 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLewis acidic stiborafluorenes for the fluorescence turn-on sensing of fluoride in drinking water at ppm concentrations†
Masato
Hirai
and
François P.
Gabbaï
*
Department of Chemistry, Texas A&M University, College Station, Texas 77843-3255, USA
First published on 11th February 2014
Abstract
Prompted by the importance of fluoride in fields such as drinking water analysis and 18F-positron emission tomography, we have synthesized and investigated three Lewis acidic organostiboranes containing the (2,2′-biphenylene)phenylantimony(V) moiety as a common subunit and a catecholate (1), a tetrachlorocatecholate (2) or an alizarin (alizarin = 1,2-dihydroxyanthraquinone) ligand (3). DFT calculations show that the Lewis acidity of these stiboranes arises from the presence of a low lying antimony-centered orbital. While 1 shows no measurable fluoride anion affinity in 7/3 vol. THF–water mixtures, spectrophotometric titrations show that the more electron deficient stiboranes 2 and 3 bind fluoride anions in the same medium with binding constants (K) of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 (±1400 M−1) for 2 and 16100 (±1100) M−1 for 3. Formation of the fluoride complexes has been confirmed by isolation of TAS[2–F] and TAS[3–F] (TAS = tris(dimethylamino)sulfonium), the structures of which have also been determined. While formation of [2–F]− does not trigger a marked photophysical response, a color change from yellow to dark red is observed upon conversion of 3 into [3–F]− in CH2Cl2. This color change, which has been investigated using TD-DFT calculations, is also accompanied by an increase in the fluorescence of the alizarin chromophore at 616 nm. Owing to its high fluoride affinity and fluorescence turn-on properties, 3 can be used in biphasic water–CH2Cl2 mixtures to assay sub-ppm fluoridation levels in tap water or bottled infant drinking waters.
500 (±1400 M−1) for 2 and 16100 (±1100) M−1 for 3. Formation of the fluoride complexes has been confirmed by isolation of TAS[2–F] and TAS[3–F] (TAS = tris(dimethylamino)sulfonium), the structures of which have also been determined. While formation of [2–F]− does not trigger a marked photophysical response, a color change from yellow to dark red is observed upon conversion of 3 into [3–F]− in CH2Cl2. This color change, which has been investigated using TD-DFT calculations, is also accompanied by an increase in the fluorescence of the alizarin chromophore at 616 nm. Owing to its high fluoride affinity and fluorescence turn-on properties, 3 can be used in biphasic water–CH2Cl2 mixtures to assay sub-ppm fluoridation levels in tap water or bottled infant drinking waters.
Introduction
The complexation of fluoride anions in protic media is a topic of intense research because of applications in the field of drinking water analysis1,2 and 18F-positron emission tomography.3 Numerous organic compounds that interact with the anion via the formation of hydrogen bonds have been considered for this purpose.4–14 However, the efficient capture of this anion in protic solvents typically necessitates the use of a Lewis acidic receptor.15–20 While ample precedents show that group 13 Lewis acids are especially well suited for this purpose,21–27 recent advances in the chemistry of organo-group 15 compounds as Lewis acids28–32 and fluorophores33–37 have led us to question whether organoantimony(V) species may also be competent for the complexation and fluorescence sensing of fluoride ions in protic solvents.38–44 With this objective in mind, we have recently investigated the 9-anthryltriphenylstibonium cation ([AntPh3Sb]+) and found that this cation captures fluoride in 9/1 vol. water–DMSO to afford the corresponding fluorostiborane AntPh3SbF.45 The fluorescence properties of [AntPh3Sb]+ as well as its fluoride affinity are such that sensing can be carried out at ppm or sub-ppm fluoride concentrations. We concluded from these initial experiments that the high fluoride affinity of [AntPh3Sb]+ arises from strong Coulombic effects which drive the ion pairing process. While the influence of such forces cannot be disputed, we have now decided to determine whether neutral organoantimony(V) compounds would be sufficiently Lewis acidic to complex fluoride anions in protic solvents. In this article, we present an initial validation of this idea.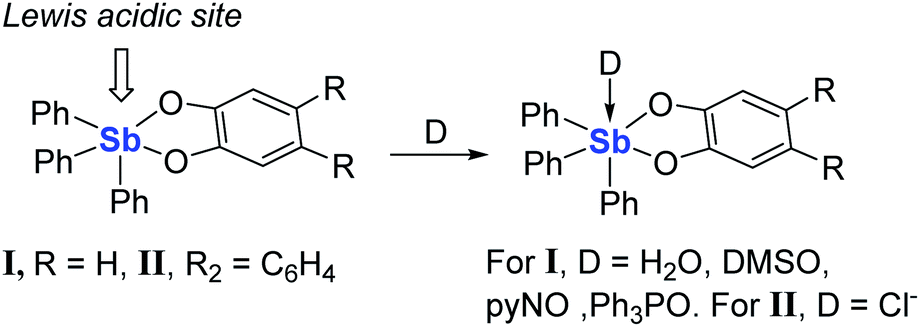
In search of a class of Lewis acidic organoantimony species that we could employ in the present study, we were drawn by a number of reports dealing with Lewis adducts of triarylantimony catecholates such as I, which adopts a square pyramidal geometry46,47 and readily forms adducts with a number of Lewis bases including water,47 DMSO, N-pyridine oxide,48 and triphenylphosphine oxide.49 Such compounds have also been shown to engage anions, as in the case of II which forms an adduct with chloride anions.50
Results and discussion
