 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the electronic environment of cations and anions using ionic liquid mixtures†
Ignacio J.
Villar-Garcia
*,
Kevin R. J.
Lovelock
,
Shuang
Men
and
Peter
Licence
*
The School of Chemistry, The University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: i.villar-garcia@imperial.ac.uk; pete.licence@nottingham.ac.uk; Fax: +44 (0)115 9513563; Tel: +44 (0)7827767149/+44 (0)115 846 6176
First published on 8th April 2014
Abstract
Electrostatic interactions are ubiquitous in ionic liquids and therefore, the electronic environment (i.e. the distribution of electron density) of their constituent ions has a determining influence on their properties and applications. Moreover, the distribution of electron density on atoms is at the core of ionic liquid molecular dynamics simulations. In this work, we demonstrate that changing the composition of ionic liquid mixtures can tune the electronic environment of their constituent ions, both anions and cations. The electronic environment of these ions can be monitored by measuring the characteristic electron binding energies of their constituent atoms by X-ray photoelectron spectroscopy (XPS). The possibility to fine tune, in a controlled way, the electronic environment of specific ions provides an invaluable tool to understand ionic liquid properties and allows the design of ionic liquid mixtures towards specific applications. Here, we demonstrate the power of this tool by tuning the electronic environment of a catalytic centre, and consequently its catalytic activity, by the use of ionic liquid mixtures.
Introduction
Ionic liquids, low temperature molten salts composed entirely of mobile ions, exhibit a large range of properties that make them useful in a wide variety of fields such as synthesis and catalysis,1 biocatalysis,2 electrochemistry,3 separation technology4 or materials science.5 One of the reasons for this wide span of applications is their tunability. Every combination of ions that give rise to an ionic liquid has its own set of properties. The number of combinations can reach the order of trillions if ionic liquids mixtures are considered.6 In the last years, several applications and fundamental studies of ionic liquid mixtures have appeared.7 Very promising examples can be found in their application in lithium battery cells,8 magnesium-based rechargeable batteries,9 dye-sensitised solar cells,10 and in the electrodeposition of metals including magnesium.11 The use of tuneable ionic liquid mixtures as chromatographic stationary phases,12 and solvents for the controlled self-assembly of amphiphilic materials,13 or enzymatic mediated reactions14 also highlights the potential of mixtures to improve the performance of existing ionic liquid-based systems.The potential to fine tune the properties of ionic liquid media by the use of mixtures is clear and the ability to understand and predict these properties would constitute an invaluable tool. However, for applications of ionic liquid mixtures to become widespread, a deep understanding of their properties and the molecular level structure that determines this is needed. The physical and chemical properties of mixture systems have begun to be consistently measured and simulated. The properties of several ionic liquid mixtures have been found to follow an ideal or quasi-ideal mixing behaviour.15 Ionic liquid mixtures have also been found to exhibit enhanced properties when compared to the neat ionic liquids used in their preparation.15c,16 Structurally, ionic liquid mixtures seem to consist of mixtures of randomly distributed ions.7,17 In more detail, their molecular level structure may be dominated by one of the constituent ionic liquids.7,17d,18 The electronic environment of the ions constituting ionic liquids plays a dominant role in determining the molecular level structure, macroscopic properties and the interaction of ionic liquids with other molecules. For instance, accurate reproduction of molecular charge distributions using point charges is critical in classical molecular simulations. In ionic liquids this charge distribution plays an essential role. There is on-going debate as to which force fields most adequately describe ionic liquids.19 Models that allow for polarisation and charge transfer in ionic liquids have recently gained recognition. Traditionally, the total charge on individual ions has been assumed to be an integer.20 However, reduced charges have been used to account for partial charge transfer between the anion and the cation.21 Polarisable force fields, which can artificially reproduce charge transfer effects, have also been used but at much greater computational expense.19c,22 Experimentally obtained charge distributions would be an invaluable piece of information in parameterising accurate electrostatic interactions for ionic liquid systems. The distribution of electron density also has a significant impact in the way molecules interact. Ionic liquid solute interactions can have an enormous impact on applications including catalysis or electrochemistry which depend on the electronic distribution of specific species in solution. For example, the electronic environment of the metal centre of a catalyst is a major factor controlling its catalytic activity. The nature of the ionic liquid anion has been proven to have a determining influence in the activity and stability of several cationic catalytic systems.23 In terms of electrochemical applications, ionic liquid mixtures have also been used to tune the redox potentials of different compounds in solution.24 These examples evidence that the ability to measure and control the electronic environment of ions within ionic liquids, and therefore programming its properties and potential interactions, will be of utmost importance for the ionic liquid field.
