 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of a spin-coupled Mo(III) in the nitrogenase iron–molybdenum cofactor†
Ragnar
Bjornsson‡
a,
Frederico A.
Lima‡§
a,
Thomas
Spatzal¶
b,
Thomas
Weyhermüller
a,
Pieter
Glatzel
c,
Eckhard
Bill
a,
Oliver
Einsle
*b,
Frank
Neese
*a and
Serena
DeBeer
*ad
aMax-Planck-Institut für Chemische Energiekonversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: serena.debeer@cec.mpg.de
bInstitute for Biochemistry, Albert-Ludwigs-Universität Freiburg, Albertstrasse 21, 79104 Freiburg, Germany
cEuropean Synchrotron Radiation Facility, Boĩte Postale 220, 38043 Grenoble Cedex, France
dDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA
First published on 9th April 2014
Abstract
Nitrogenase is a complex enzyme that catalyzes the formation of ammonia utilizing a MoFe7S9C cluster. The presence of a central carbon atom was recently revealed, finally completing the atomic level description of the active site. However, important prerequisites for understanding the mechanism – the total charge, metal oxidation states and electronic structure are unknown. Herein we present high-energy resolution fluorescence detected Mo K-edge X-ray absorption spectroscopy of nitrogenase. Comparison to FeMo model complexes of known oxidation state indicates that the Mo in the FeMo cofactor of nitrogenase is best described as Mo(III), in contrast to the universally accepted Mo(IV) assignment. The oxidation state assignment is supported by theoretical calculations, which reveal the presence of an unusual spin-coupled Mo(III) site. Although so far Mo(III) was not reported to occur in biology the suggestion raises interesting parallels with the known homogenous Mo catalysts for N2 reduction, where a Mo(III) compound is the N2-binding species. It also requires a reassignment of the Fe oxidation states in the cofactor.
Introduction
Nitrogenase is the enzyme responsible for the catalytic reduction of dinitrogen (N2) to ammonia. In contrast to the industrial Haber–Bosch process that reacts N2 and H2 at high temperature and pressure, the biological reduction proceeds at ambient temperature and pressure using a complex multicomponent protein system, which utilizes eight electrons, eight protons and 16 MgATP molecules.1,2 Unraveling the structural and mechanistic details of how nature activates the strong bond of dinitrogen is of fundamental importance and this knowledge could aid in the design of better catalysts.The molybdenum–iron component (MoFe) of nitrogenase of Azotobacter vinelandii includes two types of complex metal–clusters, the “P-cluster” and the FeMo cofactor (FeMoco). The P-cluster serves as an electron transfer site, while FeMoco is generally agreed to be the site of dinitrogen reduction. The FeMo cofactor consists of 7 irons, 1 molybdenum, 9 sulfides and an interstitial light atom that was recently identified as carbon by XES, ESEEM and high resolution crystallography.3,4 With the identity of the interstitial atom clear, the basic molecular structure of the cofactor can at last be considered complete. However, many questions remain about the resting form of the enzyme – namely the total charge of the cofactor, the oxidation state distribution and the electronic structure. Yet, this information is essential for any informed discussion of the molecular level mechanism of dinitrogen reduction. It is also essential knowledge for any rationally designed, bio-inspired catalysts.
Presently, three oxidation state assignments for FeMoco are discussed in the literature: (i) 6Fe(II)1Fe(III)Mo(IV);5 (ii) 4Fe(II)3Fe(III)Mo(IV);6 and (iii) 2Fe(II)5Fe(III)Mo(IV).7 These can alternately be written in terms of total charge on the cluster when sulfur and carbon are taken in their usual closed-shell form S2− and C4−, thus giving [MoFe7S9C]3−, [MoFe7S9C]1− and [MoFe7S9C]1+, respectively. For all of these oxidation state assignments, one can arrive at a total spin of S = 3/2, consistent with EPR spectroscopy.8,9 Interestingly, all of these assignments assume a closed-shell diamagnetic Mo(IV).
The Mo(IV) assignment in the FeMo cofactor is based on early Mo K-edge XAS studies and 95Mo ENDOR. As the molybdenum atom can be probed directly by either of these methods, it serves as an important starting point for understanding the complex electronic structure of the cluster. The initial XAS studies were carried out in the 1970s and 1980s, before the structure of the cofactor was known. Based on Mo–S derived bond lengths from the EXAFS region, an oxidation state of either Mo(III) or Mo(IV) was suggested.10,11 The absorption edge positions did not give a clear indication of the Mo oxidation state, largely due to the significant core hole lifetime broadening at high energies. Later, 95Mo ENDOR experiments12–14 indicated a small Mo hyperfine coupling in the protein and these data were interpreted as the molybdenum most plausibly being a closed-shell Mo(IV) rather than a S = 3/2 Mo(III) or S = 1/2 Mo(V). However, the authors acknowledged that the spin coupling schemes utilized were highly simplified and this assignment was not definitive.13 In 1988, a follow up Mo L-edge XAS study15 assigned the Mo atom in nitrogenase as Mo(IV), when taking the ENDOR data into account. However, a quantitative analysis of Mo L-edge data was prohibitive due to the proximity of the sulfur K-edge and the inadequacy of the available theoretical tools at that time. From this point on, the Mo(IV) oxidation state appeared to be set in the literature, despite the fact that the authors of both the XAS and ENDOR studies acknowledged the limitations of their methods.
In the present study, we directly address the question of the Mo oxidation state in FeMoco by using high-energy resolution fluorescence detected X-ray absorption spectroscopy (HERFD-XAS) as a means to overcome standard XAS limitations. K-edge HERFD-XAS detects fluorescent photons resulting from 2p to 1s radiative decay as a function of the incident X-ray energy scanned across an absorption edge.16,17 When this type of measurement is carried out the resultant spectra have greatly improved energy resolution (from ∼6–8 eV in standard XAS to ∼3.5 eV in the present study). Hence, HERFD-XAS at the Mo K-edge allows for more richly featured near-edge spectra than traditional XAS measurements, enabling the quantitative interpretation of these data using DFT calculations, as recently demonstrated by a calibration study.18
In this work, Mo HERFD-XAS is applied to the MoFe protein of nitrogenase and to a series of FeMo model complexes. These include the dimeric model complexes (Et4N)2[Cl2Fe(μ-S)2MoO2] (1),19 (Et4N)2[Cl2Fe(μ-S)2Mo(O)3(Cl4Cat)] (2),19 the cubane complex (Et4N)[(Tp)MoFe3S4Cl3] (3),20 and the double cubane model (Bu4N)2[[(Tp)MoFe3S4Cl3]2(μ-S)] (4).20 A schematic representation of these model compounds is given in the ESI (Fig. S1†). The structure of complex (3) is very similar to the half cubane containing the Mo atom in the FeMo cofactor of nitrogenase. A comparison of the structure of complex 3 and the FeMo cofactor is given in Fig. 1.