Contending that spirocyclic stiboranes may exhibit greater structural stability and provide less hindered access to the antimony atom, we decided to investigate the Lewis acidic behavior of the stiborafluorene 1 and its tetrachloro-analog 2, which has been previously reported.50 Compound 1 was obtained in a 72% yield by reaction of the known (2,2′-biphenylene)phenylstibine50 with tert-butyl hydroperoxide48 and catechol in toluene at 0 °C. This compound has been fully characterized. Its 1H NMR spectrum shows 9 distinct signals whose multiplicity suggests that the derivative adopts a Cs symmetry. This view is confirmed by the crystal structure of the complex which shows that the biphenylene and catecholato groups are located at the base of the square pyramidal antimony atom, with the phenyl group defining the apex (see ESI†). The structures of these two compounds have been computationally optimized using DFT methods. The LUMO of these two complexes are localized on the stiborafluorene moieties and resemble that of the parent fluorenyl cation, with a larger contribution of the atom at the 9-position, in this case the antimony atom, which participates in the π-system via an orbital of σ*(Sb–CPh) character. The energy of the LUMO in 2 (−1.73 eV) is notably lower than that of 1 (−1.50 eV), an effect that we assign to the perchlorination of the catechol group in 2 (Fig. 1). | ||
| Fig. 1 (Top) Chemical structures of 1 and 2. (bottom) Contour plot and energy of the LUMO in 1 (panel A) and 2 (panel B) (isodensity = 0.036). Panel C shows the similarity existing between the LUMO of the fluorenyl cation and that of 2. | ||
With these compounds in hand, we decided to investigate their fluoride anion affinity in aqueous solutions. To this end, we carried out a spectrophotometric fluoride titration experiment in 7/3 vol. THF–water solution (Fig. 2). While no changes are observed in the UV-Vis spectrum of 1 upon incremental addition of TBAF, we observed clear evidence of fluoride anion binding in the case of 2. Indeed, the intensity of the band centered at 287.4 nm increases with the fluoride anion concentration. While the origin of these small spectral changes is difficult to assign, they can be fitted to a 1:1 binding isotherm affording a stability constant of 13500 (±1400) M−1 for [2–F]−.51 Formation of [2–F]− was confirmed by an end-of-titration electrospray ionization mass spectroscopy (ESI-MS) measurement which showed the molecular ion at m/z = 614.7764 amu. It is worth noting that neutral Lewis acids including boranes such as Mes3B51,52 or fluorosilanes such as Ph3SiF53 fail to complex fluoride under such conditions, a difference that underscores the unusual Lewis acidic properties of the stiborafluorene 2. Also, the contrasting behavior of 1 and 2 demonstrates that the perchlorinated and thus more electron withdrawing catecholate group in 2 effectively increases the Lewis acidity of the antimony center. This conclusion is consistent with the lower energy calculated for the LUMO of 2 when compared to 1 (Fig. 1).
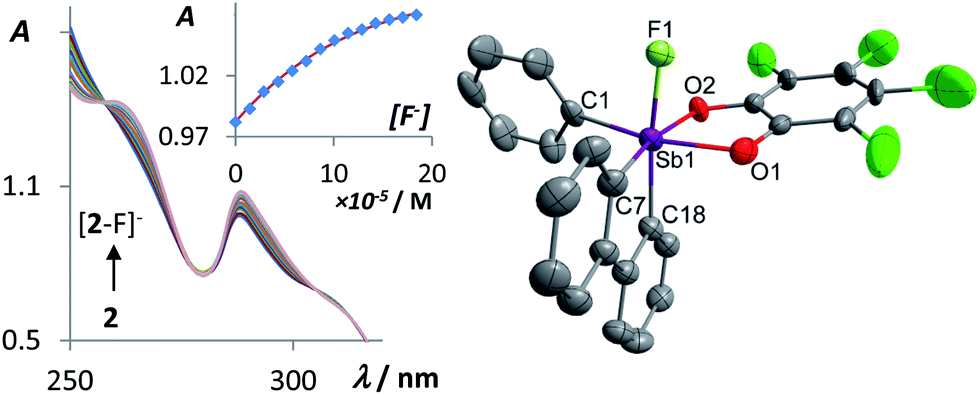 | ||
| Fig. 2 Left: absorption spectra in 7/3 vol. THF–water showing the conversion of 2 (7.2 × 10−5 M) into [2–F]− upon addition of fluoride anions. The inset shows the experimental and the calculated 1:1 fluoride binding isotherm for 2. The data were measured at 287.4 nm and fitted with K = 13500 (±1400) M−1 (ε(2) = 13600 M−1 cm−1 and ε([2–F]−) = 15300 M−1 cm−1). Right: crystal structure of TAS[2–F]. Thermal ellipsoids are drawn at the 50% probability level. The hydrogen atoms and the TAS cation are omitted for clarity. Selected bond lengths (Å) and angles (deg): Sb1–F1 1.973(4), Sb1–O1 2.105(4), Sb1–O2 2.082(4), Sb1–C1 2.131(6), Sb1–C7 2.128(6), Sb1–C18 2.141(6), F1–Sb1–C18 172.11(18), C1–Sb1–O1 168.21(19), O2–Sb1–C7 167.51(19), O1–Sb1–O2 78.65(16), C7–Sb1–C18 82.8(2). | ||
The anionic complex [2–F]− can be easily obtained as a tris(dimethylamino)sulfonium (TAS) salt by reaction with TASF in THF (Scheme 1). The 1H NMR resonances of [2–F]− show a loss of the Cs symmetry with the hydrogen atoms of the biphenylene backbone becoming unequivalent. The 19F NMR spectrum of this complex features a single resonance at −102.8 ppm for [2–F]− corresponding to the antimony-bound fluoride anions. ESI-MS of this salt shows the molecular peak of [2–F]− at 614.7764 amu. The crystal structure of the salt TAS[2–F] shows that the anion and the cation are well separated. The anionic component [2–F]− displays an antimony atom in a slightly distorted octahedral geometry (Fig. 2). The fluoride anion, arbitrarily denoted as an axial ligand, is located trans from a phenylene ring of the biphenylene backbone. The tetrachlorocatecholate and the phenyl group both lie in the equatorial plane. The fluorine antimony distance for [2–F]− is 1.973(4) Å, which is slightly longer than the average Sb–F bond length in SbF6− (1.844 Å).54
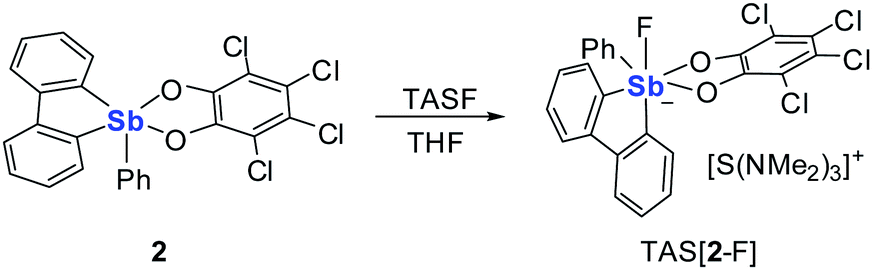 | ||
| Scheme 1 Synthesis of TAS[2–F]. | ||
Although the above results demonstrate that stiborafluorenes are competent molecular recognition units for fluoride anions, the photophysical response accompanying fluoride binding is very weak. This lack of an adequate signaling response makes compounds such as 2 poorly suited for sensing applications. In order to overcome this limitation, we questioned whether the tetrachlorocatecholate ligand of 2 could be replaced by a 1,2-dihydroxybenzene derivative with comparable electron-withdrawing properties, yet more prevalent photophysical properties. These considerations led us to focus on alizarin red (1,2-dihydroxyanthraquinone),55–57 a chromophore that has been previously used in tandem with phenylboronic acid for the fluorescence detection of fluoride anions.58–60 The alizarin red chromophore could be conveniently incorporated into the stiborafluorene platform by the route depicted in Scheme 2 to afford compound 3 as a dark yellow derivative. The proton spectrum of 3 confirms the presence of the 1,2-dihydroxyanthraquinone. Despite the unsymmetrical nature of the 1,2-dihydroxyanthraquinone ligand, only four C–H resonances from the stiborafluorene backbone are observed, which is suggestive of a fluxional structure. Although we have not been able to obtain a crystalline sample of this complex, we assume that it adopts a square pyramidal geometry analogous to that observed for 1 and 2. DFT calculations reveal that the HOMO and LUMO of 3 are based on the 1,2-dihydroxyanthraquinone ligand. The LUMO+1 of 3 is localized on the stiborafluorene moiety and resembles the LUMO of 1 and 2 with a large lobe on the antimony atom (Fig. 3). The energy of this stiborafluorene-based orbital (−1.61 eV) suggest that the anthraquinone backbone exerts an electron withdrawing effect intermediate between that of the catecholate and tetrachlorocatecholate ligands.