X-ray photoelectron spectroscopy (XPS) can be used to monitor the electronic environment of the constituent atoms of a sample. Binding energies measured by XPS can be correlated to calculated atomic charges.25 Binding energy shifts have been compared to quantum mechanically calculated atomic charges of different samples and useful relationships were extracted.26 Due to their low vapour pressure, XPS can be used to analyse ionic liquids and study the electronic environment of their constituent ions.27 Recent XPS studies for neat ionic liquids have demonstrated that the characteristic binding energies from the carbon and nitrogen atoms within the cation headgroup correlate with the calculated amount of charge transferred from the anion to the cation.28 For anions such as halides or acetate, [OAc]−, the electron density on the cation was found to be higher than for low basicity anions such as [Tf2N]− or [TfO]− (where [Tf2N]− = bis[(trifluoromethane)sulfonyl]imide and [TfO]− = trifluoromethanesulfonate). These changes in electron density were positively correlated to anion basicity (β), determined by solvatochromic methods.28a,b
In this publication, we study the use of ionic liquid mixtures to tune the electronic environment of their constituent ions. We present a detailed XPS study of three different ionic liquid mixtures systems composed of a common cation and two different anions at different molar ratios (a total of 16 mixtures). Two mixture systems contain a strongly coordinating, high basicity anion and a weakly coordinating, low basicity anion: [C8C1Im]Cl + [C8C1Im][Tf2N] (denominated as [C8C1Im]Clx[Tf2N]1−x) and [C8C1Im]Cl + [C8C1Im][TfO] ([C8C1Im]Clx[TfO]1−x) and one of the mixture systems contains two strongly coordinating, high basicity anions: [C8C1Im][OAc] + [C8C1Im]Cl ([C8C1Im]Clx[OAc]1−x). The measured binding energies confirm changes in the electronic environment of the ions with composition. These changes can be used to tune the properties and interactions of a particular ionic liquid system. To demonstrate the power of this tool, we use an ionic liquid mixture to fine tune the electronic environment of a catalytic metal centre in an ionic liquid solution and achieve distinct turnover frequencies.
Results and discussion
Tuning the electronic environment of ions
Fig. 1 illustrates how the electronic environment of both anions and cations within ionic liquids can be tuned using mixtures, showing the N 1s and Cl 2p3/2 high resolution XP spectra (characteristic of the electronic environment of the cation and anion respectively) of all the different [C8C1Im]Clx[Tf2N]1−x, [C8C1Im]Clx[TfO]1−x and [C8C1Im]Cl0.5[OAc]0.5 mixtures and respective neat ionic liquids. It can be clearly seen that for mixtures of anions of different basicity, i.e. [C8C1Im]Clx[Tf2N]1−x and [C8C1Im]Clx[TfO]1−x, the binding energy of the Ncation 1s peak (Fig. 1a and b) changes in a quasi-linear behaviour from the characteristic energy of one of the neat ionic liquids to the other. It should be noted at this point that the full width at half maximum (FWHM) of the photoemission envelopes in the mixtures spectra were