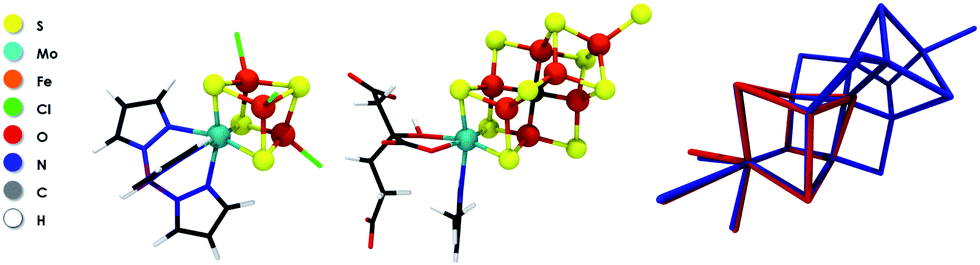 | ||
| Fig. 1 Molecular structures of 3 (left), FeMoco (center) and an overlay of the two experimental X-ray structures (right). | ||
These model complexes provide an excellent basis for an oxidation state assignment of FeMoco since they all represent Fe/S/Mo systems with known Mo oxidation states as derived through analysis of 57Fe Mössbauer data. Hence, from this information and the total charge of the compounds, as well as X-ray diffraction data, the dimeric complexes are assigned as Mo(V),21 while the cubane compounds contain Mo(III).20,22 For [MoFe3S4]3+ cores, as in 3 and 4, the Mo(III) assignment is further supported by theory.23
In this work, we show that the detailed comparison of protein and model Mo-HERFD XAS data together with the results of density functional theory (DFT) calculations requires a revised oxidation state assignment for molybdenum in the resting state of FeMoco. Further, we demonstrate that the Mo(III) assignment holds for any of the proposed [MoFe7S9C]3−, [MoFe7S9C]1− or [MoFe7S9C]1+ total charges on the cluster. These results thus support Fe-based redox over this four electron series and suggest that a Mo(III) site could be mechanistically relevant.
Results and discussion
Fig. 2 shows a comparison of the normalized Mo K-edge HERFD-XAS data for compounds 1–4 and the intact MoFe protein. As the only Mo present in the MoFe protein is associated with FeMoco, the spectrum corresponds to a unique Mo site. Compounds 1 and 2 clearly have the most intense pre-edges (at ∼20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 003.6 and 20004.0 eV) and the highest energy rising edges (at 20009.9 and 20011.5 eV), respectively, consistent with the tetrahedral local geometry and a Mo(V) oxidation state assignment. Complexes 3 and 4 have weaker pre-edge intensities, consistent with the octahedral local site symmetry and lower energy rising edges (at 20009.1 and 20008.7 eV), consistent with a two electron reduction to Mo(III). Interestingly, the Mo HERFD-XAS data for the MoFe protein is very similar to that of 3 and 4, suggesting a similar electronic structure. The pre-edge appears at approximately the same energy (20002.1 eV) and the rising edge is similar in energy, albeit slightly lower at 20008.4 eV. This suggests a Mo(III) oxidation state in FeMoco, and is clearly inconsistent with the previous assignment of Mo(IV), in which case the pre-edge and rising edge positions should be observed at higher energies.
003.6 and 20004.0 eV) and the highest energy rising edges (at 20009.9 and 20011.5 eV), respectively, consistent with the tetrahedral local geometry and a Mo(V) oxidation state assignment. Complexes 3 and 4 have weaker pre-edge intensities, consistent with the octahedral local site symmetry and lower energy rising edges (at 20009.1 and 20008.7 eV), consistent with a two electron reduction to Mo(III). Interestingly, the Mo HERFD-XAS data for the MoFe protein is very similar to that of 3 and 4, suggesting a similar electronic structure. The pre-edge appears at approximately the same energy (20002.1 eV) and the rising edge is similar in energy, albeit slightly lower at 20008.4 eV. This suggests a Mo(III) oxidation state in FeMoco, and is clearly inconsistent with the previous assignment of Mo(IV), in which case the pre-edge and rising edge positions should be observed at higher energies.
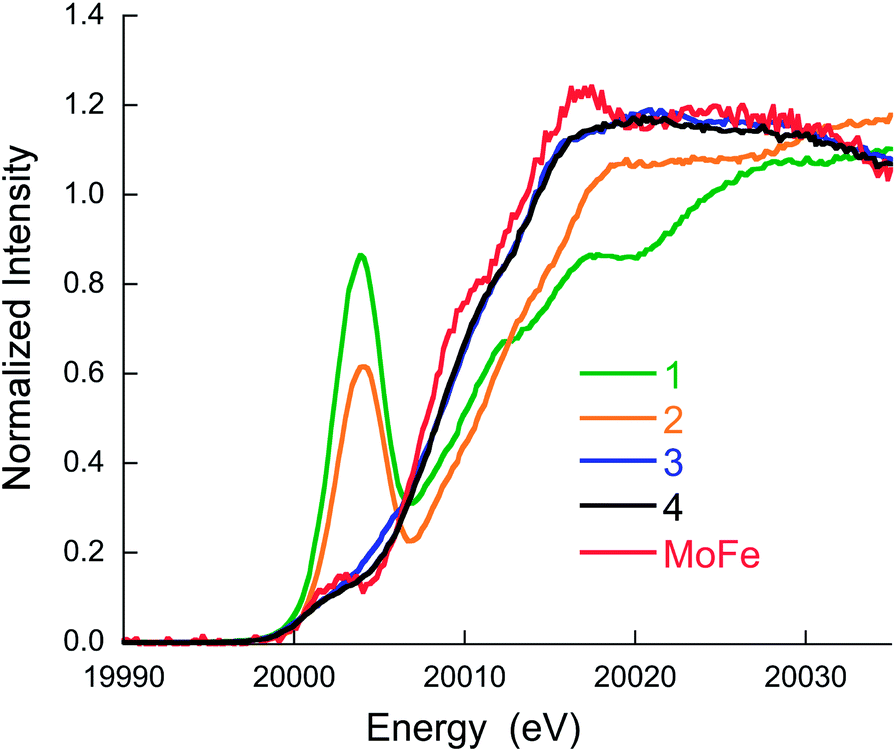 | ||
| Fig. 2 Mo K-edge HERFD-XAS data for compounds 1–4 and MoFe protein. | ||
In order to obtain more quantitative insight into the electronic structure of the model compounds and the MoFe protein, we tested the ability of our previously established time-dependent density functional theory (TD-DFT) protocol for the calculation of Mo K-pre-edge spectra of heterometal clusters. Calculations on the model systems were performed on the anions in a polarizable continuum, while calculations of FeMoco were performed on a large 225 atom cluster model, consisting of the metal–sulfur cluster and a part of the nearest protein environment. An illustration and xyz coordinates of the FeMoco cluster model used in the calculations is shown in the ESI.† As the charge of FeMoco is not confidently known, we considered the 3 possibilities: [MoFe7S9C]3−, [MoFe7S9C]1− and [MoFe7S9C]1+.