 | ||
| Scheme 2 Synthesis of 3 and TAS[3–F]. | ||
 | ||
| Fig. 3 (Top) Contour plot of the relevant orbitals in 3 and [3–F]− (isodensity = 0.036). (bottom) Structure of the crystallized enantiomer of TAS[3–F]. Thermal ellipsoids are drawn at the 50% probability level. The hydrogen atoms and the TAS cations are omitted for clarity. Selected bond lengths (Å) and angles (deg) with the corresponding metrical parameters for the second independent molecule in brackets: Sb1–F1 1.978(4) [1.979(2)], Sb1–O1 2.077(3) [2.082(3)], Sb1–O2 2.100(3) [Sb2–O6 2.100(2)], Sb1–C1 2.126(4) [2.127(4)], Sb1–C7 2.132(4) [2.128(4)], Sb1–C18 2.141(4) [2.138(4)], F1–Sb1–C18 172.14(12) [170.05(12)], C1–Sb1–O2 160.53(12) [162.58(12)], C7–Sb1–O1 172.36(12) [171.80(12)], O1–Sb1–O2 77.61(10) [77.90(10)], C7–Sb1–C18 82.02(15) [81.96(14)]. | ||
Complex [3–F]− can be isolated as a crystalline TAS salt when generated from 3 and TASF in THF (Scheme 2). This salt has been isolated and fully characterized. Its composition has been verified by elemental analysis. When this compound is dissolved in CD3CN and analyzed by 19F NMR spectroscopy, two signals are observed at −107.3 and −112.3 ppm with a 1:1 intensity ratio. We speculate that these two signals, which are close to those measured for [2–F]− (−102.8 ppm), arise from the existence of diastereomers that differ by the orientation of the unsymmetrical 1,2-dihydroxyanthraquinone with respect to the Sb–Ph bond (Scheme 2). Crystallization of TAS[3–F] lead to the isolation of single crystals which contain the two enantiomers of one of the diastereomers (Fig. 3). In these crystals, the two enantiomers, which are not related by crystallographically imposed symmetry, are found in the asymmetric unit. Their structures are, as expected, very similar with an octahedral geometry at antimony and with Sb–F bond lengths of 1.978(2) and 1.979(2) Å comparable to those in [2–F]−.
In a solution of dry CH2Cl2, the absorption spectra of 3 is dominated by a broad absorption band at λmax = 430 nm arising from the 1,2-dihydroxyanthraquinone chromophore. The energy of this band is similar to that observed in other alizarin containing derivatives.55–60 TD-DFT calculations show that this absorption band corresponds to the HOMO to LUMO transition (λmax(calculated) = 435 nm, f = 0.2598). Incremental addition of fluoride ions induces a notable redshift of the low energy band as illustrated in Fig. 4. This phenomenon is ascribed to the conversion of 3 into [3–F]−, whose formation is essentially quantitative as indicated by the shape of the 1:1 binding isotherm (Fig. 3). Inspection of the spectra also shows that the energy of the absorption band shifts by 50 nm upon conversion of 3 (λmax = 430 nm) into [3–F]− (λmax = 482 nm). This redshift is accompanied by a marked colorimetric response which can be readily detected with the naked eye when the reaction is carried out at mM concentrations. Using the same level of theory as for 3, the structure of [3–F]− has been optimized using DFT methods and subsequently subjected to TD-DFT calculations. These calculations show that the frontier orbitals remain centered on the alizarin chromophore, with the same atomic distribution as in the case of 3 (Fig. 3). These calculations also show that their energy is perturbed by the presence of an antimony-bound fluoride anion. This perturbation is reflected by the narrower HOMO–LUMO gap and the calculated wavelength of λmax(calculated) = 484 nm (f = 0.2824) (vs. the experimental value of λmax = 482 nm). These theoretical results show that the redshift observed upon conversion of 3 into [3–F]− originates from the conversion of the stiborane into a negatively charged, electron-rich fluoroantimonate, which destabilizes the alizarin-based HOMO and narrows the HOMO–LUMO gap by 0.36 eV from 3.31 eV in 3 to 2.95 eV in [3–F]− based on the computed energy of the frontier orbitals. These calculations are in good agreement with the experimentally observed 50 nm (or 0.31 eV) redshift observed upon fluoride binding. The redshift observed upon formation of the fluoroantimonate [3–F]− bears a parallel to the chemistry of some organoboron-based fluoride sensors, for which conversion of the neutral boron center into an electron-rich fluoroborate moiety also results in a redshift of the absorption band of the appended chromophore.61,62 A spectrophotometric titration carried out in 7/3 vol. THF–water shows that the stability constant of [3–F]− (16100 (±1100) M−1) is close to that of [2–F]− (13500 (±1400) M−1) (Fig. S6†). The aliquot after titration was analyzed by ESI-MS which showed the molecular peak of [3–F]− at m/z = 607.0655 amu. Under these conditions, however, the redshift of the low energy band is not as marked as in neat CH2Cl2, a difference that we assign to the coordination of water to the antimony atom.
