 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of 5f-orbital participation in unexpected inversion of the σ-bond metathesis reactivity trend of triamidoamine thorium(IV) and uranium(IV) alkyls†
Benedict M.
Gardner
a,
Peter A.
Cleaves
a,
Christos E.
Kefalidis
b,
Jian
Fang
bc,
Laurent
Maron
*b,
William
Lewis
a,
Alexander J.
Blake
a and
Stephen T.
Liddle
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: stephen.liddle@nottingham.ac.uk
bLPCNO, CNRS & INSA, Université Paul Sabatier, 135 Avenue de Rangueil, Toulouse 31077, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
cCollege of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China
First published on 26th February 2014
Abstract
We report on the role of 5f-orbital participation in the unexpected inversion of the σ-bond metathesis reactivity trend of triamidoamine thorium(IV) and uranium(IV) alkyls. Reaction of KCH2Ph with [U(TrenTIPS)(I)] [2a, TrenTIPS = N(CH2CH2NSiPri3)33−] gave the cyclometallate [U{N(CH2CH2NSiPri3)2(CH2CH2NSiPri2C[H]MeCH2)}] (3a) with the intermediate benzyl complex not observable. In contrast, when [Th(TrenTIPS)(I)] (2b) was treated with KCH2Ph, [Th(TrenTIPS)(CH2Ph)] (4) was isolated; which is notable as Tren N-silylalkyl metal alkyls tend to spontaneously cyclometallate. Thermolysis of 4 results in the extrusion of toluene and formation of the cyclometallate [Th{N(CH2CH2NSiPri3)2(CH2CH2NSiPri2C[H]MeCH2)}] (3b). This reactivity is the reverse of what would be predicted. Since the bonding of thorium is mainly electrostatic it would be predicted to undergo facile cyclometallation, whereas the more covalent uranium system might be expected to form an isolable benzyl intermediate. The thermolysis of 4 follows well-defined first order kinetics with an activation energy of 22.3 ± 0.1 kcal mol−1, and Eyring analyses yields ΔH‡ = 21.7 ± 3.6 kcal mol−1 and ΔS‡ = −10.5 ± 3.1 cal K−1 mol−1, which is consistent with a σ-bond metathesis reaction. Computational examination of the reaction profile shows that the inversion of the reactivity trend can be attributed to the greater f-orbital participation of the bonding for uranium facilitating the σ-bond metathesis transition state whereas for thorium the transition state is more ionic resulting in an isolable benzyl complex. The activation barriers are computed to be 19.0 and 22.2 kcal mol−1 for the uranium and thorium cases, respectively, and the latter agrees excellently with the experimental value. Reductive decomposition of “[U(TrenTIPS)(CH2Ph)]” to [U(TrenTIPS)] and bibenzyl followed by cyclometallation to give 3a with elimination of dihydrogen was found to be endergonic by 4 kcal mol−1 which rules out a redox-based cyclometallation route for uranium.
Introduction
Since the search for volatile compounds for uranium isotope separation in the Manhattan project,1,2 organoactinide chemistry has received continued interest, mainly focusing on uranium.3–26 Early work on uranium tetraalkyls contributed to the perception that σ-bonded actinide hydrocarbyls were unstable,2 but Marks et al. showed that with suitable supporting ligands these linkages can be kinetically stabilised.27–29 Moreover, Hayton et al. have recently shown that homoleptic uranium–alkyls can be kinetically stabilised by saturating the coordination sphere.30–33 However, although these linkages can be stabilised by ancillary ligands that resist C–H activation, their intrinsically polarised, reactive nature remains and thus they are tamed but not tempered.The extent to which f-orbital bonding interactions occur at uranium is still hotly debated, as is their influence on reactivity and potential nuclear waste separation applications.34–37 However, studies have shown that uranium bonding can involve moderate 5f orbital participation leading to modification of reactivity with consequences for the nature of the chemical outcome.38–40 In contrast, the thorium(IV) ion is generally perceived as being unable to access the 5f-orbital manifold. Thus, its bonding is viewed as being restricted to electrostatics resulting in thorium–ligand bonds that are generally more labile than their uranium(IV) counterparts. Added to the greater ionic radius of Th(IV) (94 pm) vs. U(IV) (89 pm),41 thorium–ligand bonds are generally expected to be more reactive compared to uranium–ligand bonds.
The above arguments suggest that the reactivity of thorium and uranium should be different, but the picture is complicated and not well understood and reports showing divergent experimental reactivity patterns of closely comparable uranium and thorium systems are rare.38,42–45 Intramolecular amino-alkene hydroamination reactions report a trend of thorium being more reactive than uranium; however, there are some notable exceptions for which there is no explanation, but similar trends have been observed for lanthanide complexes suggesting that f-orbital participation is not a key factor.39,46 Studies by Marks on bis(cyclopentadienyl) actinide dialkyls showed that thorium derivatives are often more stable than their uranium analogues.2 However, in some cases reduction of uranium(IV) to uranium(III) provided a decomposition route for uranium not available to thorium. Whilst bond polarities and accessible oxidation states were considered, f-orbital participation was not examined.29 Recent studies of bis(cyclopentadienyl) actinide dialkyl reactivity by Kiplinger et al. have shown varied reactivity trends for thorium vs. uranium,43,47,48 but their origins were not resolved. Indeed, the factors that contribute to an isostructural pair of uranium and thorium complexes exhibiting similar or different reactivities are legion and a recent computational study could not derive any clear conclusion.49 Where f-orbital participation has been probed, this has compared the differences of the f-block to d-block systems.40 Recently, the formation of a uranium benzyne complex vs. a thorium tetraaryl derivative was reported.38 In that report greater f-orbital participation in the bonding for the uranium product was found and it was postulated that this provided kinetic facilitation of the benzyne.
As part of our work on triamidoamine chemistry,50–61 our attention turned to comparing f-orbital participation in the bonding of uranium and thorium and how this might affect reactivity. Since certain cyclometallated actinide complexes,62–68 in particular those with triamidoamine ligands, have novel properties,61,62 uranium and thorium metallacyclic complexes supported by N(CH2CH2NSiPri3)33− (TrenTIPS) were targeted since this ligand has stabilised reactive uranium complexes in multiple oxidation states.50,52 It should be noted that facile cyclometallation of N-silyl-alkyl groups is always observed for Tren thorium and uranium alkyls because the metallocycle alkyl groups are stabilised by negative hyperconjugation to silicon as well as the entropic effects from liberating an alkane. Indeed, in a wider context out of dozens of examples of Tren N-silyl metal complexes only one benzyl derivative has been authenticated making this ligand combination very rare.69 Uranium and thorium benzyl derivatives are known,21,30,45,64,70–76 but where stable ligand metal benzyl combinations are known this is with ligands that do not cyclometallate; where Tren N-silylalkyl ligands are concerned this is facile.61
Here, we report the synthesis of cyclometallated and benzyl triamidoamine derivatives of uranium and thorium, which surprisingly display inverted reactivity trends. Although the thorium system would be expected to be the most ionic and hence most reactive it is the least reactive and a cyclometallate and its benzyl precursor can both be isolated. In contrast, the uranium system, which would be predicted to be most stable, is the most reactive and only the cyclometallate and not the benzyl intermediate can be isolated. These observations are surprising on two counts. Firstly, the thorium benzyl complex should be unstable with respect to cyclometallation and thus its stability is notable. Secondly, of either of the two complexes uranium should, from an a priori assessment, present the best chance for a Tren metal benzyl complex but this is not the case. We show by an experimentally calibrated computational study that: (i) decomposition involving reduction of uranium(IV) to uranium(III) with elimination of bibenzyl is not operative; (ii) the reversal of anticipated reactivity is due to the σ-bond metathesis transition state for uranium being stabilised by uranium 5f-orbital participation that is not present for thorium. Thus, the additional involvement of uranium 5f orbitals actually enhances the reactivity. This study provides a rare account of how 5f-orbital participation in reactive intermediate transition states can alter the chemical reactivity of isostructural systems in counter-intuitive ways that cannot be predicted but which are important to understand.
Results and discussion
Synthesis and structures of thorium precursors
With [U(TrenTIPS)(X)] (X = Cl, 1a; I, 2a) species having previously been reported by some of us,50,52 we targeted the analogous thorium halide complexes required to access the cyclometallated species. Complex [Th(TrenTIPS)(Cl)] (1b) was readily prepared on multi-gram scales from [Li3(TrenTIPS)] and [ThCl4(THF)3.5]77 in THF and after work-up was isolated in near-quantitative yields as an essentially pure solid (Scheme 1). Crystals of 1b suitable for a single crystal X-ray diffraction study were obtained from toluene. The molecular structure of 1b is shown in Fig. 1 with selected bond lengths and angles.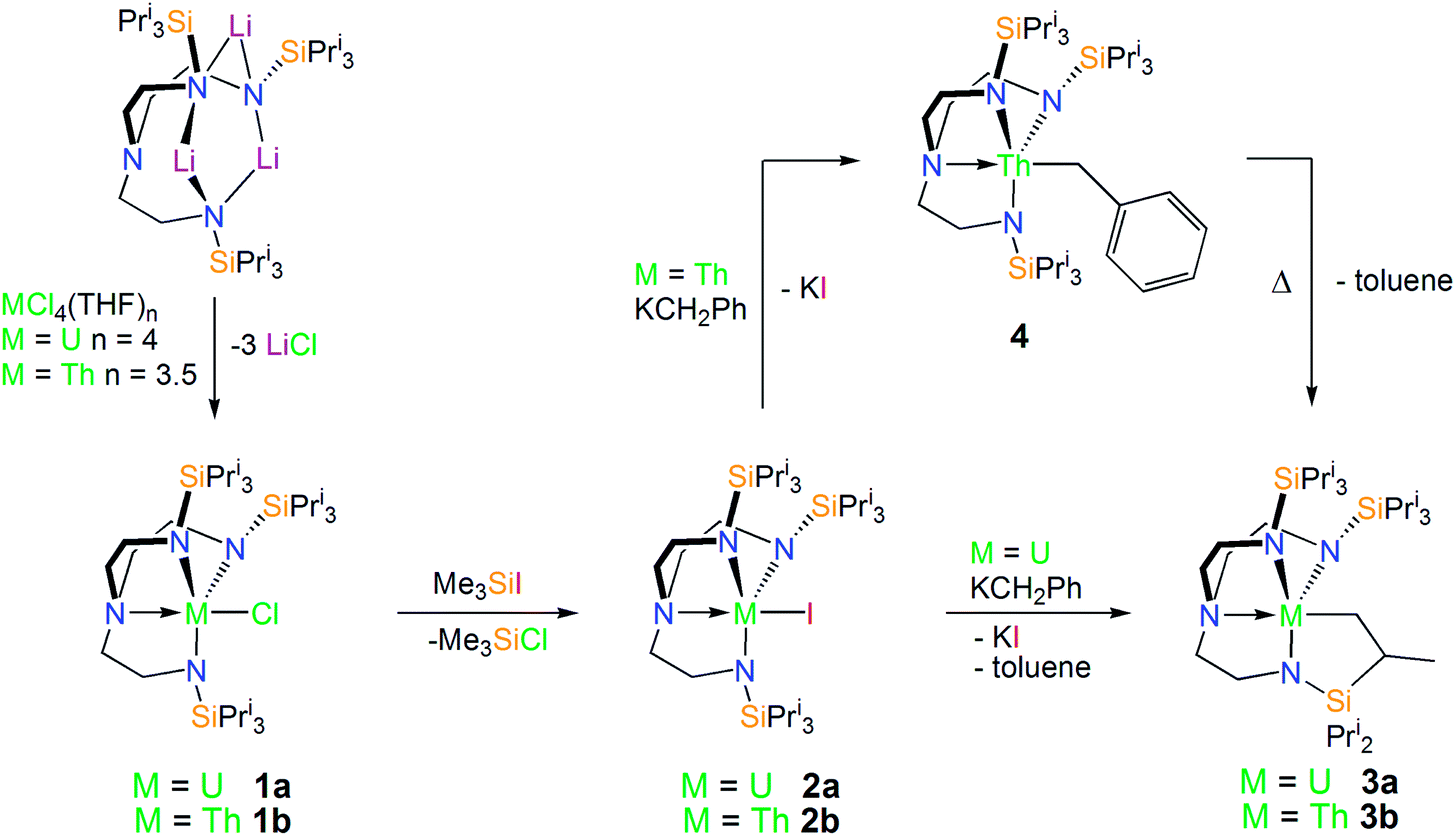 | ||
| Scheme 1 Experimental synthesis of thorium and uranium halide, benzyl, and cyclometallated complexes 1–4. | ||
 | ||
| Fig. 1 Molecular structure of 1b with displacement ellipsoids set at 30% probability and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Th1–Cl1, 2.693(2); Th1–N1, 2.309(3); Th1–N2, 2.305(3); Th1–N3, 2.284(3); Th1–N4, 2.668(3); N4–Th1–Cl1, 173.51(6). | ||
The Th centre in 1b is five-coordinate, adopting a distorted trigonal bipyramidal geometry with the N4 and Cl1 centres occupying the axial sites [N4–Th1–Cl1 = 173.51(6)°]. The bond lengths and angles in 1b are unremarkable, with Th–Namide, Th–Namine and Th–Cl bond lengths of 2.299 (av.), 2.668(3) and 2.693(2) Å, respectively, typical of Th(IV)–Namide Th(IV)–Namine and Th(IV)–Cl bond distances.78
The iodide derivative of 1b, [Th(TrenTIPS)(I)] (2b), was targeted since iodide elimination from actinide centres is often easier to achieve than elimination of chloride.61,79,80 Accordingly, a toluene solution of 1b was treated with a slight excess of Me3SiI to afford [Th(TrenTIPS)(I)] (2b) and Me3SiCl (Scheme 1). Despite numerous attempts the structure of 2b could not be unambiguously confirmed by single crystal X-ray diffraction due to persistently poor data. However, the proposed formulation is supported by the characterisation data which additionally confirms the absence of coordinated donor solvents (full details are provided in the ESI†).
Synthesis and structures of uranium and thorium cyclometallate complexes
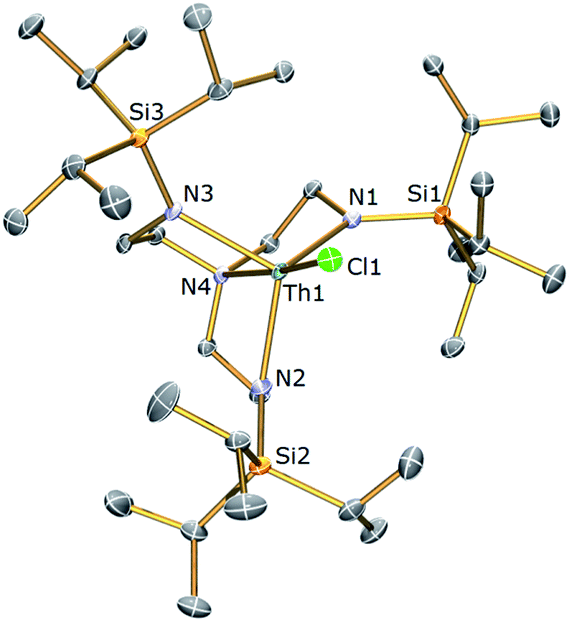
Cyclometallated Tren–uranium and –thorium compounds have been prepared previously61,62via treatment of a suitable halide precursor with one equivalent of benzylpotassium. The reaction most probably proceeds via a reactive benzyl intermediate which C–H activates the silylalkyl arm of Tren eliminating one equivalent of toluene and forming an An–C σ-bond. However, these putative benzyls are very reactive since none have ever been observed with only the cyclometallated products being isolated. An analogous methodology was employed here to access uranium and thorium TrenTIPS-cyclometallates.A green-brown toluene solution of [U(TrenTIPS)(I)]52 (2a) reacted with KCH2Ph to give a dark orange-red solution. Following work-up and recrystallisation from hexane, a single crystal X-ray diffraction study was performed, which confirmed the structure of the anticipated alkyl complex to be [U{N(CH2CH2NSiPri3)2(CH2CH2NSiPri2C[H]MeCH2)}] (3a). The molecular structure of 3a is illustrated in Fig. 2 with selected bond lengths and angles.
 | ||
| Fig. 2 Molecular structure of 3a with displacement ellipsoids set at 30% probability and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): U1–N1, 2.258(16); U1–N2, 2.298(15); U1–N3, 2.289(15); U1–N4, 2.514(15); U1–C2, 2.55(2); N4–U1–C2, 146.1(7); N1–U1–C2, 76.1(7). | ||
The U centre in 3a is five-coordinate, adopting a distorted trigonal bipyramidal geometry with the carbon C2 and amine centre N4 occupying the axial positions. The U–Namide and U–Namine bond lengths of 2.282 (av.) and 2.514(15) Å, respectively, are typical of U(IV)–Namide and U(IV)–Namine bond distances. The U1–C2 bond length of 2.55(2) Å is within the reported range of U–C single bonds [2.40–2.55 Å].78 In contrast, the cyclometallate [U{N(CH2CH2NSiMe2But)2(CH2CH2NSiMeButCH2)}]62 features a longer U–C bond [2.752(11) Å], attributed to increased geometric strain relative to 3a due to its incorporation within a four-membered [U–N–Si–C] ring as opposed to the five membered ring present in 3a. This is supported by the less acute bite angle in 3a [76.1(7)° vs. 66.1(2)°].
The 1H NMR spectrum of 3a in C6D6 at room temperature consists of 27 resonances spanning the chemical shift range +47 to −35 ppm. This is consistent with the expected unsymmetrical C1 Tren environment, which is also highlighted by the observation that three resonances are observed in the 29Si{1H} NMR spectrum of 3a, and confirms the presence of a paramagnetic uranium centre. The room temperature Evans method magnetic moment of 3a in C6D6 was found to be 2.90 μB, which is lower than the theoretical free ion value for the formal 3H4 ground state of 5f2 U(IV) of 3.58 μB,81 but uranium(IV) compounds usually fall within the range 2.50–3.20 μB.14,30,82,83
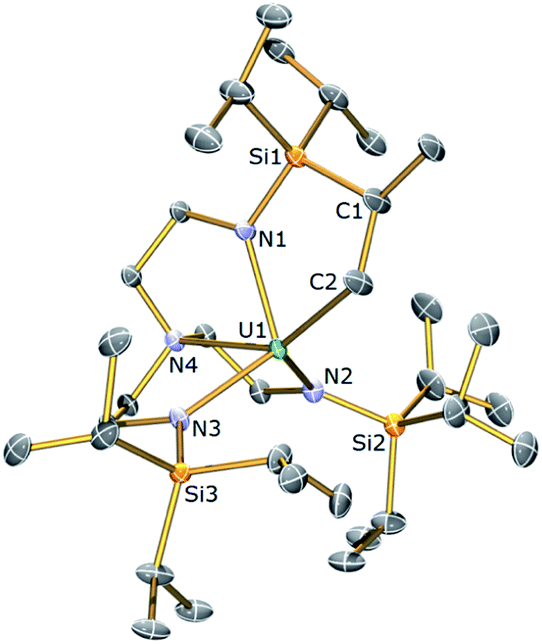
The thorium(IV) analogue of 3a was targeted via the treatment of 2b with KCH2Ph. Following this reaction, 1H NMR revealed the consumption of 2b along with the formation of a single new product; however the 1H NMR spectrum of this product consisted of only 8 resonances. Following work-up and crystallisation from methylcyclohexane, colourless crystals suitable for single crystal X-ray diffraction analysis were isolated. These revealed the new compound to be the Tren–thorium benzyl complex [Th(TrenTIPS)(CH2Ph)] (4), which is notable for being stable under ambient conditions rather than undergoing cyclometallation. The molecular structure of 4 is illustrated in Fig. 3 with selected bond lengths and angles.
 | ||
| Fig. 3 Molecular structure of 4 with displacement ellipsoids set at 30% probability. Hydrogen atoms and three of the four unique molecules in the asymmetric unit are omitted for clarity. Selected bond lengths (Å) and angles (°): Th1–N1, 2.262(6); Th1–N2, 2.309(6); Th1–N3, 2.313(6); Th1–N4, 2.701(6); Th1–C34, 2.590(8); N4–Th1–C34, 170.2(2); N1–Th1–N4, 69.2(2). | ||
The asymmetric unit of the solid state structure of 4 comprises four crystallographically independent molecules, but in each case the thorium centre is 5-coordinate adopting a distorted trigonal bipyramidal geometry where the amine resides trans to a terminal η1-benzyl group. The mean Th–Namide bond lengths within each independent molecule of 4 range from 2.294(6) to 2.311(7) Å and the four Th–Namine bond lengths are statistically invariant with a mean value of 2.706 Å. The Th–C bond lengths in 4 range from 2.536(8) to 2.622(7) Å (mean Th–C−Cispo ∠ = 123.2°). All the Th–Namide, Th–Namine and Th–C bond lengths in 4 lie within the reported ranges for Th(IV) complexes.78 In addition the 1H NMR spectrum of 4 in C6D6 at room temperature consists of 8 resonances located in the chemical shift range of 0–10 ppm and the 13C{1H} NMR spectrum of 4 comprises 9 resonances with the signal for the benzyl-CH2 group observed at 113.7 ppm.
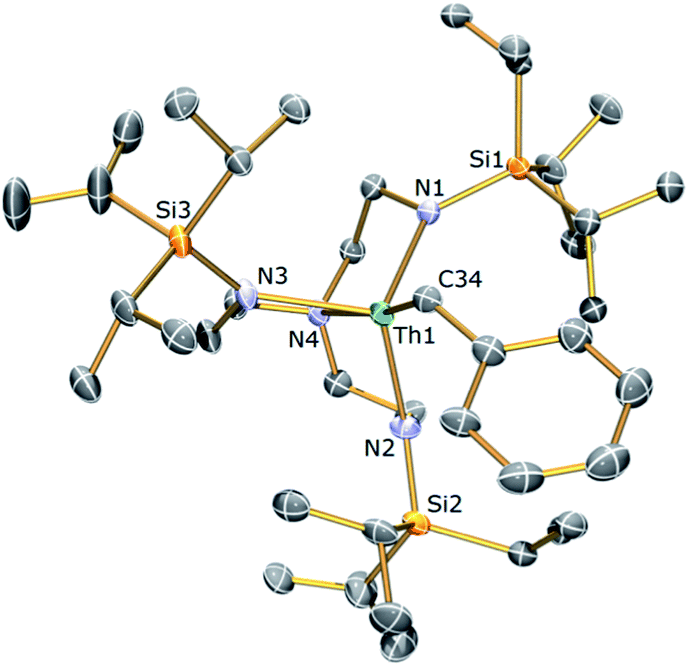
With 4 in-hand, it was envisaged that C–H activation to produce the desired Tren–thorium metallacycle might require thermolytic activation since a benzyl complex has been proposed to be an intermediate en route to reported Tren–uranium cyclometallates.61,62 It was observed that warming NMR samples of 4 at 80 °C produced a markedly different 1H NMR spectrum after 2 hours consistent with: (i) complete consumption of 4; (ii) production of a new Tren-containing product; and (iii) elimination of one equivalent of toluene. This reaction was reproduced on a bulk scale and following work-up and recrystallisation from hexane, colourless single crystals were subjected to an X-ray diffraction analysis that revealed the new complex to be [Th{N(CH2CH2NSiPri3)2(CH2CH2NSiPri2C[H]MeCH2)}] (3b). The molecular structure of 3b is illustrated in Fig. 4 with selected bond lengths and angles.
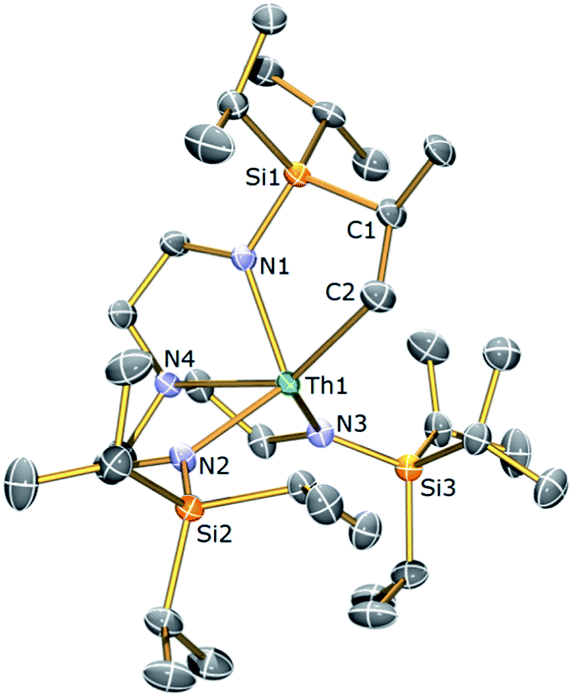 | ||
| Fig. 4 Molecular structure of 3b with displacement ellipsoids set at 30% probability and H atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Th1–N1, 2.309(3); Th1–N2, 2.337(3); Th1–N3, 2.339(3); Th1–N4, 2.607(3); Th1–C2, 2.563(4); N4–Th1–C2, 145.64(11); N1–Th1–N4, 68.54(10). | ||
Complex 3b is essentially isostructural to 3a, with any significant deviation between the metal–ligand bond distances presented by the two complexes ascribed to the smaller ionic radius of U(IV) versus Th(IV).41 Similar to 3a, complex 3b exhibits a highly unsymmetrical ligand environment with its room temperature 1H NMR spectrum revealing 16 resonances spanning the narrow range 0.68 to 3.90 ppm. The large reduction in the number of signals in the 1H NMR spectrum of 3b compared to 3a almost certainly arises from the large difference in chemical shift range of 3avs.3b leading to multiple overlapped resonances in the latter. The metallated carbon exhibits the expected downfield chemical shift (105.7 ppm) in the 13C{1H} NMR spectrum of 3b and like 3a three resonances are observed in the 29Si{1H} NMR spectrum of 3b.
Given the generally electrostatic bonding of Th(IV) and the larger ionic radius leading to an overall increase in thorium–ligand bond lability, it would be expected that 4 would be more unstable toward cyclometallation than its hypothetical uranium congener. Counter-intuitively, what is experimentally observed is the reverse of this where 4 is a stable crystallisable intermediate, albeit one that will convert to the metallacycle upon thermolysis.
Mechanistic studies
Since 4 converts to 3b in solution under thermolytic conditions, the mechanism of this cyclometallation was probed by a kinetic NMR study. The reaction was found to follow well-defined first order kinetics with respect to 4, with rate constants showing the expected increase from 2.25 ± 0.1 × 10−5 s−1 at 40 °C to 5.13 ± 0.3 × 10−4 s−1 at 70 °C, Fig. 5. Arrhenius analysis revealed an activation energy of 22.3 ± 0.1 kcal mol−1, which lies within the range of those reported for thermal cyclometallation reactivity profiles of thorium complexes, e.g. [ThCp*2(R)2] (Cp* = η5-C5Me5, R = alkyl).29 Eyring analysis determined ΔH‡ to be 21.7 ± 3.6 kcal mol−1, which is in line with the elimination of one molecule of toluene per Th centre. ΔS‡ was found to be −10.5 ± 3.1 cal K−1 mol−1, which is consistent with this type of reaction;84–86 the value for ΔS‡ is negative and of moderate magnitude, inferring that the transition state is sterically constrained and well-defined, and is consistent with the exclusive product selectivity observed experimentally. Such a transition state would be consistent with a four-membered [Th–C–H–C] ring during σ-bond metathesis as observed for lanthanides and early or high oxidation state transition metals, which are incapable of supporting classical oxidative addition–reductive elimination sequences.87 Closely related transition metal Tren complexes readily cyclometallate, presumably due to the availability of d-orbitals and their much greater ability to engage in covalent interactions.69,88–90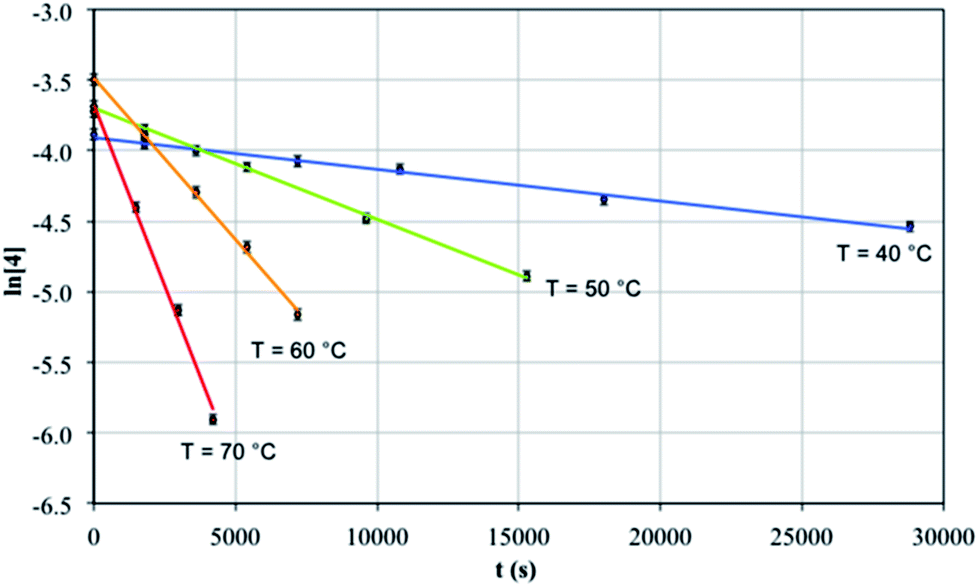 | ||
| Fig. 5 Plot of ln[4] vs. time (s) for the conversion of 4 to 3b. These data fit well-defined first order kinetics with respect to 4 with rate constants of 2.25 ± 0.1 × 10−5 s−1 (40 °C), 7.89 ± 0.1 × 10−5 s−1 (50 °C), 2.29 ± 0.1 × 10−4 s−1 (60 °C), and 5.13 ± 0.3 × 10−4 s−1 (70 °C). | ||
The observation that 4 is isolable in the reaction from 2b to 3b raises the question of whether “[U(TrenTIPS)(CH2Ph)]” is stable. However, variable-temperature NMR measurements of the reaction between 2a and KCH2Ph always proceed smoothly to give 3a with no intermediates observable over the temperature range 193 to 298 K and thus it was not possible to obtain any experimental mechanistic data on the conversion of 2a to 3a. Attempts to trap the putative benzyl intermediate with the known benzyl traps73,74 [Ph3C][BArf4] [Arf = 3,5-(CF3)2-C6H3] and B(C6F5)3 were also unsuccessful but it should be noted that benzylpotassium is not capable of deprotonating the N-silyl alkyl groups of Tren compounds.
Marks and coworkers showed that bis(cyclopentadienyl) uranium(IV) dialkyls can decompose to uranium(III) derivatives,29 so we considered the possibility that addition of benzyl potassium to 2a affords “[U(TrenTIPS)(CH2Ph)]” which then reductively decomposes to [U(TrenTIPS)] and half an equivalent of bibenzyl; the former could in principle then cyclometallate with concomitant elimination of dihydrogen to form 3a. However, [U(TrenTIPS)] has been prepared by us previously and it does not cyclometallate to give 3a, even at elevated temperatures. Also, we did not observe any resonances in the 1H NMR spectra from reactions of 2a with benzyl potassium that would be consistent with the formation of bibenzyl or dihydrogen.
We considered the possibility that steric effects could be driving the observed reactivity since uranium is smaller than thorium. However, we note that [U(TrenTMS)(Cl)(THF)] [TrenTMS = N(CH2CH2NSiMe3)3], which is open enough to leave space for a THF molecule to coordinate to uranium, also spontaneously cyclometallates when treated with benzyl potassium61 so we conclude that steric effects are not a factor (see below).
Since a redox-based reaction mechanism seemed highly unlikely, the observed contrast between the reactivity modes for uranium and thorium was probed computationally to determine the origin of this discrepancy since a σ-bond metathesis reaction would intuitively be the most likely mechanism by which cyclometallation of the N-silyl alkyl groups could occur.
Computational investigations
In order to propose a plausible reaction profile, DFT (at B3PW91 level of theory) calculations were carried out. The calculations show that although the reductive extrusion of benzyl from “[U(TrenTIPS)(CH2Ph)]” to give [U(TrenTIPS)] and half a molar equivalent of bibenzyl is thermodynamically favorable (−12 kcal mol−1), subsequent cyclometallation to give 3a and half a molar equivalent of dihydrogen is unfavorable (16 kcal mol−1) and thus this route, which has no experimental support, is disfavoured by 4 kcal mol−1 and can thus be ruled out. From a thermodynamic standpoint, there is little preference for the formation of the benzyl complexes (labelled 4a and 4b hereafter) from complexes 2a and 2b (Scheme 2). Thus, the experimental conversion of 2a to 3a but isolable sequence of 2b to 4 to 3b should originate from kinetic factors that derive from orbital facilitation present for uranium but not thorium.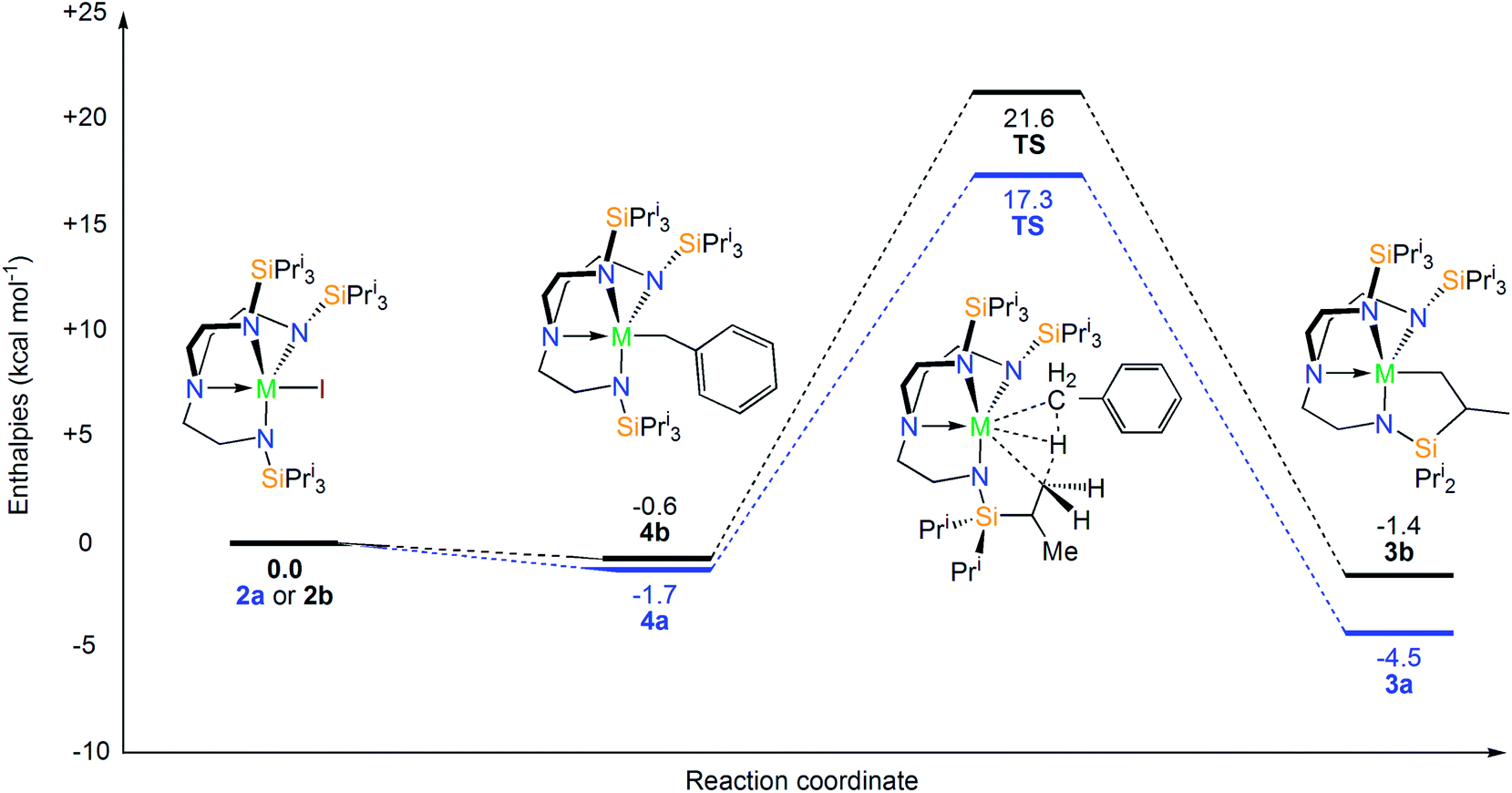 | ||
| Scheme 2 Enthalpy energy profiles (at 298 K) for the gas phase cyclometallation reaction for the complexes 2a (uranium, blue profile) and 2b (thorium, black profile) computed at the B3PW91 level of theory. | ||
The cyclometallation transition states (TS) were located on the potential energy surface and are depicted in Fig. 6. The geometries of the TSs are essentially identical to the classical σ-bond metathesis arrangement,91 with the hydrogen atom that is ultimately transferred lying between the two alkyl groups as reported in previous studies of σ-bond metathesis for U–Th catalyzed C–H activation.92 In particular, the M–H distance is the shortest interatomic distance and the C–H–C angle is close to 180° (176° for U and 172° for Th). Interestingly, Natural Bond Orbital (NBO) analyses at the transition states indicate that the metal–benzyl bonds are already broken (no interactions are found either at the first nor at the second order donor–acceptor level) and that a M–CH2 bond is formed (found at the first order NBO level). The activation barriers were computed to be 19.0 and 22.2 kcal mol−1 for the uranium and thorium cases, respectively. It is germane to note that the calculated activation barrier for the Th case is in excellent agreement with the experimental value of 22.3 kcal mol−1 and compares well to computed f-block σ-bond metathesis reaction activation barriers of ∼20 kcal mol−1.93–95
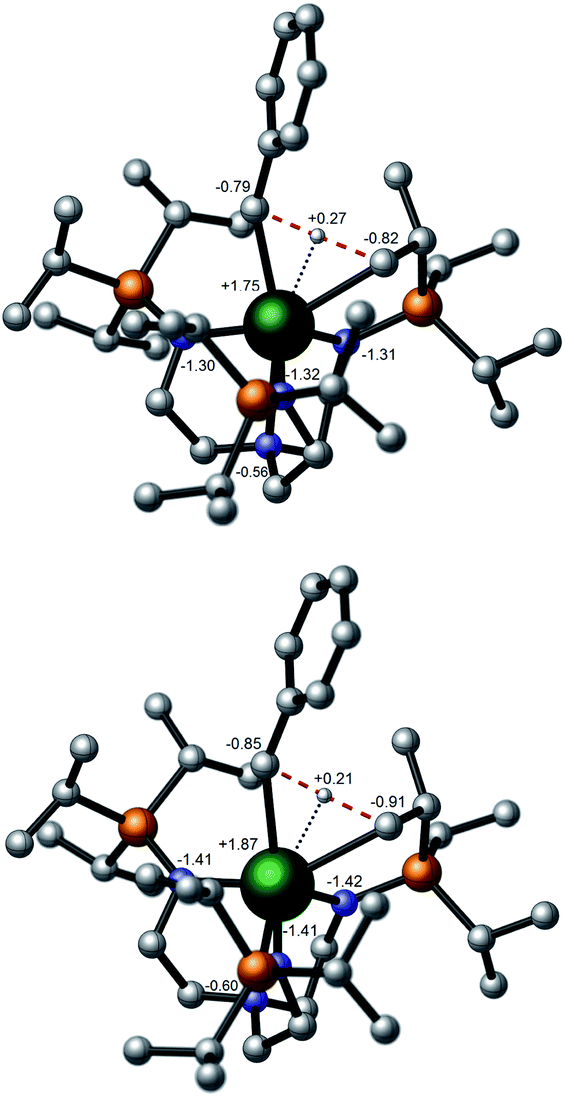 | ||
| Fig. 6 Calculated transition states for the cyclometallation step of 4a (top, uranium) and 4b (bottom, thorium) with selected atom natural population charge labelling and peripheral hydrogen atoms omitted for clarity. | ||
In order to rationalise the slight kinetic preference for U over Th, natural population analysis (NPA) was carried out (Fig. 6). The charge is slightly lower at the uranium centre than at thorium, which is consistent with a slightly more ionic TS for the latter than for the former. This is further highlighted by scrutinising the natural charges of the carbon centres of the alkyl groups that are somewhat higher in the thorium case than for uranium. Overall, the description of the charge distribution shows the characteristic hallmarks of a σ-bond metathesis TS.94 It has been proposed that the observed difference between uranium and thorium can be attributed to the lack of 5f involvement in the bonding for the latter, hence inducing a difference in covalency between the two metals (with uranium being more covalent).38 To further test the hypothesis of the difference in ionicity/covalency due to the 5f electron involvement at the TS, the molecular orbitals (MOs) were analyzed (see ESI†). In the uranium case, four occupied orbitals describe metal–alkyl interactions (mainly employing 5f orbitals) whereas only two are found in the thorium case (mainly involving 6d orbitals). This is in-line with reports38,40 of the involvement of 5f orbitals in bonding but there is no clear correlation between this and a difference in covalency between thorium and uranium. The cyclometallation reaction step was computed to be slightly exothermic for uranium by 2.8 kcal mol−1 overall whereas it is almost athermic for Th overall (0.8 kcal mol−1).
This reactivity might be explained by either the steric hindrance around the metal centre (sterically induced reactivity) or the participation of the 5f orbitals. In order to test the hypothesis of the importance of 5f orbital participation and their effect on the observed reactivity, we computed the enthalpy profiles for both metals without including the 5f electrons and orbitals in the calculation using large core relativistic effective core potentials (RECPs) that were thus treated as 6d transition metals. The use of large core RECPs has been demonstrated to be appropriate to describe U(IV) and Th(IV) chemistry95–97 and it has even been possible to show previously that the major effect was the core polarisation rather than the 5f involvement. When f-orbitals and electrons are excluded from the calculation, the profiles for uranium and thorium are found to be the same, with the same activation barrier (see ESI†). Moreover, the barrier for thorium is exactly the same with both approaches, in-line with a lack of 5f involvement for this atom. For uranium, the barrier is approximately 3 kcal mol−1 higher without 5f electrons than when these electrons are included in the calculation. In this case, the charge at the uranium centre is higher and similar to the thorium example. This suggests that the lack of 5f orbitals increases the charges and thus makes the bonds more ionic. Furthermore, calculations were carried out on the hypothetical Zr and Hf complexes. The computed barriers were also found to be around 24–26 kcal mol−1, which is higher than for uranium. As the steric congestion is really enhanced in the two latter complexes compared to the thorium or uranium ones, these results rule out sterically induced reactivity. Thus, the calculations suggest that 5f orbital participation in the bonding indeed induces a clear change in the bonding, which can superficially be interpreted as covalency,98–100 and this in turn has a profound effect on the experimentally observed reactivity.
Conclusions
To conclude, we have reported the synthesis of cyclometallated and benzyl triamidoamine derivatives of uranium and thorium, which surprisingly exhibit reactivity trends that are the reverse of what would be predicted. Although the thorium system would be anticipated to be the most ionic and therefore reactive, it is the least reactive and a cyclometallated complex and its benzyl precursor can both be isolated. In contrast, the uranium system, which would be expected to be less reactive, is more reactive and only the cyclometallated complex can be isolated. Experimentally supported calculations suggest that this reversal of expected reactivity can be attributed to the σ-bond metathesis transition state for uranium being stabilised by 5f orbital participation in the interatom interactions in the TS, which thus enhances the reactivity. In contrast, the thorium case lacks 5f orbital stabilisation and thus the transition state is more ionic and kinetically less accessible and therefore this system is surprisingly less reactive. The calculations also suggest that a cyclometallation mechanism for uranium involving redox chemistry or steric effects can be discounted. This study represents an example of how 5f orbital participation can steer chemical reactivity in an unexpected yet profound manner.Acknowledgements
We are grateful to the Royal Society, Engineering and Physical Science Research Council, European Research Council, COST Action CM1006, UK National Nuclear Laboratory, University of Nottingham, Institut Universitaire de France, Humboldt Foundation, CINES, and CalMip for generous support of this work and to Dr Jonathan McMaster (University of Nottingham) for valuable discussions.Notes and references
- M. Tsutsui, N. Ely and R. Dubois, Acc. Chem. Res., 1976, 9, 217 CrossRef CAS.
- T. J. Marks, Acc. Chem. Res., 1976, 9, 223 CrossRef CAS.
- M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137 CrossRef CAS PubMed.
- B. M. Gardner and S. T. Liddle, Eur. J. Inorg. Chem., 2013, 3753 CrossRef CAS.
- D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266–267, 2 CrossRef CAS PubMed.
- T. W. Hayton, Chem. Commun., 2013, 49, 2956 RSC.
- M. Ephritikhine, Organometallics, 2013, 32, 2464 CrossRef CAS.
- H. S. La Pierre and K. Meyer, Inorg. Chem., 2013, 52, 529 CrossRef CAS PubMed.
- O. P. Lam, C. Anthon and K. Meyer, Dalton Trans., 2009, 9677 RSC.
- C. R. Graves and J. L. Kiplinger, Chem. Commun., 2009, 3831 RSC.
- A. R. Fox, S. C. Bart, K. Meyer and C. C. Cummins, Nature, 2008, 455, 341 CrossRef CAS PubMed.
- T. Andrea and M. S. Eisen, Chem. Soc. Rev., 2008, 37, 550 RSC.
- M. Ephritikhine, Dalton Trans., 2006, 2501 RSC.
- I. Castro-Rodriguez and K. Meyer, Chem. Commun., 2006, 1353 RSC.
- A. J. Lewis, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 13185 CrossRef CAS PubMed.
- M. R. MacDonald, M. E. Fieser, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 13310 CrossRef CAS PubMed.
- J.-C. Berthet, P. Thuéry, N. Garin, J.-P. Dognon, T. Cantat and M. Ephritikhine, J. Am. Chem. Soc., 2013, 135, 10003 CrossRef CAS PubMed.
- D. Patel, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Commun., 2013, 4, 2323, DOI:10.1038/ncomms3323.
- N. Tsoureas, O. T. Summerscales, F. G. N. Cloke and S. M. Roe, Organometallics, 2013, 32, 1353 CrossRef CAS.
- O. J. Cooper, D. P. Mills, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, Chem. - Eur. J., 2013, 19, 7071 CrossRef CAS PubMed.
- S. J. Kraft, P. E. Fanwick and S. C. Bart, J. Am. Chem. Soc., 2012, 134, 6160 CrossRef CAS PubMed.
- A. S. P. Frey, F. G. N. Cloke, M. P. Coles, L. Maron and T. Davin, Angew. Chem., Int. Ed., 2011, 123, 7013 CrossRef.
- C. R. Graves, P. Yang, S. A. Kozimor, A. E. Vaughn, D. L. Clark, S. D. Conradson, E. J. Schelter, B. L. Scott, J. D. Thompson, P. J. Hay, D. E. Morris and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 5272 CrossRef CAS PubMed.
- C. P. Lam, C. Anthon, F. W. Heinemann, J. M. O'Connor and K. Meyer, J. Am. Chem. Soc., 2008, 130, 6567 CrossRef PubMed.
- A. S. P. Frey, F. G. N. Cloke, P. B. Hitchcock, I. J. Day, J. C. Green and G. Aitken, J. Am. Chem. Soc., 2008, 130, 13816 CrossRef CAS PubMed.
- I. Castro-Rodriguez, H. Nakai, P. Gantzel, L. N. Zakharov, A. L. Rheingold and K. Meyer, J. Am. Chem. Soc., 2003, 125, 15734 CrossRef CAS PubMed.
- T. J. Marks, A. M. Seyam and J. R. Kolb, J. Am. Chem. Soc., 1973, 95, 5529 CrossRef CAS.
- T. J. Marks and W. A. Wachter, J. Am. Chem. Soc., 1976, 98, 703 CrossRef CAS.
- P. J. Fagan, J. M. Manriquez, E. A. Maatta, A. M. Seyam and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 6650 CrossRef CAS.
- S. Fortier, B. C. Melot, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2009, 131, 15512 CrossRef CAS PubMed.
- S. Fortier, J. R. Walensky, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 11732 CrossRef CAS PubMed.
- L. A. Seaman, P. Hrobárik, M. F. Schettini, S. Fortier, M. Kaupp and T. W. Hayton, Angew. Chem., Int. Ed., 2013, 52, 3259 CrossRef CAS PubMed.
- L. A. Seaman, J. R. Walensky, G. Wu and T. W. Hayton, Inorg. Chem., 2013, 52, 3556 CrossRef CAS PubMed.
- W. W. Lukens, N. M. Edelstein, N. Magnani, T. W. Hayton, S. Fortier and L. A. Seaman, J. Am. Chem. Soc., 2013, 135, 10742 CrossRef CAS PubMed.
- L. P. Spencer, P. Yang, S. G. Minasian, R. E. Jilek, E. R. Batista, K. S. Boland, J. M. Boncella, S. D. Conradson, D. L. Clark, T. W. Hayton, S. A. Kozimor, R. L. Martin, M. M. MacInnes, A. C. Olson, B. L. Scott, D. K. Shuh and M. P. Wilkerson, J. Am. Chem. Soc., 2013, 135, 2279 CrossRef CAS PubMed.
- L. A. Seaman, G. Wu, N. Edelstein, W. W. Lukens, N. Magnani and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 4931 CrossRef CAS PubMed.
- S. A. Kozimor, P. Yang, E. R. Batista, K. S. Boland, C. J. Burns, D. L. Clark, S. D. Conradson, R. L. Martin, M. P. Wilkerson and L. E. Wolfsberg, J. Am. Chem. Soc., 2009, 131, 12125 CrossRef CAS PubMed.
- L. A. Seaman, E. A. Pedrick, T. Tsuchiya, G. Wu, E. Jakubikova and T. W. Hayton, Angew. Chem., Int. Ed., 2013, 52, 10589 CrossRef CAS PubMed.
- C. E. Hayes, R. H. Platel, L. L. Schafer and D. B. Leznoff, Organometallics, 2012, 31, 6732 CrossRef CAS.
- T. Cantat, C. R. Graves, K. C. Jantunen, C. J. Burns, B. L. Scott, E. J. Schelter, D. E. Morris, P. J. Hay and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 17537 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- D. J. Grant, T. J. Stewart, R. Bau, K. A. Miller, S. A. Mason, M. Gutmann, G. J. McIntyre, L. Gagliardi and W. J. Evans, Inorg. Chem., 2012, 51, 3613 CrossRef CAS PubMed.
- J. A. Pool, B. L. Scott and J. L. Kiplinger, J. Am. Chem. Soc., 2005, 127, 1338 CrossRef CAS PubMed.
- D. E. Morris, R. E. Da Re, K. C. Jantunen, I. Castro-Rodriguez and J. L. Kiplinger, Organometallics, 2004, 23, 5142 CrossRef CAS.
- K. C. Jantunen, C. J. Burns, I. Castro-Rodriguez, R. E. Da Re, J. T. Golden, D. E. Morris, B. L. Scott, F. L. Taw and J. L. Kiplinger, Organometallics, 2004, 23, 4682 CrossRef CAS.
- B. D. Stubbert and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 6149 CrossRef CAS PubMed.
- J. A. Pool, B. L. Scott and J. L. Kiplinger, J. Alloys Compd., 2006, 418, 178 CrossRef CAS PubMed.
- J. L. Kiplinger, B. L. Scott, E. J. Schelter, J. A. Pool and D. Tournear, J. Alloys Compd., 2007, 444–445, 477 CrossRef CAS PubMed.
- P. Yang, I. Warnke, R. L. Martin and P. J. Hay, Organometallics, 2008, 27, 1384 CrossRef CAS.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482 CrossRef CAS PubMed.
- D. M. King, F. Tuna, J. McMaster, W. Lewis, A. J. Blake, E. J. L. McInnes and S. T. Liddle, Angew. Chem., Int. Ed., 2013, 52, 4921 CrossRef CAS PubMed.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717 CrossRef CAS PubMed.
- B. M. Gardner, J. C. Stewart, A. Davis, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9265 CrossRef CAS PubMed.
- B. M. Gardner, D. Patel, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 123, 10624 CrossRef.
- B. M. Gardner, D. Patel, A. D. Cornish, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Chem. - Eur. J., 2011, 17, 11266 CrossRef CAS PubMed.
- B. M. Gardner, J. McMaster, F. Moro, W. Lewis, A. J. Blake and S. T. Liddle, Chem. - Eur. J., 2011, 17, 6909 CrossRef CAS PubMed.
- D. Patel, D. M. King, B. M. Gardner, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Chem. Commun., 2011, 47, 295 RSC.
- B. M. Gardner, W. Lewis, A. J. Blake and S. T. Liddle, Inorg. Chem., 2011, 50, 9631 CrossRef CAS PubMed.
- B. M. Gardner, J. McMaster, W. Lewis and S. T. Liddle, Chem. Commun., 2009, 2851 RSC.
- S. T. Liddle, J. McMaster, D. P. Mills, A. J. Blake, C. Jones and W. D. Woodul, Angew. Chem., Int. Ed., 2009, 48, 1077 CrossRef CAS PubMed.
- B. M. Gardner, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2009, 131, 10388 CrossRef CAS PubMed.
- R. Boaretto, P. Roussel, N. W. Alcock, A. J. Kingsley, I. J. Munslow, C. J. Sanders and P. Scott, J. Organomet. Chem., 1999, 591, 174 CrossRef CAS.
- R. Boaretto, P. Roussel, A. J. Kingsley, I. J. Munslow, C. J. Sanders, N. W. Alcock and P. Scott, Chem. Commun., 1999, 1701 RSC.
- S. J. Simpson, H. W. Turner and R. A. Andersen, Inorg. Chem., 1981, 20, 2991 CrossRef CAS.
- S. Fortier, J. R. Walensky, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 6894 CrossRef CAS PubMed.
- J. W. Bruno, T. J. Marks and V. W. Day, J. Organomet. Chem., 1983, 250, 237 CrossRef CAS.
- C. M. Fendrick and T. J. Marks, J. Am. Chem. Soc., 1984, 106, 2214 CrossRef CAS.
- J. W. Bruno, G. M. Smith, T. J. Marks, C. K. Fair, A. J. Schultz and J. M. Williams, J. Am. Chem. Soc., 1986, 108, 40 CrossRef CAS.
- C. Morton, I. J. Munslow, C. J. Sanders, N. W. Alcock and P. Scott, Organometallics, 1999, 18, 4608 CrossRef CAS.
- P. G. Edwards, R. A. Andersen and A. Zalkin, Organometallics, 1984, 3, 293 CrossRef CAS.
- A. Zalkin, R. A. Andersen and J. Brennan, Acta Crystallogr., Sect. A: Found. Crystallogr., 1987, 43, 421 Search PubMed.
- C. A. Cruz, D. J. H. Emslie, L. E. Harrington and J. F. Britten, Organometallics, 2008, 27, 15 CrossRef CAS.
- C. A. Cruz, D. J. H. Emslie, C. M. Robertson, L. E. Harrington, H. A. Jenkins and J. F. Britten, Organometallics, 2009, 28, 1891 CrossRef CAS.
- C. E. Hayes and D. B. Leznoff, Organometallics, 2010, 29, 767 CrossRef CAS.
- E. M. Broderick, N. P. Gutzwiller and P. L. Diaconescu, Organometallics, 2010, 29, 3242 CrossRef CAS.
- M. J. Monreal and P. L. Diaconescu, J. Am. Chem. Soc., 2010, 132, 7676 CrossRef CAS PubMed.
- T. Cantat, B. L. Scott and J. L. Kiplinger, Chem. Commun., 2010, 46, 919 RSC.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed.
- P. Roussel, R. Boaretto, A. J. Kingsley, N. W. Alcock and P. Scott, J. Chem. Soc., Dalton Trans., 2002, 1423 RSC.
- J. Maynadié, J.-C. Berthet, P. Thuéry and M. Ephritikhine, Organometallics, 2006, 25, 5603 CrossRef.
- I. Castro-Rodriguez, K. Olsen, P. Gantzel and K. Meyer, J. Am. Chem. Soc., 2003, 125, 4565 CrossRef CAS PubMed.
- J. L. Brown, C. C. Mokhtarzadeh, J. M. Lever, G. Wu and T. W. Hayton, Inorg. Chem., 2011, 50, 5105 CrossRef CAS PubMed.
- G. Nocton, J. Pécaut, Y. Filinchuk and M. Mazzanti, Chem. Commun., 2010, 46, 2757 RSC.
- M. D. Fryzuk, T. S. Haddad and S. J. Rettig, Organometallics, 1991, 10, 2026 CrossRef CAS.
- P. G. Hayes, W. E. Piers and M. Parvez, Organometallics, 2005, 24, 1173 CrossRef CAS.
- K. R. D. Johnson and P. G. Hayes, Chem. Soc. Rev., 2013, 42, 1947 RSC.
- M. E. Thompson, S. M. Baxter, A. R. Bulls, B. J. Burger, M. C. Nolan, B. D. Santarsiero, W. P. Schaefer and J. E. Bercaw, J. Am. Chem. Soc., 1987, 109, 203 CrossRef CAS.
- A. J. Roering, A. F. Maddox, L. T. Elrod, S. M. Chan, M. B. Ghebreab, K. L. Donovan, J. J. Davidson, R. P. Hughes, T. Shalumova, S. N. MacMillan, J. M. Tanski and R. Waterman, Organometallics, 2008, 28, 573 CrossRef.
- C. C. Cummins, R. R. Schrock and W. M. Davis, Organometallics, 1992, 11, 1452 CrossRef CAS.
- R. R. Schrock, S. W. Seidel, N. C. Mösch-Zanetti, K.-Y. Shih, M. B. O'Donoghue, W. M. Davis and W. M. Reiff, J. Am. Chem. Soc., 1997, 119, 11876 CrossRef CAS.
- R. Waterman, Organometallics, 2013, 32, 7249 CrossRef CAS.
- A. Yahia and L. Maron, Organometallics, 2009, 28, 672 CrossRef CAS.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS PubMed.
- Z. Lin, Coord. Chem. Rev., 2007, 251, 2280 CrossRef CAS PubMed.
- L. Castro, A. Yahia and L. Maron, Dalton Trans., 2010, 39, 6682 RSC.
- L. Castro, A. Yahia and L. Maron, C. R. Chim., 2010, 13, 870 CrossRef CAS PubMed.
- L. Castro, A. Yahia and L. Maron, ChemPhysChem, 2010, 11, 990 CrossRef CAS PubMed.
- N. Kaltosyannis, Inorg. Chem., 2013, 52, 3407 CrossRef PubMed.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394 CrossRef CAS PubMed.
- J. R. Walensky, R. L. Martin, J. W. Ziller and W. J. Evans, Inorg. Chem., 2010, 49, 10007 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental considerations, thermodynamic plots, crystallographic and computational details for compounds 1b, 3a, 3b, and 4. CCDC 981878–981881. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc00182f |
| This journal is © The Royal Society of Chemistry 2014 |