A comparison of the experimental and calculated pre-edges is shown in Fig. 3A and B, respectively. It is evident from Fig. 3B that the calculations reproduce the energies and intensities of the experimental spectra with excellent accuracy. Fig. S9† shows the energy and intensity correlations between experiment and theory for a total of 14 model complexes studied by Mo HERFD (8 from a previous study and an additional 6 from the current study, full details are provided in the ESI†). R-values of 0.95 and 0.98 are obtained for the energy and intensity correlations, respectively. Furthermore, the calculations demonstrate that the Mo K-pre-edge spectra are not sensitive to the assumed total charge of FeMoco. Hence, the total charge on the FeMo cofactor cannot be unambiguously assigned on the basis of these data.
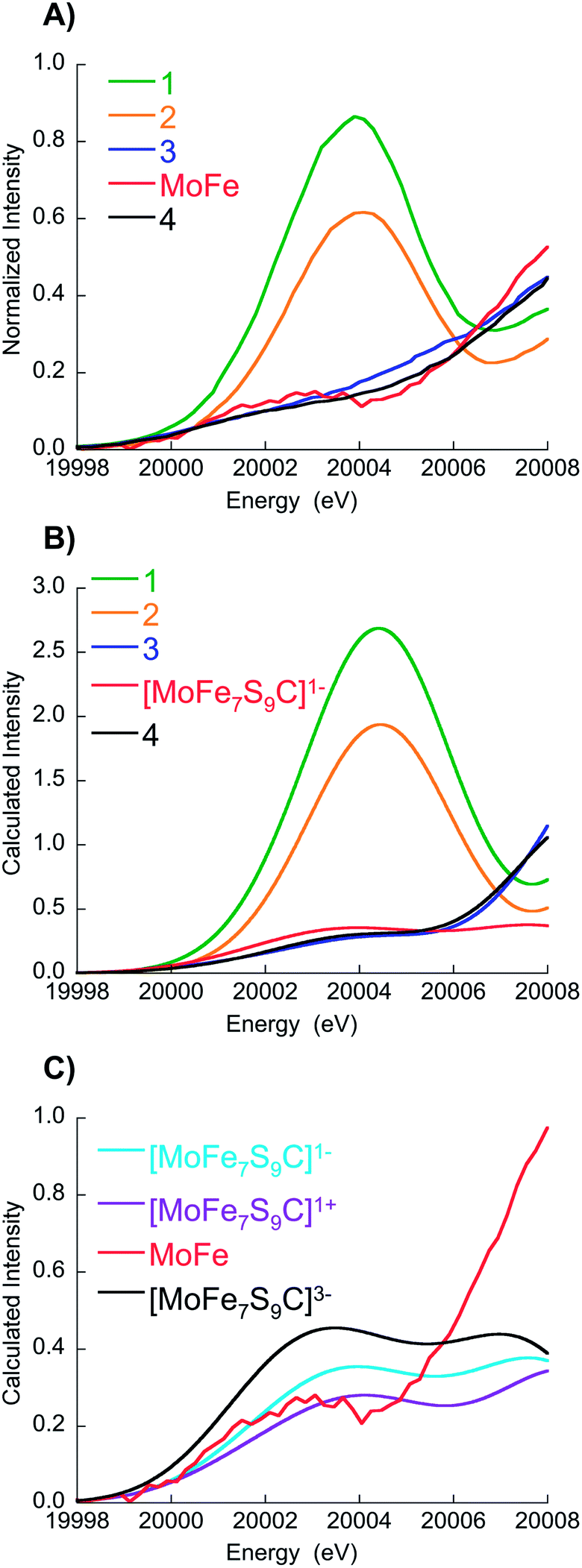 | ||
| Fig. 3 (A) Expansion of experimental data over the pre-edge region, (B) corresponding TD-DFT calculations for 1–4 and [MoFe7S9C]1− (C) experimental MoFe protein data vs. FeMoco calculations in three different charge states. Note in (C), the experimental MoFe data have been scaled by a factor of 1.85 in order to facilitate comparison with the calculated spectra. This scaling factor was established in a previous Mo HERFD XAS calibration study (see ref. 18 for details). | ||
Importantly, the strong correlation between the experimental and computed spectra demonstrates the suitability of our protocol to obtain more insight into the nature of the Mo oxidation state of FeMoco. However, since the electronic structure of FeMoco is extremely complex, it turns out to be most revealing to first analyze the electronic structure of 3, the closest structural analogue of FeMoco. In fact, the root-mean-square-deviation (RMSD) for the Fe, Mo, S/C and O/N/C atoms is ∼0.15 Å (Fig. 1), demonstrating that 3 is a reasonable structural analogue for the molybdenum coordination environment in FeMoco. Furthermore, both, the experimental and calculated XAS edges indicate strong electronic similarities (Fig. 3).
Iron–sulfur clusters are known to have valence-delocalized electronic structures involving complex spin couplings.24 Hence, in order to obtain clean reference points for the analysis we have studied a hypothetical model of 3 where the iron ions were substituted by closed-shell diamagnetic Ga3+ ions, but without relaxing the structure. This in silico experiment allows one to study the properties of the Mo(IV) or Mo(III) state in the ‘native’ coordination environment without any complications arising from spin coupling. The analysis of the (canonical or localized) Kohn–Sham orbitals obtained for the singlet state of [Mo(IV)Ga3S4]5+ (4d2) demonstrates spin pairing in the t2g-shell of molybdenum while the triplet state has 2 t2g orbitals occupied as expected. The lowest energy Mo XAS transitions (Mo 1s → 4d t2g) from unrestricted TD-DFT calculations confirm the closed-shell d2 configuration of a singlet Mo(IV) as 4 transitions to the t2g shell are observed (and α and β transitions are of almost the same energy) as shown in Fig. 4. The triplet [Mo(IV)Ga3S4]5+ on the other hand shows 4 unequal transitions (1α and 3β), consistent with the 4d t2g configuration expected for a triplet. The triplet [Mo(IV)Ga3S4]5+ is 3.6 kcal mol−1 lower in energy than the singlet (consistent with Hund's rule for an octahedral ion). A calculation on the lowest MS = 3/2 state of [Mo(III)Ga3S4]4+ (quartet 4d3) results in single occupation of all 3 t2g orbitals (↑↑↑). The Mo-dxy based MO is 0.4 eV lower in energy than the almost degenerate dxz and dyz-based set. Interestingly, the lowest MS = 1/2 state of [Mo(III)Ga3S4]4+, features a configuration without spin-pairing. Careful inspection reveals that all Mo-t2g based orbitals are singly occupied; the dxy-based MO is 0.19 eV lower in energy than dxz and dyz. The MS = 1/2 state of [Mo(III)Ga3S4]4+ arises as one electron-spin is aligned oppositely to the others, i.e. (↑↑↓). This state is merely 5.9 kcal mol−1 higher in energy than the MS = 3/2 state. These results are consistent with the d3 Tanabe–Sugano diagram where a 2E state, involving no spin pairing, is the lowest excited state for medium to strong ligand fields.
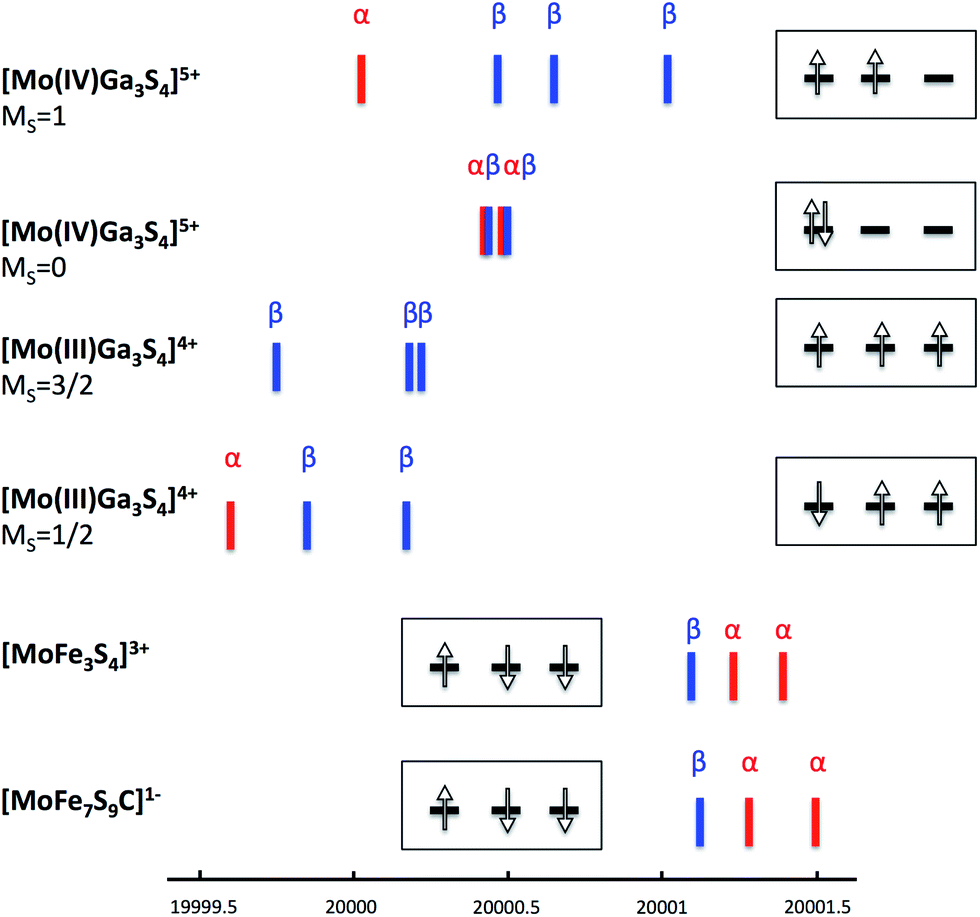 | ||
| Fig. 4 TD-DFT calculated Mo 1s → 4d t2g transitions for [Mo(IV)Ga3S4]5+ (MS = 1 and MS = 0), [Mo(III)Ga3S4]4+ (MS = 3/2 and MS = 1/2), [MoFe3S4]3+ complex 3 and FeMoco model [MoFe7S9C]1−. Insets show the 4d t2g electron configuration for each compound based on the TD-DFT-calculated 4d transitions (the minor splitting of the orbitals is ignored for simplicity). | ||
Adding back in the irons, i.e., calculating the real [MoFe3S4]3+ system, 3, results in a much more complicated electronic structure as spin coupling between all four open-shell metal atoms occurs. Starting from several possible high-spin solutions and performing spin flips on individual centers, we find the lowest state of the whole four-spin system to be a broken-symmetry solution with total MS = 3/2 (consistent with the experimentally observed S = 3/2 spin state22 of the full system).
In this state, the local spin on one iron is aligned antiparallel to the other iron spins. Curiously, considerable spin population (−0.6 unpaired electrons) on the molybdenum is found, yet lower than expected for hypothetically isolated Mo(III) with local spin S = 3/2 (and higher than expected for Mo(IV), S= 0). A localized orbital analysis reveals three singly occupied t2g orbitals on the molybdenum, clearly indicating Mo(III) (see Fig. 5). However, surprisingly, these singly occupied orbitals demonstrate the same type of configuration as in the previously described MS = 1/2 state in the Ga-substituted cluster (1 α and 2 β spin orbitals in this case, i.e. ↑↓↓). This either suggests that the Mo(III) ion in [MoFe3S4]3+ has a non-Hund ground state with S = 1/2, arising due to strong coupling to the irons (but without a doubly occupied t2g orbital), or possibly the spin coupling is even more complex owing to canting of all the metal spins involved. The orbital analysis also reveals electron delocalization between two iron atoms with parallel majority spin alignment, thus indicating a mixed-valence delocalized Fe(II)–Fe(III) pair, a common feature in iron–sulfur chemistry.24–26 Despite the delocalization present, a formal oxidation state assignment of Mo(III)–Fe(III)2Fe(II) seems appropriate.