comparable to those exhibited by the neat ionic liquid spectra, see the ESI† (Fig. S7a, b and c). This observation clearly illustrates that the XP spectra of the mixtures are not composed of a superposition of the XP spectra of each neat ionic liquid, but a new signal corresponding to a series of entirely new electronic environments. This data agrees with the well reported structure of ionic liquid mixtures consisting of a randomly distributed mixture of all component ions.17a–c Consequently, the cations in the ionic liquid mixture are all interacting with a statistically similar environment which is composed of an average number of both anions at the same time.17d As a result of this mixed interaction, the net amount of charge transferred to each cation in the mixture is identical, i.e. the electronic environment of the cation is now distinct to that of the two neat ionic liquids. The binding energies acquired for the carbon atoms within the cationic headgroup (C2, C4, C5, C6 and C7 1s) and Ncation 1s peaks suggest that the final average electron density associated with the cation is a weighted average of the electron densities of the same cation in the two neat ionic liquids, where the weighting is reflective of the molar ratio of the two neat ionic liquids employed. These results are also consistent with preliminary analyses made on a pyrrolidinium-based ionic liquid mixture.28cFig. 2 and S8† show the measured binding energies for each component of the [C8C1Im]Clx[Tf2N]1−x and [C8C1Im]Clx[TfO]1−x mixtures respectively (extracted from suitable fitting models, see Experimental section in the ESI†) plotted as a function of composition (molar fraction of [C8C1Im]Cl) to allow trends to be easily identified. All the measured binding energies for the carbon and nitrogen atoms within the cationic headgroup (C2, C4, C5, C6 and C7 1s and Ncation 1s: Figures 2a–d and S8a–d) shift progressively, in a near-linear behaviour, between the binding energies characteristic of the corresponding neat ionic liquids. | ||
| Fig. 1 High resolution XP spectra of the different mixture systems: (a) N 1s and (d) Cl 2p3/2 for [C8C1Im]Clx[Tf2N]1−x, (b) N 1s and (e) Cl 2p3/2 for [C8C1Im]Clx[TfO]1−x and (c) N 1s and (f) Cl 2p3/2 for [C8C1Im]Clx[OAc]1−x. Data recorded for the neat ionic liquids is also included for comparison. In this case the intensity of the photoemission peaks have been normalised to the intensity of the Ncation 1s peak, so the natural change in peak areas with composition can be also visualised. All XP spectra are charge corrected by setting the aliphatic carbon signal, Calkyl 1s, to 285.0 eV as in reference 28d (see supplementary information for fitting models) and offset vertically for clarity. | ||
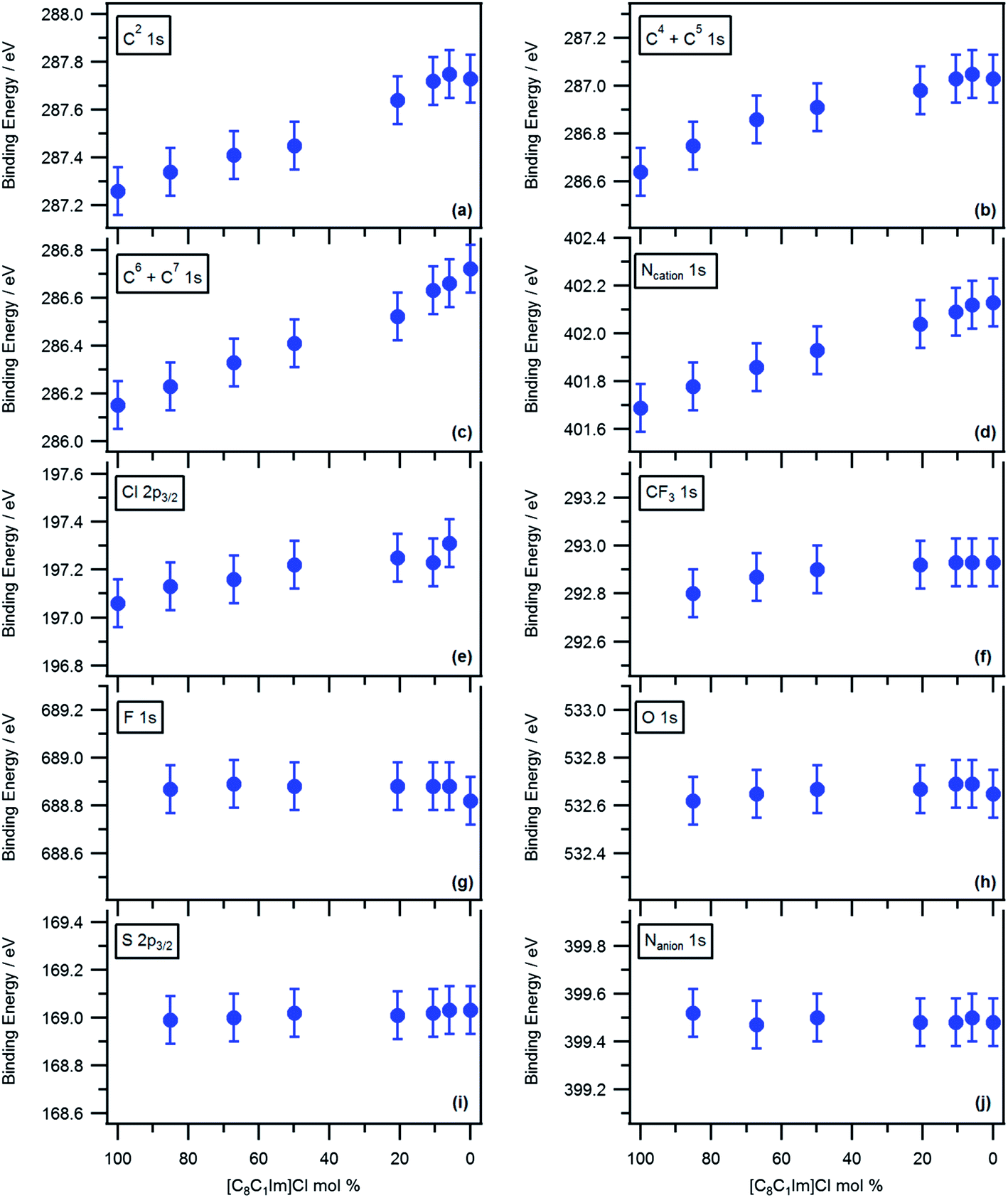 | ||
| Fig. 2 (a) C 1s C2, (b) C 1s C4 + C5, (c) C6 + C7 1s, (d) Ncation 1s, (e) Cl 2p3/2, (f) CF3 1s, (g) F 1s, (h) O 1s, (i) S 2p3/2 and (j) Nanion 1s, binding energies for [C8C1Im]Clx[Tf2N]1−x mixtures. The binding energies were measured after charge correcting all XP spectra by setting the carbon aliphatic signal, Calkyl 1s, to 285.0 eV as in reference 28d (see supplementary information for fitting models). The y-axis scales for binding energy are all the same range, making comparisons easier; hence, the error bars (±0.1 eV) are all the same height. | ||
The Cl 2p3/2 peak in the [C8C1Im]Clx[Tf2N]1−x and [C8C1Im]Clx[TfO]1−x mixtures are observed at higher binding energies than the Cl 2p3/2 peak in [C8C1Im]Cl (Fig. 1d and e and 2e and S8e† respectively), i.e. the higher the amount of [C8C1Im][Tf2N] or [C8C1Im][TfO], the higher the binding energy of the Cl 2p3/2 peak. This variation in binding energy can only be explained by a change in the interactions between the Cl− and other components in the mixture, in comparison to those in neat [C8C1Im]Cl. In a mixture, both anions are competing for more favourable interaction sites17d and on average, each cation will be interacting with both types of anions at the same time. The presence of poorly coordinating [Tf2N]− will clearly lead to a reduction in the number of basic anions in the coordination sphere of the cation. As [Tf2N]− anions transfer less electronic charge to the cation than Cl− anions, the cations within the mixture will have more capacity to accept electron density from Cl− anions. Consequently, the Cl− anions will be able to transfer more electronic charge to these cations. This larger donation of charge will leave the Cl− anion with less electron density and therefore, their electrons will exhibit higher binding energies than the Cl− anion electrons in neat [C8C1Im]Cl. In Fig. 2f–j and S8f–i,† it can be seen that there is no significant perturbation in the measured binding energies of the elements within the [Tf2N]− and [TfO]− in the mixtures. [Tf2N]− and [TfO]− are low basicity anions and they do not participate in significant charge transfer.28a Moreover, as the negative charge associated with [Tf2N]− is delocalised across many atoms, any change in electron density will be distributed around many atoms and therefore, will be too small to be measured directly by XPS, i.e. it will be within the error of the measurement.
In Fig. 1c and f present a study of a mixture of two ionic liquids containing two strongly coordinating, high basicity anions. The exhibited binding energy of the Ncation 1s peak for the [C8C1Im]Cl0.5[OAc]0.5 mixture and neat [C8C1Im]Cl and [C8C1Im][OAc] are the same within acceptable experimental error (see Fig. S9† as well). Both anions are highly coordinating, highly basic in nature and transfer a significant amount of electron density to the cation. The Cl 2p3/2 and the O 1s peaks (Fig. S9†) for [C8C1Im]Cl0.5[OAc]0.5 are also observed at the same binding energy (within error) as in the neat [C8C1Im]Cl and [C8C1Im][OAc] respectively. In summary, the binding energies of all of the elements in the mixture are all comparable to those in the neat ionic liquids (Fig. S9 in the ESI†), i.e. any difference in binding energies is smaller than detectable by XPS. It can be concluded that the electronic environment of the cations and anions in [C8C1Im]Cl0.5[OAc]0.5 is very similar to that in the neat ionic liquids themselves, electronically it could be stated that they are almost identical.
The binding energy data presented in this study evidences the tunable nature of the electronic environment of the ions within ionic liquid mixtures. The electronic environment of the ions of a particular ionic liquid can be effectively modified by the addition of other ions. The amount of fine tuning of the electronic environment of a particular cation is determined by the initial electronic environments of the cations in the neat ionic liquids used for the mixture. The electronic environments of the cations in [C8C1Im]Cl and [C8C1Im][OAc] are very similar and therefore, mixtures containing the two will not have a measurable effect in the electronic environment of the ions in the mixture. [C8C1Im]Clx[Tf2N]1−x and [C8C1Im]Clx[TfO]1−x mixtures offer a wider range of fine tuning for the electronic environment of both the cation and the anion. The tunability range is slightly higher in the [C8C1Im]Clx[Tf2N]1−x system as the difference in the electronic environment of the cations in the neat ionic liquids is the largest. Finding a combination of ionic liquids with larger differences in electronic environment of the cation will widen the possibilities. For example, anions such as tris(pentafluoroethyl)trifluorophosphate, [FAP]−, and bis[(pentafluoroethylane)sulfonyl]imide, [Pf2N]−, with the lowest investigated basicity,28a could allow even wider tunability for the [C8C1Im]+ cation electronic environment. The change in the chloride electronic environment for the different ionic liquid mixtures means that ionic liquid mixtures with a common cation can also be used to tune the anion's electronic environment. It is expected that the tunability range of the anion can be further increased by ionic liquid mixtures containing different cations.
This data on binding energies also offers invaluable information to computational simulations of ionic liquids. It is especially relevant in order to evaluate point charges in computational force field models. XPS data shows that charge transfer from anion to cation occurs for ionic liquid mixtures as well as for neat ionic liquids and it is characteristic of every combination of ions. Therefore, when carrying out simulations of ionic liquids and ionic liquid mixtures, reduce charge or polarisable force fields will more accurately describe the real charge distribution within ionic liquids. In addition, the distinct binding energies and narrow peaks (with similar FWHM as for neat ionic liquids) obtained for ionic liquid mixtures suggest that the electronic environment of the ions within ionic liquid mixtures is distinct and unique. This cannot be the consequence of a mixture of different local distribution of ions but the result of the sum of long range anion–cation interactions and charge transfer events. Consequently, simulations based on small clusters (maybe <10 ions) will not be able to capture the real electronic environment of ionic liquid mixtures as it is the combination of several solvent shells that appears to influence the electron density on the ions. Moreover, binding energies could be used as a useful solvation parameter. For neat ionic liquids, the binding energy of Ncation 1s has been found to linearly correlate with the hydrogen bond donor ability of the anion, β, calculated by solvatochromic methods.28a,b For mixtures, β has not been measured as determining β for mixtures is more challenging than for pure samples as one of the ions normally preferentially interacts with the probing dye giving a wrong measure of the average hydrogen donor ability of the mixture.29 XPS binding energies could also be used as an alternative to β in both neat ionic liquids and ionic liquid mixtures.
Tuning the electronic environment of a metal catalyst in solution
The electronic environment of the ions that constitute ionic liquids also has a determining influence in the interaction of these ions with solutes.30 Changing the interaction of ions with starting materials, reactive intermediates or catalysts can have an impact upon the outcome of a chemical reaction.31 Ionic liquids can act as ligands of charged catalysts.23b–d Some cationic catalyst have been found to be stable, unstable, active or even change coordination depending on the constituent anion of the ionic liquid media.23b–d For example, it has been shown that a mixture of Pd(PPh3)4/NaCl/[C8C1Im][A] and aryl bromide renders a stable phosphine–imidazolydidene palladium complex that successfully catalyses the Suzuki cross-coupling reaction of the aryl bromide with phenylboronic acid over different cycles.23b,c The formation of the final catalytic complex and its activity have been found to depend on the nature of the anion. The complex is not formed when [PF6]− and [SbF6]− anions are used and different turnover frequencies (TOF) are obtained for different ionic liquids in which the active complex is formed. It has been suggested that anions of high basicity interact more strongly with the catalyst and change the electronic environment of the catalyst and hence its activity and TOF.The electronic environment of the metal centre can also be probed directly using XPS.32 We have measured the Pd 3d5/2 binding energies of the solutions produced after mixing aqueous Na2CO3/aryl bromide/Pd(PPh3)4/NaCl/[C8C1Im][Tf2N], aqueous Na2CO3/aryl bromide/Pd(PPh3)4/NaCl/[C8C1Im][OAc] and aqueous Na2CO3/aryl bromide/Pd(PPh3)4/NaCl/[C8C1Im]Cl0.5[Tf2N]0.5 (see Experimental information). The measured binding energy for the Pd 3d5/2 in [C8C1Im]Cl0.5[Tf2N]0.5 containing a highly basic Cl− anion and a non-coordinating [Tf2N]− anion rendered a palladium metal centre with a Pd 3d5/2 binding energy value (338.4 eV) that lies approximately equidistant between the binding energy values observed for the same palladium catalyst dissolved in a high basicity [OAc]− ionic liquid (338.7 eV) and a low basicity [Tf2N]−-based ionic liquid (337.6 eV). These binding energies are a measure of the electronic density of the palladium metal centre of the catalyst. Subsequently, Suzuki cross-coupling reactions of phenylboronic acid with the aryl bromide in the three systems were performed under the same conditions and their TOF calculated. Fig. 3 shows that there is a lineal correlation between the Pd 3d5/2 binding energy and the TOF for these systems. These measurements exemplify how the combination of two different ionic liquids can be used to fine tune the anion–catalyst interactions and therefore, the electronic environment of the metal centre of a catalyst, in order to modify the TOF of the reaction. XPS is also proven as an ideal technique to measure these changes in electronic environment in ionic liquid media.
 | ||
| Fig. 3 Pd 3d5/2 binding energies of the solutions produced after mixing aqueous Na2CO3/aryl bromide/Pd(PPh3)4/NaCl/[C8C1Im][Tf2N], aqueous Na2CO3/aryl bromide/Pd(PPh3)4/NaCl/[C8C1Im][OAc] and aqueous Na2CO3/aryl bromide/Pd(PPh3)4/NaCl/[C8C1Im]Cl0.5[Tf2N]0.5vs. turnover frequency (TOF) of the Suzuki cross-coupling reaction of phenylboronic acid with the aryl bromide in these mixtures at the same conditions. | ||
Conclusions
Changes in the composition of ionic liquid mixtures can be used to tune the electronic environment (i.e. the distribution of electron density) of their ions, both anions and cations. XPS is ideally suited to investigate these changes in electronic environment by the measurement of binding energies of specific atoms. We have shown that the final electronic environment of the cation depends upon the nature and relative percentages of the anions present in the mixture. Moreover, the electronic environment of the anion is also affected by the presence of other anions. We also show that the observed electronic environments are the sum of long range anion–cation interactions and charge transfer events and are consequently, distinct and unique for every mixture. Therefore, simulations should use large clusters in order to capture the real electronic environment within ionic liquids and ionic liquid mixtures. Binding energy data is also an invaluable experimental starting point to aid in the parameterisation of point charges in force field models used in simulations of ionic liquids and ionic liquid mixtures. The electronic environment within ionic liquids is one of the dominating factors governing the properties exhibited by ionic liquids and the interactions of their constituting ions with solutes. The ability to fine tune the electronic environment of ions within ionic liquid systems signifies a step forward towards the ultimate goal of ionic liquids systems: delivery of a material with a very specific series of physical and chemical properties to match a given application. We demonstrate this potential by finely tuning the interactions of an ionic liquid mixture with a metal catalyst in order to modify the electronic environment of the metal centre and consequently the TOF of the catalysed reaction.Acknowledgements
PL acknowledges the EPSRC for the award of an ARF (EP/D073014/1). KRJL acknowledges Imperial College London for the award of a JRF. The authors are grateful to Ms Emily F. Smith and Prof. Robert G. Jones, The University of Nottingham, and Richard P. Matthews, Imperial College Nottingham, for helpful discussions and critical advice.References
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed.
- F. van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757 CrossRef CAS PubMed.
- Electrochemical Aspects of Ionic Liquids, ed. H. Ohno, Wiley, Hoboken, New Jersey, 2nd edn, 2011 Search PubMed.
- A. Berthod, M. Ruiz-Angel and S. Carda-Broch, J. Chromatogr. A, 2008, 1184, 6 CrossRef CAS PubMed.
- T. Torimoto, T. Tsuda, K. Okazaki and S. Kuwabata, