the corresponding metal complexes M-1 were readily assembled with Cd(OAc)2, Ni(OAc)2, and Cu(OAc)2 (Table 1). Spectral analysis indicated formation of Ni-1 to be effectively slower. A first fast interaction of Ni(II)-ions with 1 results in a complex with similar absorbance properties in the visible region as 1, followed by a clean, slower conversion to the blue metal complex Ni-1 (kobs = 10 M−1 s−1, Fig. 8).
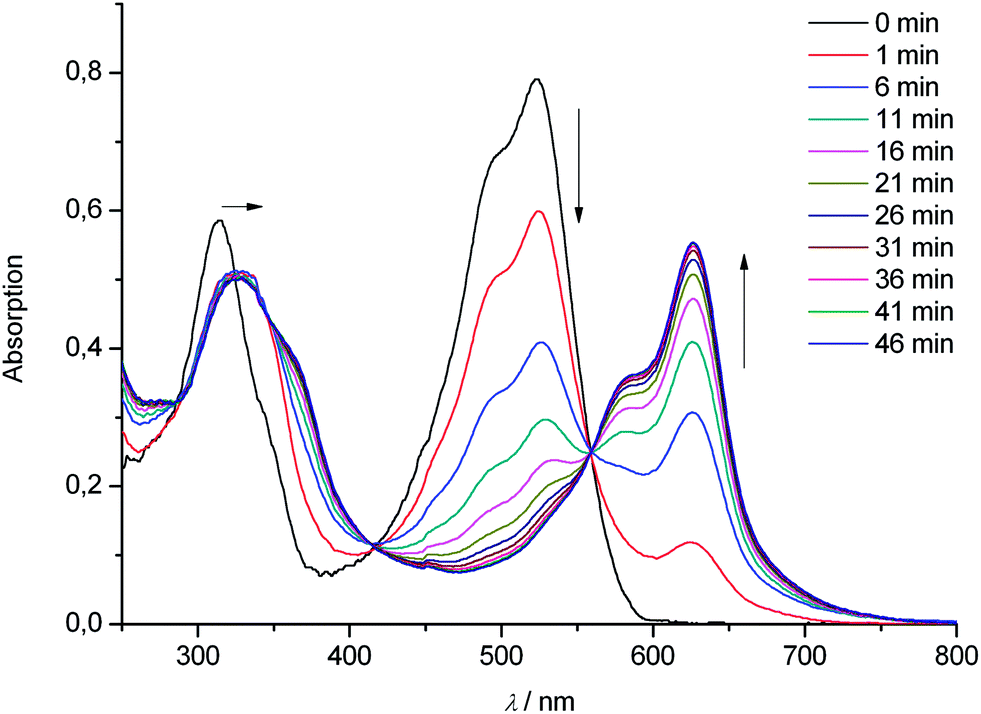 | ||
| Fig. 8 UV/Vis-spectroscopic analysis of Ni-incorporation into the PiCC 1. Reaction of 1 (20 μM) with 10 eq. of Ni(OAc)2 in MeOH at 22 °C gives Ni-1 cleanly. | ||
The strongly luminescent complexes Zn-1 and Cd-1 provided the opportunity to use fluorescence to analyze the formation of these metal complexes from the PiCC 1. A solution of the PiCC 1 in MeOH exhibited only very weak luminescence with a maximum at 620 nm, with a roughly two orders of magnitude lower intensity than that of the luminescence of Zn-1 or Cd-1 (emission maxima at 650 or 648 nm, resp., see ESI, Fig. S6†). Thanks to a rapid rate of formation of Zn-1 and Cd-1, and a high affinity of Zn(II)- and Cd(II)-ions for 1, the observation of the luminescence of Zn-1 and Cd-1 allowed for the quantitative detection of these ions at concentrations down to about 1 nM (see Fig. 9, and ESI, Fig. S7 and S8†). The nearly linear dependence of the fluorescence upon the total concentration of Zn- and Cd-ions indicated 1:1 stoichiometry in their complexes with 1 and strong binding of these metal ions (with an equilibrium constant >109 M−1). Cu(II)-ions displaced Zn(II) and Cd(II) from the complexes Zn-1 and Cd-1, consistent with even better binding of Cu(II) by 1.
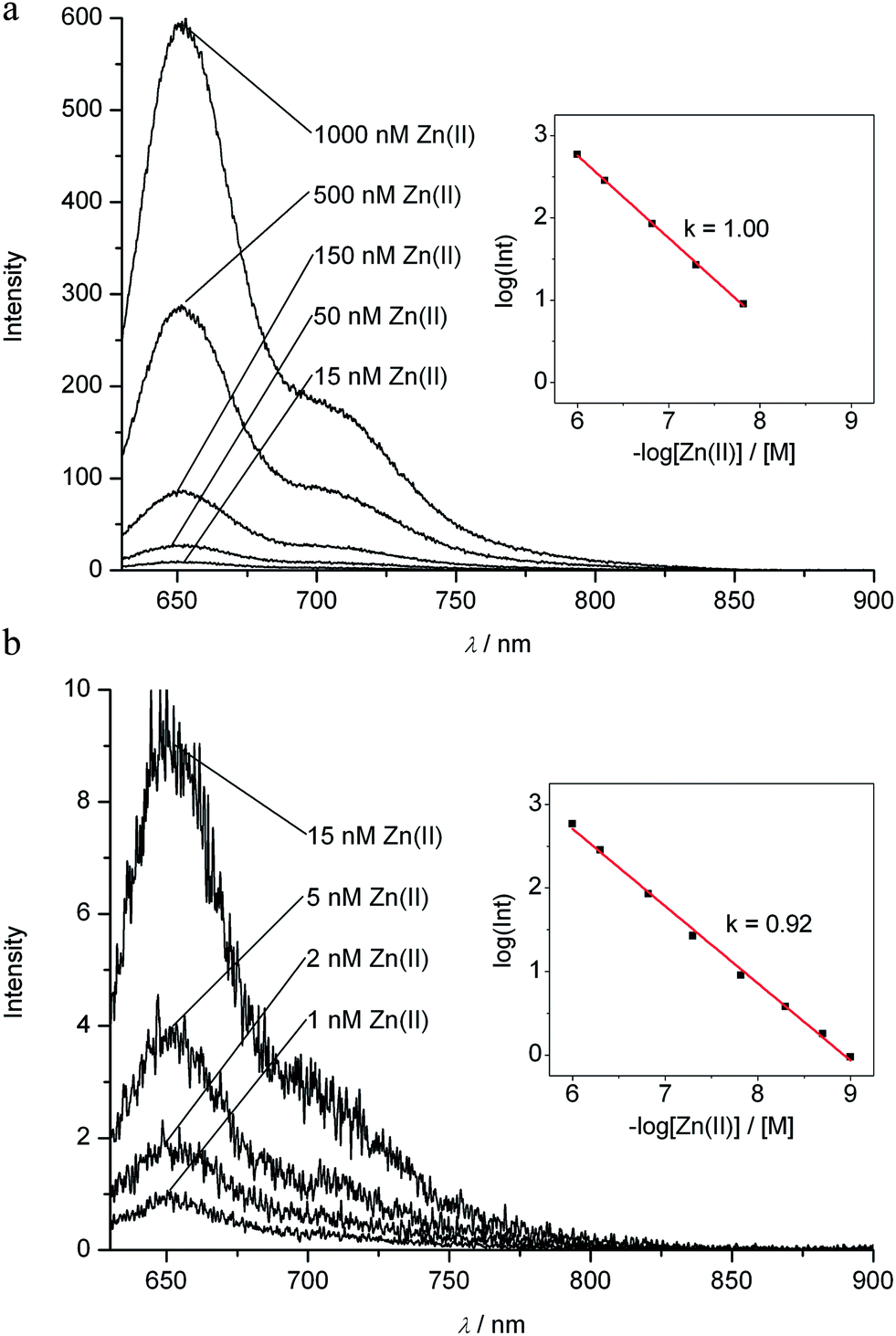 | ||
| Fig. 9 Assay for Zn(II)-ions in solution by detection of the red fluorescence of the zinc complex Zn-1. Top: fluorescence spectra for solutions of PiCC (1, 7.0 μM) in MeOH and concentrations of Zn(II)-ions (total) from 1000 nM down to 15 nM. Bottom: fluorescence spectra for solutions of PiCC (1, 7.0 μM) in MeOH and concentrations of Zn(II)-ions (total) from 15 nM down to 1 nM; insets: linear log/log plots of the fluorescence intensity at 650 nm vs. total concentration of zinc ions (see Experimental part and ESI† for details). Fluorescence spectra and values of intensities at 650 nm were corrected for weak background luminescence of the solution of pure 1 (for uncorrected spectra and further details, see ESI, Fig. S7†). | ||
Discussion
Phyllobilins are linear tetrapyrroles from catabolism of chlorophyll.3,5,6 Chlorophyll breakdown is easily seen in fall leaves and in ripening fruit by their characteristic colour changes.7,17,18 This process has aroused interest, not only from the point of basic human curiosity,19 but also for ecological and economic reasons.20 It has been estimated to involve about 1000 million tons each year,1,20,21 indeed, implying the formation of a similarly huge amount of phyllobilins.5,7 In senescent leaves, colourless, ‘nonfluorescent’ bilane-type catabolites typically accumulate, classified as NCCs and DNCCs,5 and suggested to represent the ‘final’ tetrapyrrolic products of chlorophyll breakdown.20,22Colourless NCCs were shown to be effective antioxidants,15 and to be readily oxidized to more unsaturated, yellow catabolites (YCCs), such as 2.13 YCC 2 also proved to be an excellent antioxidant,23 even slightly more effective than bilirubin.24 Further oxidation of YCCs (e.g.2) furnished PiCCs, e.g.1 (ref. 11) (Fig. 1). As reported here, YCC 2 is also oxidized cleanly to Zn-1 at room temperature in air saturated solution in DMF and in the presence of Zn(II)-ions. Decomplexation of Zn-1 gives 1 cleanly and provides an alternative path to the PiCC 1. Oxidation of NCCs to yellow phyllobilenes-c, such as YCC 2, extends the chromophore by formal dehydrogenation at their C15 meso-carbon. Subsequent oxidation of 2 by formal dehydrogenation at the C10 meso-position produces PiCC 1, a pink-red phyllobiladiene-b,c. The coloured phyllobilins 2 and 1 were observed in senescent leaves, e.g. of Cercidiphyllum japonicum, where they are accessible from the NCC 3 by a yet unspecified endogenous oxidation process.5,11
The crystal structure of the (potassium complex K-1 of) PiCC 1 is the first crystal structure of a chlorophyll catabolite from a higher plant. It provided detailed insights into the bonding of 1, confirming, first of all, the basic NMR-derived structure of 1, besides indicating significant double bond alternation in the main parts of the chromophore, compatible with the formula shown (e.g. in Fig. 1 and 3). The structure of a related red tetrapyrrole isolated from the green alga Auxenochlorella protothecoides is also available,25 which crystallized in an H-bonded dimeric structure. The crystal structure of K-1 also revealed the remarkable association of two PiCC molecules in the crystal into H-bonded and K-bridged pairs of enantiomers (see ESI, Fig. S1†). This crystallographic finding contrasts the absence of spectroscopic evidence, so far, for such an association of 1 in (methanolic) solution.
NCCs are bilane-type tetrapyrroles, which are not expected to coordinate well to transition metal ions. However, the main chromophore of YCCs, such as 2, is similar to that of bilirubin,16,26 which forms complexes with a variety of metal ions.27 Our preparative studies (see above) suggest weak bi-dentate coordination of 2 to Zn(II)-ions. As shown here, the still more unsaturated phyllobilin PiCC 1 represents an effective, chelate-type ligand for metal ions, comparable to the related heme-derived bilins.27,28 The Zn(II)-, Cd(II)-, Ni(II)- and Cu(II)-complexes of PiCC (1) were prepared in a remarkably straightforward way from 1 and the corresponding metal acetylacetonates. In contrast to 1, in which the C10–C11 double bond is E-configurated,11 the metal complexes M-1, such as Zn-1, require the C10–C11 and C15–C16 bonds to be Z, in order to allow for the inferred tri-dentate primary coordination of the tetrapyrrolic ligand to the metal ion (Fig. 4). According to extensive NMR analyses of the solution structures of Zn-1, Cd-1 and Ni-1, these three complexes are monomeric and the metal ions are coordinated by 1 in a similar tridentate fashion involving the N-atoms of rings B, C and D. Likewise, the NMR-derived data provide no evidence for intra-molecular N-coordination of ring A. Indeed, other polar functional groups present at ring A may engage in weak intra-molecular ligation of the central metal ion. The 1H-NMR spectrum of Ni-1 in d6-DMSO suggests a specific interaction of the 32-OH group. This Ni(II)-complex is diamagnetic in DMSO and acetonitrile, compatible with strong equatorial binding of the Ni(II)-ion by 4 ligand atoms. Coordination of further ligands (e.g. polar solvent molecules) to the bound divalent transition metal ions in M-1 is also likely, as these metal ions have a strong preference for being (at least) 4-coordinate.27,28 Thus, solutions of the Ni(II)-complex Ni-1 in MeOH are indicated by the 1H-NMR spectrum to be paramagnetic, a phenomenon also observed in other tetrapyrrolic Ni(II)-complexes.29 However, no evidence for a significant formation of dimeric assemblies in solution was seen in the spectra. Clearly, the chlorophyll-derived bilin-type tetrapyrrole PiCC represents a new type of natural three-dentate ligand for transition metal-ions.
In all four complexes investigated here, metal ion coordination resulted in a notable red shift of the absorbance maximum at long wavelength by roughly 100 nm. This was most pronounced for the copper complex Cu-1, with an absorbance maximum at 635 nm. The deduced double bond E to Z isomerisation may come up for part of the red shift; typically, it would mainly affect transition intensities, rather than their energies.11,16 Indeed, related spectral properties were also found in blue transition metal complexes of linear tetrapyrroles obtained via photooxygenolysis of porphyrins.30–32 The spectroscopic behaviour of 1, and its metal complexes, also reminds of that of some artificial tripyrrones,16 as well as of prodigiosenes, natural ‘tripyrrolic’ alkaloids with interesting antibacterial and antifungal properties,33 which are also capable of binding a variety of metal ions.16,33 In methanolic solution, the diamagnetic Cd(II)- and Zn(II)-complexes of 1 exhibited strong red luminescence, whereas the paramagnetic Cu(II)-complex Cu-1 and the Ni(II)-complex Ni-1 showed negligible emission, as expected (see Fig. 10). Metal chelation not only induced a significant ‘red’ shift (to give bright blue complexes), but also led to intense luminescence with the closed-shell Zn- and Cd-ions, which do not induce rapid quenching of the excited singlet state of the coordinated tetrapyrrolic ligand. From the closely spaced (0–0)-transitions in absorption and luminescence spectra of Zn-1 near 625 nm, the singlet excited state can be estimated to be situated above the ground state by about 47 kcal mol−1. Coordination of Zn(II)-ions, or of Cd(II)-ions, induces a dramatically increased fluorescence intensity, when compared to that of the very weakly luminescent, free PiCC 1. This apparently results from rigidifying the ligand-chromophore (the tridentate B–C–D section) in the complex, which then inhibits fast radiation-less deactivation paths that appear to operate in the pink-red phyllobiladiene-b,c1. Further studies are planned to also explore the expected capacity of some coordinated metal ions in M-1 to induce intersystem crossing and to assist the formation of triplet excited states, leading to phosphorescence and sensitization of the formation of singlet oxygen.34
 | ||
| Fig. 10 Solutions of metal complexes M-1 of the PiCC 1 in MeOH (with M = Zn(II), Cd(II), Cu(II), Ni(II)), as observed under day light (top), or under UV light at a wavelength of 366 nm (bottom). | ||
In contrast to the colourless NCCs, the more unsaturated phyllobiladiene-b,c1 is a kinetically and thermodynamically effective chelator of a variety of transition metal ions. However, for tridentate binding of the four transition metal ions investigated, the tetrapyrrolic ligand must undergo E to Z isomerisation. This appears to be an effectively fast, not separately observed step in the full assembly of the complexes Zn-1, Cd-1 and Cu-1. In contrast, it was indicated to be a slow (rate limiting) step, and the formation of an intermediate was observed, during complexation of 1 with Ni(II)-ions, which still had spectral features similar to those of 1 (Fig. 8). Formation of the complexes M-1 (M = Zn(II), Cd(II), Ni(II) and Cu(II)) from 1 and Zn(II)-, Cd(II)-, Ni(II)- and Cu(II)-acetates was found to occur with effective rate constants of roughly 600, 200, 10 and 400 M−1 s−1, respectively (Table 1).
A dilute solution of a crystallized sample of the only weakly luminescent PiCC (1), when exposed to (a sub-stoichiometric amount of) Zn(II)- or Cd(II)-ions, rapidly developed the typical luminescence of Zn-1 or Cd-1. A linear dependence of the luminescence intensity at 650 nm on the total concentration of these metal-ions, down to roughly 1 nM, was observed. A log/log plot had a slope of 1.00 in the concentration range above about 10 nM, indicating formation of Zn-1 (or Cd-1) in a 1:1 stoichiometry of the bound metal-ion and PiCC ligand. The slope decreased slightly at lower metal-ion concentrations, probably due to very weak background luminescence (see Fig. 9 and ESI, Fig. S7 and S8†). Thus, the PiCC 1 may serve as a luminescence reporter for Zn- and Cd-ions in a range of concentrations between >1000 μM and nM. However, 1 was revealed to be a veritable sponge for transition metals, and the analytical luminescence observations at low concentrations of Zn(II)- and Cd(II)-ions were reliable only with very pure solutions of crystallized 1.
Binding (transition) metal ions is a prime capacity of the natural cyclic tetrapyrroles, furnishing Nature with the important class of metallo-porphyrinoid cofactors.35 Metal-binding is considered a less typical feature, biologically, of (the heme-derived) linear tetrapyrroles.27,28 The pink-coloured chlorophyll catabolite 1 represents a new class of bilin-type biological pigments, and of amphiphilic ligands binding metal ions.36 Indeed, PiCCs (such as 1) bind various transition metal ions with high affinity. In this capacity, the phyllobilin 1 reminds of the prodigiosenes, tripyrrolic alkaloids that are suspected to owe their biological activities to their natural transition metal complexes.33 PiCCs may find application in the specific detection of some closed-shell metal ions, such as Zn(II)- and Cd(II)-ions. Potentially, the PiCC 1 and related oxidized phyllobilins may also bind transition metal ions inside plant cells, and induce diagnostic optical effects there. Detection of closed-shell transition metal ions by fluorescence, such as of Zn(II)- and Cd(II)-ions, could be particularly useful for in vivo and ex vivo studies of their accumulation in plants. Indeed, a variety of plants are ‘heavy metal hyper-accumulators’, and concentrate transition metal ions in the leaf vacuoles, such as the divalent Zn, Cd, Ni and Cu ions, investigated here.37,38 The vacuoles are also believed to accumulate the ‘late’ stages of the tetrapyrrolic chlorophyll catabolites.3,39 Formation of transition metal complexes of some phyllobilins (such as of 1) may thus be relevant under physiological conditions. The possible competition by closed and open shell transition metal ions for complexation of 1 could thus be important, inside the plant, and remain to be studied. In conclusion, formation of complexes M-1 by coordination of transition metal-ions by the PiCC 1 could be a biologically significant capacity of PiCCs and of related oxidized phyllobilins. This may give such phyllobilins and their transition metal complexes unsuspected physiological functions in plants, e.g. as sensitizers for singlet oxygen,34 as toxins against pathogens.33 or in ‘heavy metal transport and detoxification’.38 Roles related to the latter ones in animals and humans have been discussed for heme-derived bilins, for which transition metal complexes were shown to occur in (oxidized) bile.27,40
Experimental part
See ESI† for general experimental details, spectroscopy and HPL-chromatography. Spectroscopy: UV/Vis: HITACHI U-3000 spectrophotometer; λmax in nm (logε). Fluorescence: Varian Cary Eclipse Fluorescence Spectrophotometer. ESI-MS: ESI source, flow rate 2 ml min−1, spray voltage 1.4 kV, solvent water–methanol 1:1 (v/v).
Synthesis of the PiCC 1 from 2
See ESI for details.†For crystallization of PiCC ( 1 ), ca. 3 mg of 1 were dissolved in MeOH (2 ml), the solution was filtered through a tight plug of cotton wool, and the filtrate was collected in a vial, which was put into a jar that contained CH2Cl2. Clear rhombus-shaped single crystals formed on the wall of the vial (see‡ and ESI,† for further crystallographic details).
Synthesis of metal complexes of the PiCC 1
Synthesis of metal complexes from the PiCC 1
Spectroscopic data
ε, MeOH): 523 (4.56), 495sh (4.49), 313 (4.40). ESI-MS: m/z (%) = 687.3 (18), 686.2 (42), 685.2 (100, [M + 2Na − H]+).
ε, MeOH): 620 (4.43), 578 (4.23), 349 (4.31), 318 (4.35). Fluorescence (excitation at 620 nm, rel. int.): 704 (0.32), 650 (1.00). Excitation spectrum (emission at 651 nm, rel. int.): 621 (1.00), 576 (0.60), 348 (0.42). ESI-MS: m/z (%) = 728.1 (16), 727.1 (36), 726.1 (66), 725.1 (100, [M + Na]+); 707.1 (9), 706.0 (7), 705.1 (10), 704.1 (9), 703.1 (18, [M + H]+). See ESI† for 1H-NMR and further ESI-MS data.
ε, MeOH): 613 (4.52), 570 (sh, 4.35), 347 (4.29), 317 (4.33). ESI-MS: m/z (%) = 778.3 (21), 777.1 (35), 776.1 (61), 775.1 (100), 774.1 (66), 773.1 (72), 772.1 (40), 771.1 (28, [M + Na]+). See ESI† for fluorescence, 1H-NMR and further ESI-MS data.
ε, MeOH): 635 (4.38), 589 (4.16), 360 (4.26), 323 (4.33). ESI-MS: m/z (%) = 743.2 (18), 742.0 (52), 741.1 (30), 740.1 (100, [M + K]+). See ESI† for further ESI-MS data.
ε, MeOH): 626 (4.41), 585 (4.20), 356 (4.17), 326 (4.23). ESI-MS: m/z (%) = 700.1 (18), 699.1 (45), 698.1 (38), 697.1 (100, [M + H]+). See ESI† for 1H-NMR and further ESI-MS data.
Rates of formation of complexes M-1 were determined at 22 °C for Zn(II)-, Cd(II)-, Cu(II)- and Ni(II)-acetates in MeOH from measurements of absorbance changes. To 3 ml of a solution of the PiCC 1 in MeOH in a UV-cell the desired amount of a solution of M(OAc)2 in MeOH was added and the cell was immediately placed into the spectrophotometer. Changes of the absorption at 620 nm were analyzed in a time scan model. A calculated fit of the experimental absorptions at 620 nm at 22 °C by pseudo first order kinetics (y = y0 + Ae−kt) gave the values for the rate constants k.
Detection of Zn 2+ -ions and of Cd 2+ -ions in solution of PiCC (1) at ambient temperature via luminescence of the complexes Zn-1 and Cd-1: for details, see ESI.†
Acknowledgements
We would like to thank Gerhard Scherzer for excellent technical assistance, and we are grateful to the Austrian National Science Foundation (FWF, projects P-19596 and I-563) for financial support.References
- Chlorophylls, ed. S. B. Brown, J. D. Houghton and G. A. F. Hendry, CRP-Press, Boca Raton, USA, 1991 Search PubMed.
- P. Matile, S. Hörtensteiner, H. Thomas and B. Kräutler, Plant Physiol., 1996, 112, 1403–1409 CAS.
- S. Hörtensteiner and B. Kräutler, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 977–988 CrossRef PubMed.
- B. Kräutler, B. Jaun, K. Bortlik, M. Schellenberg and P. Matile, Angew. Chem., Int. Ed., 1991, 30, 1315–1318 CrossRef.
- B. Kräutler and S. Hörtensteiner, in Handbook of Porphyrin Science, ed. G. C. Ferreira, K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing, USA, 2013, vol. 28, pp. 117–185 Search PubMed.
- S. Moser, T. Müller, M. Oberhuber and B. Kräutler, Eur. J. Org. Chem., 2009, 21–31 CAS.
- B. Kräutler, Photochem. Photobiol. Sci., 2008, 7, 1114–1120 Search PubMed.
- T. Müller, M. Rafelsberger, C. Vergeiner and B. Kräutler, Angew. Chem., Int. Ed., 2011, 50, 10724–10727 CrossRef PubMed.
- F. G. Losey and N. Engel, J. Biol. Chem., 2001, 276, 8643–8647 CrossRef CAS PubMed.
- I. Süßenbacher, B. Christ, S. Hörtensteiner and B. Kräutler, Chem.–Eur. J., 2014, 20, 87–92 CrossRef PubMed.
- M. Ulrich, S. Moser, T. Müller and B. Kräutler, Chem.–Eur. J., 2011, 17, 2330–2334 CrossRef CAS PubMed.
- M. Scherl, T. Müller and B. Kräutler, Chem. Biodiversity, 2012, 9, 2605–2617 CAS.
- S. Moser, M. Ulrich, T. Müller and B. Kräutler, Photochem. Photobiol. Sci., 2008, 7, 1577–1581 CAS.
- C. Curty and N. Engel, Phytochemistry, 1996, 42, 1531–1536 CrossRef CAS.
- T. Müller, M. Ulrich, K.-H. Ongania and B. Kräutler, Angew. Chem., Int. Ed., 2007, 46, 8699–8702 CrossRef PubMed.
- H. Falk, Chemistry of Linear Oligopyrroles and Bile Pigments, Springer, Wien, 1989 Search PubMed.
- C. S. Barry, Plant Sci., 2009, 176, 325–333 CrossRef CAS PubMed.
- S. Hörtensteiner, Plant Mol. Biol., 2013, 82, 505–517 CrossRef PubMed.
- C. R. Bell and A. H. Lindsey, Fall Colors and Woodland Harvests, Laurel Hill Press, Chapel Hill, USA, 1990 Search PubMed.
- B. Kräutler and P. Matile, Acc. Chem. Res., 1999, 32, 35–43 CrossRef.
- G. A. F. Hendry, J. D. Houghton and S. B. Brown, New Phytol., 1987, 107, 255–302 CrossRef CAS PubMed.
- S. Hörtensteiner, Annu. Rev. Plant Biol., 2006, 57, 55–77 CrossRef PubMed.
- M. Ulrich and B. Kräutler, unpublished, see M. Ulrich, PhD thesis, University of Innsbruck, 2011.
- R. Stocker, Y. Yamamoto, A. F. McDonagh, A. N. Glazer and B. N. Ames, Science, 1987, 235, 1043–1046 CAS.
- N. Engel, A. Gossauer, K. Gruber and C. Kratky, Helv. Chim. Acta, 1993, 76, 2236–2238 CrossRef CAS.
- D. Lightner and A. F. McDonagh, Acc. Chem. Res., 1984, 17, 417–424 CrossRef CAS.
- M. Bröring, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, 2010, vol. 8, pp. 343–501 Search PubMed.
- A. L. Balch and F. L. Bowles, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, 2010, vol. 8, pp. 293–342 Search PubMed.
- C. Kratky, A. Fässler, A. Pfaltz, B. Kräutler, B. Jaun and A. Eschenmoser, J. Chem. Soc., Chem. Commun., 1984, 1368–1371 RSC.
- C. Jeandon, B. Krattinger, R. Ruppert and H. J. Callot, Inorg. Chem., 2001, 40, 3149–3153 CrossRef CAS PubMed.
- J. A. S. Cavaleiro, M. J. E. Hewlins, A. H. Jackson and M. G. P. M. S. Neves, Tetrahedron Lett., 1992, 33, 6871–6874 CrossRef CAS.
- A. Jauma, A. Escuer, J. A. Farrera and J. M. Ribo, Monatsh. Chem., 1996, 127, 1051–1062 CrossRef CAS.
- A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582–3603 CrossRef PubMed.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publ. Co., Singapore, 2010 Search PubMed.
- B. Kräutler and M. Ulrich, Patent Application WO 2012016255, 2012.
- N. Rascio and F. Navari-Izzo, Plant Sci., 2011, 180, 169–181 CrossRef CAS PubMed.
- U. Krämer, Annu. Rev. Plant Biol., 2010, 61, 517–534 CrossRef PubMed.
- P. Matile, S. Ginsburg, M. Schellenberg and H. Thomas, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 9529–9532 CrossRef CAS.
- R. J. Garner, Nature, 1954, 173, 451–452 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details on crystal structure, for syntheses, spectral analyses of new compounds, kinetic analysis of metal incorporation and detection of Zn and Cd-ions in solution. CCDC 981318. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc00348a |
| ‡ Crystal data of K-1: C35H35KN4O8·CH3OH·2.5H2O; M = 755.85, ρ = 1.303 g cm−3. Triclinic space group P (no. 2), Z = 2, a = 11.7592(8) Å, b = 12.7470(9) Å, c = 13.3933(9) Å, α = 98.948(4)°, β = 100.210(4)°, γ = 97.418(3)° and V = 1926.2(2) Å3 at 233(2) K, 4396 independent reflections, 3127 with I > 2σ(I). Final R indices [I >2σ(I)]: R1 = 0.0769, wR2 = 0.1824. Cambridge Crystallographic Data Centre CCDC # 981318 (1, PiCC). |
| This journal is © The Royal Society of Chemistry 2014 |