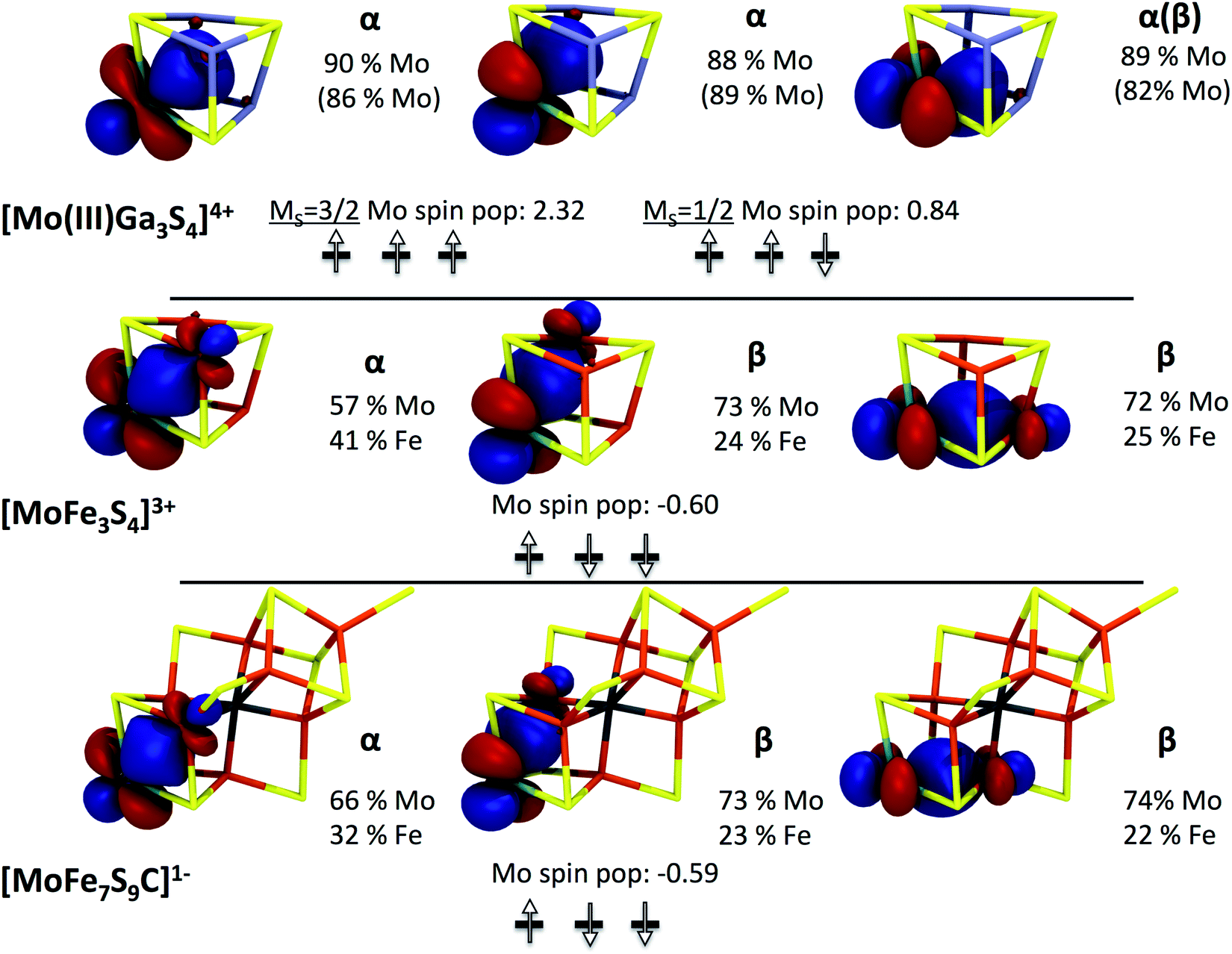 | ||
| Fig. 5 Occupied localized t2g orbitals of [Mo(III)Ga3S4]4+ in MS = 3/2 and MS = 1/2 states, [MoFe3S4]3+ complex 3 and FeMoco [MoFe7S9C]1− (calculations for [MoFe7S9C]3− and [MoFe7S9C]1+ show very similar Mo orbitals). Orbital compositions and Mulliken spin populations on Mo are shown as well (orbital compositions in parentheses refer to the MS = 1/2 solution of [Mo(III)Ga3S4C]4+). | ||
In order to substantiate this electronic structure description the Mössbauer parameters of 3 were calculated using previously established protocols27,28 (see ESI†). Mössbauer spectra of 3 show two distinct signals with an intensity ratio of 1:2 that are characterized by isomer shifts of δ = 0.46 and 0.51 mm s−1 and quadrupole splittings of ΔEQ of |0.61| and |1.09| mm s−1 respectively.20 These signals can be plausibly assigned to a single high-spin Fe(III) ion and a delocalized Fe(2.5)–Fe(2.5) mixed-valence pair, in agreement with our electronic structure calculations as well as earlier results.23 Our calculated isomer shifts for the three iron ions are 0.36, 0.45 and 0.45 mm s−1 respectively and the calculated ΔEQ values are 0.66, 1.26 and 1.25 mm s−1, in good agreement with experiment. The iron atoms with the calculated higher δ and ΔEQ values are the irons forming the mixed-valence delocalized pair. Magnetic Mössbauer measurements of similar [MoFe3S4]3+ core compounds have additionally revealed antiparallel alignment of the two iron species,29,30 in agreement with the broken-symmetry solution.
TD-DFT computed Mo XAS transitions are consistent with the quasi non-Hund configuration at the molybdenum in 3 (see Fig. 4). However, all transitions are shifted to higher energies by ∼1.5 eV compared to the Mo(III)Ga3S4 cluster, which indicates a strong Mo/Fe interaction in 3. In fact, the localized orbital analysis reveals the existence of bonding orbitals that are significantly shared (see Fig. 5) between Mo and Fe ions. This is suggestive of the formation of a partial Mo–Fe heterometal bond, or, in other words, of a double-exchange effect in 3. The double-exchange effect is a well-known phenomenon in iron–sulfur clusters,24–26 however, it has not previously been suggested between molybdenum and iron. Thus, these results strongly suggest that an effective Hamiltonian describing Mo–Fe cubane systems has to account for both Heisenberg exchange and double exchange effects between molybdenum and iron.