 | ||
| Fig. 4 Spectral changes in the UV-Vis absorption spectrum of 3 (5.5 × 10−5 M in CH2Cl2) upon addition of fluoride. The inset on the top left shows the experimental and the calculated 1:1 fluoride binding isotherms for 3 at 483 nm. The data were fitted with K > 107 M−1 (ε(3) = 1400 M−1 cm−1 and ε([3–F]− = 7850 M−1 cm−1)). The inset on the top right shows the fluorescence spectra of 3 (5.0 × 10−6 M in CH2Cl2) before and after addition of a stoichiometric amount of fluoride (λexcitation = 482 nm). The pictures of the cells were taken using a solution of 3 (5.5 × 10−5 M) in CH2Cl2, before and after addition of fluoride. The fluorescent images (labelled as Fluo) were illuminated with a hand-held UV lamp. | ||
We have also tested the fluorescence properties of 3. The fluorescence spectra of this compound in CH2Cl2 show a broad emission at 616 nm, characteristic of the alizarin red chromophore (Fig. 4 and 5).58–60 With a quantum yield of Φ = 0.2% (λexcitation = 482 nm), this emission is very weak. Gratifyingly, we found that addition of fluoride to the solution results in a drastic fluorescence increase from Φ = 0.2% for 3 to Φ = 3.0% for [3–F]−. The intensity increases linearly with the first equivalent of fluoride indicating quantitative formation of [3–F]−. The fluorescence turn-on response observed during this anion binding reaction is assigned to the increased rigidity of the hexacoordinate antimony complex [3–F]−.
 | ||
| Fig. 5 (A) Fluorescence spectra (λexcitation = 482 nm) of solutions of 3 (5.0 × 10−6 M) in CH2Cl2. For each measurement, the solution was prepared by the 100-fold dilution of a 5.0 × 10−4 M solution of 3 which had been layered with an aqueous solution of KF (0, 1.9 and 3.8 ppm) containing TPABr (20 mM) and a citrate buffer (10 mM, pH 4.68). (B) Drinking water analysis data: each fluorescence spectrum is obtained with a solution of 3 in CH2Cl2 (5.0 × 10−5 M) after layering with a standard fluoride solution or an unknown sample. The spectra drawn with dotted lines correspond to the standard fluoride solutions (0, 0.4, 0.7 and 1.0 ppm, from bottom to top). The spectra obtained for the unknown samples are drawn with solid lines (S1 = H-E-B® Baby Purified Water (with fluoride added); S2 = Nursery® Water; S3 = College Station Tap Water. (C) Naked-eye fluorescence and colorimetric response associated with the formation of [3–F]− at a concentration of 5.0 × 10−4 M. | ||
The anion binding properties of complex 3 have been evaluated under biphasic conditions. We first layered a CH2Cl2 solution of 3 (1 mL, [3] = 5.0 × 10−4 M) with an aqueous solution (5 mL) containing tetrapropylammonium bromide (TPABr; 20 mM) as a phase transfer agent.63 Upon shaking of this biphasic mixture, the color of the CH2Cl2 layer changes from pale yellow to dark red, a phenomenon assigned to hydroxide binding to the antimony center of 3. Gratifyingly, we found that this interfering reaction could be prevented by simply buffering the water layer at pH 4.68 using citric acid/citrate (10 mM). Using these conditions, we decided to interrogate the system with low concentrations of fluoride and we observed that ppm concentrations of this anion can be readily assessed with the naked eye. Indeed, addition of 1.9 ppm of fluoride (1.0 × 10−4 M KF) to the water layer results in a distinct darkening of the CH2Cl2 layer from yellow to pale orange (Fig. 5). A further intensification of the color is observed when the KF concentration is raised to 3.8 ppm (2.0 × 10−4 M KF). Formation of [3–F]− was confirmed by UV-Vis and fluorescence measurements as well as by 19F NMR measurements (13200 scans) of the CH2Cl2 layer which shows the two expected peaks at −107.2 and −112.4 ppm. No color change was observed in the presence of other anions such as Cl−, Br−, NO3−, HCO3−, H2PO4− and HSO4−, which indicates that 3 is highly selective for fluoride anions.
These fluoride sensing results suggest that 3 may be well suited for real-life applications. To put this possibility to a test, we have investigated the use of 3 for tap water and bottled water analysis. Using biphasic conditions analogous to those described above, we analyzed several drinking water samples. We found that the tap water in College Station contains 0.4(±0.05) ppm of fluoride, which is close to the concentration of 0.44 ppm documented in the most recent water quality report. We also assayed two different brands of fluoridated water marketed for infant consumption. In the first water sample, sold by the H-E-B® supermarket chain as H-E-B® Baby Purified Water (with fluoride added), we found a fluoride concentration of 0.8(±0.1) ppm which is in good agreement with the maximum fluoride content of 1 ppm advertised on the label. The second water sample was Nursery® Water with an advertised maximum concentration of 0.7 ppm. For this water sample, our method provided a concentration of 0.6(±0.7) ppm, again in good agreement with the level of fluoridation advertised on the label.
Conclusion
The results presented in this paper show that neutral organoantimony(V) species may be sufficiently Lewis acidic to overcome the high hydration energy of the fluoride anion. This is the case of compounds 2 and 3 which are readily converted into the corresponding fluoroantimonate anions [2–F]− and [3–F]−. While fluoride binding to the antimony center does not necessarily trigger a strong photophysical response as in the case of 2, the incorporation of an alizarin chromophore in 3 imparts some advantageous turn-on properties. These turn-on properties along with its elevated fluoride affinity make this derivative a useful water compatible fluoride sensor which can be used for the determination of sub-ppm concentrations of fluoride ions in bottled and tap waters.Experimental section
General considerations
Because of poor gastrointestinal uptake, oral LD50 values for antimony compounds (e.g. 0.5 g kg−1 for SbCl3 and 1.1g kg−1 for SbCl5 in rat) are relatively high. However antimony compounds are very toxic when administered intravenously. We have therefore handled these compounds with great caution and recommend any experimentalist to do the same. N,N,N′N′-Tetramethylethylenediamine (tmeda) was purchased from Aldrich and distilled from powdered CaH2 and stored under N2. Biphenyl and [S(NMe2)3][Me3SiF2] (TASF) were purchased from Aldrich and used as received. Antimony trichloride (SbCl3), triphenyl stibine (Ph3Sb), n-butyl lithium (2.3 M in hexane), 1,2-dihydroxyanthraquinone (alizarin red) were purchased from Alfa Aesar. Tetrachloro-o-benzoquinone was purchased from Acros Organics. (2,2′-Biphenylene)phenylstibine and stiborane 2 (ref. 50) were prepared according to the reported procedure. All preparations were carried out under an atmosphere of dry N2 employing either a glovebox or standard Schlenk techniques. Solvents were dried by passing through an alumina column (pentane, CH2Cl2) or refluxing under N2 over Na/K (Et2O and THF). All other solvents were ACS reagent grade and used as received. NMR spectra were recorded on a Varian Unity Inova 500 FT NMR (499.42 MHz for 1H, 469.86 MHz for 19F, 125.60 MHz for 13C) spectrometer at ambient temperature. Chemical shifts are given in ppm and are referenced to residual 1H and 13C solvent signals and external BF3·Et2O for 19F. Elemental analyses were performed by Atlantic Microlab (Norcross, GA). The pH measurements were carried out with a Radiometer PHM290 pH meter equipped with a VWR SympHony electrode. Electronic absorption spectra were recorded at ambient temperature using an Ocean Optics USB4000 spectrometer with an Ocean Optics ISS light source. Emission spectra were recorded at ambient temperature using a PTI QuantaMaster™ 30 fluorescence spectrofluorometer. Electrospray ionization mass spectra were recorded on Applied Biosystems PE SCIEX QSTAR. The NMR data was reported according to the following numbering scheme:
Synthesis of 1
To a suspension of (2,2′-biphenylene)phenylstibine (285.7 mg, 81 μmol) and catechol (110.1 mg, 81 μmol) in toluene (15 mL) at 0 °C was added a toluene solution (5 mL) of tert-butyl hydroperoxide (70 wt% in water, 104.0 mg, 0.081 mmol) dropwise over a period of 15 min. After stirring the mixture at reduced temperature for 15 min, the solvent was removed under vacuum and washed with two portions of methanol (5 mL each) to afford 1 as a yellow product (268.6 mg, 72% yield). Large yellow single crystals of 1 were obtained by slow diffusion of pentane into a chloroform solution at ambient temperature. 1H NMR (499.42 MHz, CDCl3): δ 8.08 (d, H4, 3JH–H = 7.5 Hz; 2H), 8.02 (d, o-SbPh, 3JH–H = 7.5 Hz; 2H), 7.64 (pseudo dt, H1, 3JH–H = 7.0 Hz, 4JH–H = 2.0 Hz; 2H), 7.57 (pseudo td, H2 or H3, 3JH–H = 7.5 Hz, 4JH–H = 1.5 Hz; 2H), 7.51 (pseudo td, H2 or H3, 3JH–H = 7.5 Hz, 4JH–H = 1.5 Hz; 2H), 7.45 (pseudo td, p-SbPh, 3JH–H = 7.5 Hz, 4JH–H = 1.5 Hz; 1H), 7.38 (pseudo td, H2, 3JH–H = 7.0 Hz, 4JH–H = 2.0 Hz; 2H), 6.97 (m, 2H, H6), 6.68 (m, 2H, H5). 13C{1H} NMR (125.60 MHz, CDCl3): δ 147.66 (o-C6H4), 140.78, 135.11, 133.96, 132.89, 132.51, 132.29, 132.19, 129.77, 129.71, 122.75, 118.97. The NMR spectra of this compound are provided in the ESI† as a measure of purity.Synthesis of TAS[2–F]
To a solution of 2 (41.8 mg, 70 μmol) in THF (3 mL) was added a solution of TASF (19.3 mg, 70 μmol) in THF (3 mL) at ambient temperature. After stirring for 10 min, the solvent was evaporated under vacuum and the remaining solid was washed with two portions (5 mL each) of diethyl ether to afford TAS[2–F] as a white solid (54.6 mg, 84% yield). Colorless single crystals of TAS[2–F] were obtained by slow diffusion of pentane into a THF solution at ambient temperature. 1H NMR (499.42 MHz, CD3CN): δ 8.07 (d, 3JH–H = 7.5 Hz; 1H), 8.04 (d, 3JH–H = 8.0 Hz; 1H), 7.93 (d, J = 7.5 Hz; 1H), 7.61 (dd, 3JH–H = 8 Hz, J2 = 2 Hz; 2H), 7.56 (dt, J1 = 7.5 Hz, J2 = 1.5 Hz; 1H), 7.44–7.40 (m, 2H), 7.39 (dd, J1 = 8 Hz, J2 = 2 Hz; 1H), 7.35–7.25 (m, 4H), 2.82 (s, N(CH3)2;18H). 13C{1H} NMR (125.60 MHz, CD3CN): δ 150.34, 150.50, 149.89, 149.52, 144.65, 142.87, 142.63, 142.02, 140.85, 140.83, 134.87, 134.78, 133.43, 132.03, 131.36, 130.55, 129.98, 129.90, 129.68, 123.93, 123.80, 117.83, 116.81, 116.27, 116.25, 39.36 (N(CH3)2). 19F NMR (469.86 MHz, CD3CN): δ −102.8 (s). Elemental analysis calculated (%) for C30H31Cl4FN3O2SSb: C, 46.18; H, 4.01; N, 5.39; found C, 46.90; H, 4.20; N, 5.36.Synthesis of 3
To a suspension of (2,2′-biphenylene)phenylstibine (177.4 mg, 0.5 mmol) and alizarin red (94%, 128.9 mg, 0.5 mmol) in toluene (15 mL) at 0 °C was added a toluene solution (5 mL) of tert-butyl hydroperoxide (70 wt% in water, 64.9 mg, 5.0 × 10−4 mol) dropwise over a period of 15 min. After stirring the mixture at reduced temperature for an hour, the solvent was removed under vacuum and washed with two portions (5 mL each) of methanol followed by two portions of diethyl ether (5 mL each) to afford 3 as a dark yellow product (160.8 mg, 55% yield). 1H NMR (499.42 MHz, CDCl3): δ 8.34 (d, H11, 3JH–H = 7.5 Hz; 1H), 8.26 (broad, H4 + H5 + H14; 3H), 8.06 (d, o-SbPh, 3JH–H = 7.5 Hz; 2H), 7.84 (d, H10, 3JH–H = 8.5 Hz; 1H), 7.75–7.58 (m, m-SbPh + H1 + H3 + H6 + H8 + H13; 7H), 7.50 (t, p-SbPh, 3JH–H = 8.5 Hz; 1H), 7.43 (m, H2 + H7 + H12; 3H), 7.28 (d; merged with CDCl3 resonance, H9; 1H). 13C{1H} NMR (125.60 MHz, CDCl3): δ 183.40 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 182.83 (CO), 154.21, 149.4, 140.76, 135.17, 135.04, 133.92, 133.76, 133.28, 133.01, 132.98, 132.76, 131.57, 130.75, 130.23, 130.11, 126.78, 126.69, 125.28, 123.01, 120.08, 118.62, 116.65. Elemental analysis calculated (%) for C32H19O4Sb: C, 65.23; H, 3.25; found C, 64.83; H, 3.29. The NMR spectra of this compound are provided in the ESI† as an additional measure of purity.
O), 182.83 (CO), 154.21, 149.4, 140.76, 135.17, 135.04, 133.92, 133.76, 133.28, 133.01, 132.98, 132.76, 131.57, 130.75, 130.23, 130.11, 126.78, 126.69, 125.28, 123.01, 120.08, 118.62, 116.65. Elemental analysis calculated (%) for C32H19O4Sb: C, 65.23; H, 3.25; found C, 64.83; H, 3.29. The NMR spectra of this compound are provided in the ESI† as an additional measure of purity.
Synthesis of TAS[3–F]
To a solution of 3 (44.8 mg, 76 μmol) in THF (3 mL), a solution of TASF (20.9 mg, 76 μmol) in THF (3 mL) was added dropwise at ambient temperature. After stirring for 15 min, the solvent was removed under vacuum and the remaining solid was washed with two portions of diethyl ether (5 mL each) to afford TAS[3–F] as a dark red solid (54.0 mg, 92% yield). Single crystals of TAS[3–F] were obtained as dark red platelets by slow diffusion of diethyl ether into a dimethyl formamide solution at ambient temperature. Ratio of diastereoisomer based on integration of the 19F NMR signals, 54:46. 1H NMR (499.42 MHz, CDCl3): δ 8.28 (d, 3JH–H = 8.0 Hz; 1H), 8.18 (d, 3JH–H = 8.0 Hz; 1H), 8.13–7.95 (m, 9H), 7.84–7.69 (m, 5H), 7.66–7.60 (m, 4H), 7.58–7.21 (m, 18H), 7.17 (t, 3JH–H = 7.5 Hz; 1H), 7.01 (d, 3JH–H = 8.5 Hz; 1H), 6.61 (d, J = 8.5 Hz; 1H), 2.82 (s, N(CH3)2; 18H). 13C{1H} NMR (125.60 MHz, CDCl3): δ 183.67, 183.24, 183.22, 182.99, 160.18, 154.83, 154.27, 149.86, 144.79, 142.5, 141.81, 141.53, 140.36, 136.81, 135.26, 135.02, 134.48, 134.26, 133.78, 133.67, 133.38, 133.18, 132.96, 132.76, 131.34, 130.65, 130.53, 129.81, 129.36, 129.21, 129.11, 129.03, 128.95, 127.3, 126.85, 126.64, 124.32, 123.24, 123.19, 123.12, 120.50, 119.52, 115.41, 115.29, 38.72 (N(CH3)2). 19F NMR (469.86 MHz, CD3CN): δ −107.9 (s), −111.9 (s). Elemental analysis calculated (%) for C38H37FN3O4SSb: C, 59.07; H, 4.83; N, 5.44; found C, 58.62; H, 4.86; N, 5.33.
Fluoride ion complexation in water–CH2Cl2 biphasic mixture
In a typical experiment, a solution of 3 in CH2Cl2 (1.0 mL, 5.0 × 10−4 M) was layered with an aqueous solution (5 mL) containing TPABr as a phase transfer agent and a citrate buffer (10 mM, pH 4.68). In two separate experiment, 25 μL and 50 μL of a concentrated KF solution (0.02 M) were added to the aqueous layer, leading to a final fluoride concentration of 1.9 ppm (1.0 × 10−4 M) and 3.8 ppm (2.0 × 10−4 M), respectively. After shaking these mixtures for 5 minutes, the colors of the organic layer changed from yellow to pale orange for the solution containing 1.9 ppm of fluoride and to orange for the solution containing 3.8 ppm of fluoride. After separating the two layers, 50 μL aliquots of the CH2Cl2 were diluted to a total volume of 3 mL to make a 5 × 10−6 M solution. The UV-Vis spectra of these solutions were recorded and showed a redshift of the lowest energy absorption band from λmax = 423 nm (vs. 430 in dry CH2Cl2) to λmax = 475 nm (vs. 482 in dry CH2Cl2) (Fig. 5 and S7†). The fluorescence spectra were also recorded and showed an increase in a broad intensity band at λmax = 616 nm. Both UV-Vis and fluorescence measurements support the formation of [3–F]− which was also confirmed by recording the 19F NMR spectrum of the CH2Cl2 layer obtained with 3.8 ppm of fluoride. Analogous experiments with NaCl, NaBr, NaNO3, NaHCO3, NaH2PO4, NaHSO4 (4.0 × 10−4 M) did not result in a visible color change.Drinking water analysis
In a typical experiment, a 5 mm NMR tube was filled with a solution of 3 in CH2Cl2 (1.0 mL, 5.0 × 10−5 M) and layered with an aqueous solution containing TPABr (20 mM) and a citrate buffer (10 mM, pH 4.6). To obtain a calibration curve, the aqueous layer was doped with different amounts of fluoride (0, 0.4, 0.7, 1.0, and 1.9 ppm). After vigorous shaking (1 min), the tube was inserted into the cavity of the fluorometer such that only the CH2Cl2 layer was positioned in the optical path. The exact positioning of each tube in the cavity of the fluorometer was facilitated by the use of a custom made insert. The fluorescence intensity was recorded at λmax = 610 nm (λexcitation = 482 nm). A plot shows that the fluorescence intensity at λmax = 610 nm increases linearly with the fluoride concentration in the 0–1.9 ppm range (Fig. S8†). Drinking water samples (Nursery® Water, H-E-B® Baby Purified Water, and tap water of College Station) where combined with TPABr (20 mM) and buffered with citrate (10 mM, pH 4.6). The resulting solutions were transferred into a 5 mm NMR tube filled with a solution of 3 in CH2Cl2 (1.0 mL, 5.0 × 10−5 M). The fluorescence intensity was measured as described above and correlated to a fluoride concentration using the calibration described above. The measurements were reproduced two times.Computational details
Density functional theory (DFT) structural optimizations with the Gaussian 09 program.64 In all cases, the structures were optimized using the B3LYP functional65,66 and the following mixed basis set: Sb, aug-cc-pVTZ-PP;67 F, 6-31g(d′);68 C/O/H, 6-31g).69 Each structure was subsequently subjected to TD-DFT calculation using the B3LYP functional and the SMD implicit solvation model with CH2Cl2 as a solvent. The orbitals plotted in Fig. 1 and 3 as well as their energies are obtained from the TD-DFT output (with solvation). For all optimized structures, frequency calculations were carried out to confirm the absence of imaginary frequencies. The molecular orbitals were visualized and plotted in Jimp 2 program.70 The LUMO of the fluorenyl cation show in Fig. 1 was generated using the The Simple Huckel Molecular Orbital Theory Calculator program available at http://www.chem.ucalgary.ca/SHMO/.Crystallographic measurements
The crystallographic measurements were performed at 110(2) K using a Bruker APEX-II CCD area detector diffractometer, with a graphite-monochromated Mo-Kα radiation (λ = 0.71069 Å). A specimen of suitable size and quality was selected and mounted onto a nylon loop. The semi-empirical method SADABS was applied for absorption correction. The structure was solved by direct methods, which successfully located most of the non-hydrogen atoms. Subsequent refinement on F2 using the SHELXTL/PC package (version 6.1) allowed location of the remaining non-hydrogen atoms. All H-atoms were geometrically placed and refined using a standard riding model.Acknowledgements
This work was supported by the National Science Foundation (CHE-1300371), the Welch Foundation (A-1423), and Texas A&M University (Davidson Professorship, Laboratory for Molecular Simulation).Notes and references
- K. Sebelius, Fed. Regist., 2011, 76, 2383–2388 Search PubMed.
- B. E. Erickson, Chem. Eng. News DOI:10.1021/cen010611145659.
- T. A. D. Smith, J. Labelled Compd. Radiopharm., 2012, 55, 281–288 CrossRef CAS.
- M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC.
- Anion Coordination Chemistry, ed. K. Bowman-James, A. Bianchi and E. Garcia-Espana, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, p. 559 Search PubMed.
- P. A. Gale, Chem. Commun., 2011, 47, 82–86 RSC.
- S.-K. Kim and J. L. Sessler, Chem. Soc. Rev., 2010, 39, 3784–3809 RSC.
- S.-O. Kang, J. M. Llinares, V. W. Day and K. Bowman-James, Chem. Soc. Rev., 2010, 39, 3980–4003 RSC.
- P. Anzenbacher, Jr, Top. Heterocycl. Chem., 2010, 24, 205–235 Search PubMed.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- M. Cametti and K. Rissanen, Chem. Commun., 2009, 2809–2829 RSC.
- J. Yoon, S. K. Kim, N. J. Singh and K. S. Kim, Chem. Soc. Rev., 2006, 35, 355–360 RSC.
- J. L. Sessler, S. Camiolo and P. A. Gale, Coord. Chem. Rev., 2003, 240, 17–55 CrossRef CAS.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS.
- N. Chaniotakis, K. Jurkschat, D. Mueller, K. Perdikaki and G. Reeske, Eur. J. Inorg. Chem., 2004, 2283–2288 CrossRef CAS.
- I. H. A. Badr and M. E. Meyerhoff, J. Am. Chem. Soc., 2005, 127, 5318–5319 CrossRef CAS PubMed.
- M. Cametti, A. Dalla Cort, L. Mandolini, M. Nissinen and K. Rissanen, New J. Chem., 2008, 32, 1113–1116 RSC.
- C. A. D'Souza, W. J. McBride, R. M. Sharkey, L. J. Todaro and D. M. Goldenberg, Bioconjugate Chem., 2011, 22, 1793–1803 CrossRef CAS PubMed.
- Z. Liu, Y. Li, J. Lozada, P. Schaffer, M. J. Adam, T. J. Ruth and D. M. Perrin, Angew. Chem., Int. Ed., 2013, 52, 2303–2307 CrossRef CAS PubMed.
- R. Bhalla, C. Darby, W. Levason, S. K. Luthra, G. McRobbie, G. Reid, G. Sanderson and W. Zhang, Chem. Sci., 2014, 5, 381–391 RSC.
- F. Cheng, E. M. Bonder and F. Jäkle, J. Am. Chem. Soc., 2013, 135, 17286–17289 CrossRef CAS PubMed.
- H. Zhao, L. A. Leamer and F. P. Gabbai, Dalton Trans., 2013, 42, 8164–8178 RSC.
- T. S. De Vries, A. Prokofjevs and E. Vedejs, Chem. Rev., 2012, 112, 4246–4282 CrossRef CAS PubMed.
- C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed.
- T. W. Hudnall, C.-W. Chiu and F. P. Gabbaï, Acc. Chem. Res., 2009, 42, 388–397 CrossRef CAS PubMed.
- Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS PubMed.
- T. Agou, M. Sekine, J. Kobayashi and T. Kawashima, Chem. – Eur. J., 2009, 15, 5056–5062 CrossRef CAS PubMed.
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS PubMed.
- F. Dielmann, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 14071–14073 CrossRef CAS PubMed.
- L. J. Hounjet, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 4714–4717 CrossRef CAS PubMed.
- E. Conrad, N. Burford, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2009, 131, 17000–17008 CrossRef CAS PubMed.
- Y. Kim, T. W. Hudnall, G. Bouhadir, D. Bourissou and F. P. Gabbaï, Chem. Commun., 2009, 3729–3731 RSC.
- Y. Ren, W. H. Kan, V. Thangadurai and T. Baumgartner, Angew. Chem., Int. Ed., 2012, 51, 3964–3968 CrossRef CAS PubMed.
- J. Ohshita, S. Matsui, R. Yamamoto, T. Mizumo, Y. Ooyama, Y. Harima, T. Murafuji, K. Tao, Y. Kuramochi, T. Kaikoh and H. Higashimura, Organometallics, 2010, 29, 3239–3241 CrossRef CAS.
- T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed.
- Y. Matano and H. Imahori, Org. Biomol. Chem., 2009, 7, 1258–1271 CAS.
- M. Hissler, P. W. Dyer and R. Réau, Coord. Chem. Rev., 2003, 244, 1–44 CrossRef CAS.
- L. H. Bowen and R. T. Rood, J. Inorg. Nucl. Chem., 1966, 28, 1985–1990 CrossRef CAS.
- M. Jean, Anal. Chim. Acta, 1971, 57, 438–439 CrossRef CAS.
- C. R. Wade and F. P. Gabbaï, Organometallics, 2011, 30, 4479–4481 CrossRef CAS.
- C. R. Wade, I.-S. Ke and F. P. Gabbaï, Angew. Chem., Int. Ed., 2012, 51, 478–481 CrossRef CAS PubMed.
- T.-P. Lin, R. C. Nelson, T. Wu, J. T. Miller and F. P. Gabbaï, Chem. Sci., 2012, 3, 1128–1136 RSC.
- C. R. Wade, T.-P. Lin, R. C. Nelson, E. A. Mader, J. T. Miller and F. P. Gabbaï, J. Am. Chem. Soc., 2011, 133, 8948–8955 CrossRef CAS PubMed.
- H. Zhao and F. P. Gabbaï, Nat. Chem., 2010, 2, 984–990 CrossRef CAS PubMed.
- I.-S. Ke, M. Myahkostupov, F. N. Castellano and F. P. Gabbaï, J. Am. Chem. Soc., 2012, 134, 15309–15311 CrossRef CAS PubMed.
- M. Shindo and R. Okawara, Inorg. Nucl. Chem. Lett., 1969, 5, 77–80 CrossRef CAS.
- M. Hall and D. B. Sowerby, J. Am. Chem. Soc., 1980, 102, 628–632 CrossRef CAS.
- G. K. Fukin, L. N. Zakharov, G. A. Domrachev, A. Y. Fedorov, S. N. Zaburdyaeva and V. A. Dodonov, Russ. Chem. Bull., 1999, 48, 1722–1732 CrossRef CAS.
- V. A. Dodonov, A. Y. Fedorov, G. K. Fukin, S. N. Zaburdyaeva, L. N. Zakharov and A. V. Ignatenko, Main Group Chem., 1999, 3, 15–22 CrossRef CAS.
- R. R. Holmes, R. O. Day, V. Chandrasekhar and J. M. Holmes, Inorg. Chem., 1987, 26, 157–163 CrossRef CAS.
- S. Solé and F. P. Gabbaï, Chem. Commun., 2004, 1284–1285 RSC.
- M. Melaïmi and F. P. Gabbaï, J. Am. Chem. Soc., 2005, 127, 9680–9681 CrossRef PubMed.
- The [Ph3SiF2]−anion as a tetrabutyl ammonium salt undergoes full dissociation when dissolved in 7/3 vol. THF–H2O solution.
- G. J. Krüger, C. W. F. T. Pistorius and A. M. Heyns, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 2916–2918 CrossRef.
- S. Martinez-Vargas, P. Gomez-Tagle and A. K. Yatsimirsky, Inorg. Chim. Acta, 2011, 373, 226–232 CrossRef CAS PubMed.
- S.-i. Shoda, K. Shintate, M. Ishihara, M. Noguchi and A. Kobayashi, Chem. Lett., 2007, 16–17 CrossRef CAS.
- R. S. Sathish, M. R. Kumar, G. N. Rao, K. A. Kumar and C. Janardhana, Spectrochim. Acta, Part A, 2007, 66, 457–461 CrossRef PubMed.
- Y. Kubo, T. Ishida, T. Minami and T. D. James, Chem. Lett., 2006, 35, 996–997 CrossRef CAS.
- Y. Kubo, A. Kobayashi, T. Ishida, Y. Misawa and T. D. James, Chem. Commun., 2005, 2846–2848 RSC.
- Y. Kubo, T. Ishida, A. Kobayashi and T. D. James, J. Mater. Chem., 2005, 15, 2889–2895 RSC.
- C. R. Wade and F. P. Gabbaï, Dalton Trans., 2009, 9169–9175 RSC.
- N. DiCesare and J. R. Lakowicz, Anal. Biochem., 2002, 301, 111–116 CrossRef CAS PubMed.
- We found that tetrapropylammonium bromide is a better choice than tetramethyl- and tetraethyl-ammonium bromide which do not efficiently support fluoride phase transfer. We also observed that tetra-n-butylammonium bromide is too lipophilic and promotes uncontrolled hydroxide transfer to the organic phase, leading to neutralization of the Lewis acidic receptor.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- K. A. Peterson, J. Chem. Phys., 2003, 119, 11099–11112 CrossRef CAS PubMed.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS PubMed.
- J. Manson, C. E. Webster, L. M. Pérez and M. B. Hall, http://www.chem.tamu.edu/jimp2/index.html.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental and computational details. Crystallographic data in cif format. CCDC 984280–984282. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc00343h |
| This journal is © The Royal Society of Chemistry 2014 |