To summarize the electronic structure analysis of the model compound 3, we conclude that the molybdenum is indeed a Mo(III) as has been proposed for [MoFe3S4]3+ cores, but has a quasi non-Hund (↑↓↓) configuration, somehow induced by the spin coupling in the complex. The latter may be described as an excited atomic state of an octahedrally coordinated Mo(III) ion, which is forced to be the lowest state through the spin coupling between molybdenum and iron or alternatively arises due to a more complex spin canting mechanism. We note that non-Hund states have previously been proposed to explain non-Heisenberg behaviour in magnetically coupled systems.31 An important question is now whether this curious electronic structure established for 3 is also found in FeMoco.
Among the different total charge states investigated for our model of FeMoco, we focus here on the [MoFe7S9C]1− results. The analogous calculations for [MoFe7S9C]3− and [MoFe7S9C]1+ lead to very similar results which have been collected in the ESI.† The lowest energy solution found belongs to a broken-symmetry state with total MS = 3/2. A qualitatively similar Fe spin coupling pattern has been previously reported by Noodleman and co-workers32,33 and has been referred to as BS-7. Importantly, analogous to what has been found for the model compound 3, significant spin population is found on the molybdenum (∼−0.6 unpaired electrons) and a careful investigation of the localized orbitals of [MoFe7S9C]1− reveals closely analogous electronic structure (Fig. 5). Thus, FeMoco features a similar pattern involving electron delocalization between Mo and Fe and the same putative non-Hund configuration (↑↓↓) at the molybdenum center. This result also nicely explains why the calculated Mo K-pre-edge spectra of 3 and FeMoco are highly similar. In fact, inspection of the transitions that contribute to the calculated pre-edge features reveals an almost identical series of 1s → 4d t2g transitions in 3 and in [MoFe7S9C]1− (Fig. 4).
On the basis of this analysis we conclude that an almost identical electronic structure around the molybdenum is present in FeMoco and in the model compound 3. Thus, the molybdenum in FeMoco is best described as consisting of a strongly spin-coupled Mo(III) center. This description holds irrespective of the assumed charge state of FeMoco.
It is interesting to further explore the electronic structure similarities by focusing on the iron centers of FeMoco. Analysis of the localized orbitals of [MoFe7S9C]1− indicates a delocalized mixed-valence Fe(2.5)–Fe(2.5) pair in the [MoFe3S3C]1+ part of the cluster. This implies that the [MoFe3S4]3+ and [MoFe3S3C]1+ cores can all be described by the generalized spin-coupling diagram shown in Fig. 6. Importantly, since the same electronic configuration on Mo is found for all three possible charge states, it follows that all redox events must be primarily iron centered. However, what is not understood is the nature of the quasi non-Hund d3 configuration at molybdenum that we obtain from the broken-symmetry DFT calculations or the overall spin coupling in either the [MoFe3S4]3+ or the [MoFe7S9C]n cores. Provisionally, one might arrive at a S = 3/2 total spin ground state of the cubane by first antiferromagnetically coupling the mixed-valence pair with S = 9/2 to a high-spin Fe(III) ion to give an intermediate spin of S = 2 which then engages in the strong interaction with the S = 1/2 state at the molybdenum to give the final S = 3/2 result. But it is also possible that spin canting plays a major role. In the actual FeMoco the situation must be even more complex due to the presence of the other four iron ions and the central carbon. This important subject will be addressed in detail in future studies. Until the spin coupling is understood in detail, a quantitative interpretation of the Mo hyperfine coupling is unfortunately not possible.13
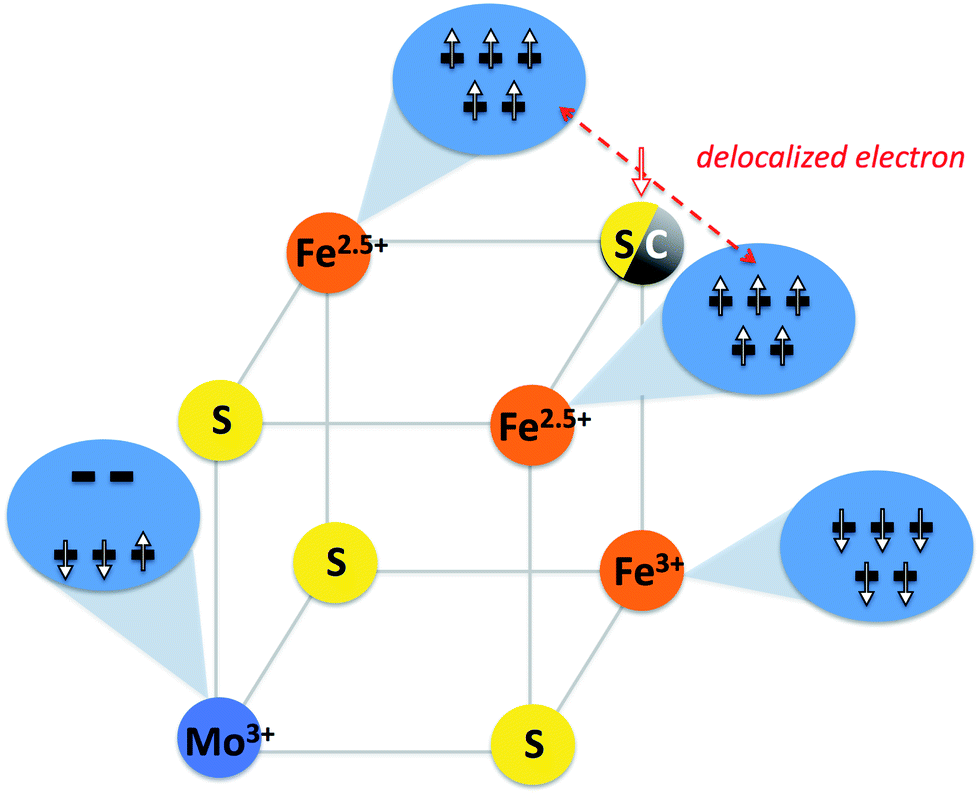 | ||
| Fig. 6 Provisional spin coupling diagram for [MoFe3S3C]1+ and [MoFe3S4]3+ cubanes based on broken-symmetry DFT calculations. Top iron atoms couple ferromagnetically and form a mixed-valence delocalized pair which is antiferromagnetically coupled to a high-spin Fe(III). The Mo(III) ion, here shown in a provisional non-Hund doublet state, couples to the irons to give a ground state spin of S = 3/2 for the cubane. | ||
We note that despite the novelty of a Mo(III) assignment, our calculations do not appear to be inconsistent with previous DFT studies on nitrogenase.7,32–35 The work of Noodleman & Case, Dance and Szilagyi revealed non-zero spin populations on the Mo atom, which are in fact inconsistent with a diamagnetic Mo(IV) assignment. At the time these studies were carried out, however, the primary focus was on the identity of the central atom and a detailed evaluation of the Mo oxidation state assignment was not undertaken. Nonetheless, we note that the reported Mo spin populations vary (from 0.02 to 0.6). These differences can largely be attributed to the amount of Hartree–Fock exchange (HF) in the used functionals, with the lowest Mo spin populations arising when pure functionals are employed. Regardless of the absolute value of the Mo spin population, however, we find that the electronic structure remains qualitatively the same. A decrease in the spin population (when using pure functionals) does not indicate a closed-shell Mo(IV) in our calculations of FeMoco (or 3), but arises instead due to the differing amount of Mo–Fe delocalization of the Mo electrons in the non-Hund configuration. Spin-pairing, as required by a closed-shell Mo(IV) configuration is never observed. A localized orbital analysis of [MoFe7S9C]1− calculated with different functionals (resulting in different Mo spin populations), support this view, as shown in the ESI.†
The interpretation of FeMoco as containing a Mo(III) evidently requires a reevaluation of the iron oxidation state assignments commonly found in the literature. In order to be consistent with the S = 3/2 ground state spin, the previously proposed FeMoco electronic structure have to be reassigned as follows:
| 6Fe(II)1Fe(III)Mo(IV) → 5Fe(II)2Fe(III)Mo(III) |
| 4Fe(II)3Fe(III)Mo(IV) → 3Fe(II)4Fe(III)Mo(III) |
| 2Fe(II)5Fe(III)Mo(IV) → 1Fe(II)6Fe(III)Mo(III) |
Thus, the present results suggests that a reevaluation of the magnetic Mössbauer data of the MoFe protein6 is necessary. It also highlights the inherent complexity of analyzing the Mössbauer data from many contributing irons in the complex delocalized electronic structure of FeMoco, especially without complete knowledge of its total charge and structure. A detailed reevaluation of these data is the subject of ongoing studies.
We note that a molybdenum(III) assignment in an enzyme cofactor is unprecedented in biology. Prior to the present study, molybdenum active sites in biology had been assigned as Mo(IV), Mo(V) or Mo(VI).36 Interestingly, a low-valent Mo active site raises parallels with the known Mo homogeneous catalysts for nitrogen reduction: the Schrock37 and Nishibayashi38 catalysts. In the more well-characterized Schrock system,39–41 it is the Mo(III) state that binds nitrogen and becomes part of a redox cycle leading to the formation of 2 equivalents of NH3. The discovery that the resting state of the nitrogenase enzyme contains a cofactor with a Mo(III) ion raises the question of whether the Mo site may play a direct role in the mechanism and this is currently being explored. It is well known that the resting state of the enzyme does not bind N2 but rather a 3- or 4-electron reduced state. Our computations have revealed that adding electrons to FeMoco generally does not result in reduction of the Mo ion. Experimental results42,43 also suggests the Mo ion remains at the same oxidation state when the cofactor is reduced. The reduction most likely takes place at the Fe ions and Fe-bound hydrides have also been suggested as carriers of the added electrons.44 The N2-binding site remains a subject of intense debate44–47 but it does not seem inconceivable that a Mo(III) ion, bound to the electron reservoir, could be the site of N2 reduction and we consider this important question far from settled.
Conclusions
Using a combined experimental and theoretical approach we have demonstrated that the molybdenum atom in the iron–molybdenum cofactor of Mo-dependent nitrogenase is best described as a Mo(III) coupled to the iron atoms in the cofactor. This is in sharp contrast to the previous description of the molybdenum as a closed-shell Mo(IV). Crucial to this oxidation state assignment was utilizing high-energy resolution fluorescence detected Mo XAS as well as a direct comparison of the MoFe protein with synthetic [MoFe3S4]3+ model compounds. The electronic structure of the FeMo cofactor, however, is still not fully understood. Understanding the spin coupling between not only the irons but also molybdenum and irons will be an important topic of future studies. Similarly, understanding the effect of the interstitial carbon atom on the electronic structure remains an open question. Ultimately a rigorous understanding of the molecular and electronic structure should aid in uncovering the basis of biological nitrogen reduction.Acknowledgements
The authors acknowledge the European Synchrotron Radiation Facility (ESRF) and the ID26 staff for the technical assistance during the experiments. SD and FN acknowledge the Max Planck Society for funding.Notes and references
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983–3012 CrossRef CAS PubMed.
- J. B. Howard and D. C. Rees, Chem. Rev., 1996, 96, 2965–2982 CrossRef CAS PubMed.
- K. M. Lancaster, M. Roemelt, P. Ettenhuber, Y. Hu, M. W. Ribbe, F. Neese, U. Bergmann and S. DeBeer, Science, 2011, 334, 974–977 CrossRef CAS PubMed.
- T. Spatzal, M. Aksoyoglu, L. Zhang, S. L. A. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Science, 2011, 334, 940 CrossRef CAS PubMed.
- H.-I. Lee, B. J. Hales and B. M. Hoffman, J. Am. Chem. Soc., 1997, 119, 11395–11400 CrossRef CAS.
- S. J. Yoo, H. C. Angove, V. Papaefthymiou, B. K. Burgess and E. Münck, J. Am. Chem. Soc., 2000, 122, 4926–4936 CrossRef CAS.
- T. V. Harris and R. K. Szilagyi, Inorg. Chem., 2011, 50, 4811–4824 CrossRef CAS PubMed.
- E. Munck, H. Rhodes, W. H. Ormejohnson, L. C. Davis, W. J. Brill and V. K. Shah, Biochim. Biophys. Acta, Protein Struct., 1975, 400, 32–53 CrossRef CAS.
- R. Zimmermann, W. H. Ormejohnson, E. Munck, V. K. Shah, W. J. Brill, M. T. Henzl and J. Rawlings, Biochim. Biophys. Acta, Protein Struct., 1978, 537, 185–207 CrossRef CAS.
- S. P. Cramer, K. O. Hodgson, W. O. Gillum and L. E. Mortenson, J. Am. Chem. Soc., 1978, 100, 3398–3407 CrossRef CAS.
- S. P. Cramer, W. O. Gillum, K. O. Hodgson, L. E. Mortenson, E. I. Stiefel, J. R. Chisnell, W. J. Brill and V. K. Shah, J. Am. Chem. Soc., 1978, 100, 3814–3819 CrossRef CAS.
- B. M. Hoffman, J. E. Roberts and W. H. Orme-Johnson, J. Am. Chem. Soc., 1982, 104, 860–862 CrossRef CAS.
- R. A. Venters, M. J. Nelson, P. A. McLean, A. E. True, M. A. Levy, B. M. Hoffman and W. H. Orme-Johnson, J. Am. Chem. Soc., 1986, 108, 3487–3498 CrossRef CAS.
- A. E. True, P. McLean, M. J. Nelson, W. H. Orme-Johnson and B. M. Hoffman, J. Am. Chem. Soc., 1990, 112, 651–657 CrossRef CAS.
- B. Hedman, P. Frank, S. F. Gheller, A. Lawrence Roe, W. E. Newton and K. O. Hodgson, J. Am. Chem. Soc., 1988, 110, 3798–3805 CrossRef CAS.
- K. Hämäläinen, D. P. Siddons, J. B. Hastings and L. E. Berman, Phys. Rev. Lett., 1991, 67, 2850–2853 CrossRef.
- P. Glatzel, T.-C. Weng, K. Kvashnina, J. Swarbrick, M. Sikora, E. Gallo, N. Smolentsev and R. A. Mori, J. Electron Spectrosc. Relat. Phenom., 2013, 188, 17–25 CrossRef CAS PubMed.
- F. A. Lima, R. Bjornsson, T. Weyhermuller, P. Chandrasekaran, P. Glatzel, F. Neese and S. DeBeer, Phys. Chem. Chem. Phys., 2013, 15, 20911–20920 RSC.
- J. Han, M. Koutmos, S. A. Ahmad and D. Coucouvanis, Inorg. Chem., 2001, 40, 5985–5999 CrossRef CAS PubMed.
- D. V. Fomitchev, C. C. McLauchlan and R. H. Holm, Inorg. Chem., 2002, 41, 958–966 CrossRef CAS PubMed.
- D. Coucouvanis, Acc. Chem. Res., 1981, 14, 201–209 CrossRef CAS.
- S. C. Lee and R. H. Holm, Chem. Rev., 2004, 104, 1135–1158 CrossRef CAS PubMed.
- M. Cook and M. Karplus, J. Chem. Phys., 1985, 83, 6344 CrossRef CAS PubMed.
- L. Noodleman, C. Y. Peng, D. A. Case and J. M. Mouesca, Coord. Chem. Rev., 1995, 144, 199–244 CrossRef CAS.
- H. Beinert, Science, 1997, 277, 653–659 CrossRef CAS.
- E. Bill, Hyperfine Interact., 2011, 205, 139–147 CrossRef PubMed.
- S. Sinnecker, L. D. Slep, E. Bill and F. Neese, Inorg. Chem., 2005, 44, 2245–2254 CrossRef CAS PubMed.
- M. Römelt, S. Ye and F. Neese, Inorg. Chem., 2009, 48, 784–785 CrossRef PubMed.
- P. K. Mascharak, G. C. Papaefthymiou, W. H. Armstrong, S. Foner, R. B. Frankel and R. H. Holm, Inorg. Chem., 1983, 22, 2851–2858 CrossRef CAS.
- M. J. Carney, J. A. Kovacs, Y. P. Zhang, G. C. Papaefthymiou, K. Spartalian, R. B. Frankel and R. H. Holm, Inorg. Chem., 1987, 26, 719–724 CrossRef CAS.
- J. P. Malrieu, R. Caballol, C. J. Calzado, C. de Graaf and N. Guihéry, Chem. Rev., 2014, 114, 429–492 CrossRef CAS PubMed.
- T. Lovell, R. A. Torres, W.-G. Han, T. Liu, D. A. Case and L. Noodleman, Inorg. Chem., 2002, 41, 5744–5753 CrossRef CAS PubMed.
- D. Lukoyanov, V. Pelmenschikov, N. Maeser, M. Laryukhin, T. C. Yang, L. Noodleman, D. R. Dean, D. A. Case, L. C. Seefeldt and B. M. Hoffman, Inorg. Chem., 2007, 46, 11437–11449 CrossRef CAS PubMed.
- T. T. Lovell, T. T. Liu, D. A. D. Case and L. L. Noodleman, J. Am. Chem. Soc., 2003, 125, 8377–8383 CrossRef CAS PubMed.
- I. Dance, Inorg. Chem., 2011, 50, 178–192 CrossRef CAS PubMed.
- M. J. Pushie and G. N. George, Coord. Chem. Rev., 2011, 255, 1055–1084 CrossRef CAS PubMed.
- D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed.
- K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2010, 3, 120–125 CrossRef PubMed.
- D. V. Yandulov and R. R. Schrock, Inorg. Chem., 2005, 44, 1103–1117 CrossRef CAS.
- R. L. McNaughton, M. Roemelt, J. M. Chin, R. R. Schrock, F. Neese and B. M. Hoffman, J. Am. Chem. Soc., 2010, 132, 8645–8656 CrossRef CAS PubMed.
- F. Studt and F. Tuczek, Angew. Chem., Int. Ed., 2005, 44, 5639–5642 CrossRef CAS PubMed.
- P. E. P. Doan, J. J. Telser, B. M. B. Barney, R. Y. R. Igarashi, D. R. D. Dean, L. C. L. Seefeldt and B. M. B. Hoffman, J. Am. Chem. Soc., 2011, 133, 17329–17340 CrossRef CAS PubMed.
- D. Lukoyanov, Z.-Y. Yang, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, J. Am. Chem. Soc., 2010, 132, 2526–2527 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2013, 46, 587–595 CrossRef CAS PubMed.
- L. C. Seefeldt, I. G. Dance and D. R. Dean, Biochemistry, 2004, 43, 1401–1409 CrossRef CAS PubMed.
- R. R. Schrock, Angew. Chem., Int. Ed., 2008, 47, 5512–5522 CrossRef CAS PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Further experimental and computational data and discussion. See DOI: 10.1039/c4sc00337c |
| ‡ These authors have contributed equally to the work presented in this manuscript. |
| § Present address: Centro Nacional de Pesquisa em Energia e Materiais, Brazilian Synchrotron Light Laboratory – LNLS, CP 6192, 13084-971 Campinas, SP, Brazil. |
| ¶ Present address: Howard Hughes Medical Institute and Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena CA 91125, USA. |
| This journal is © The Royal Society of Chemistry 2014 |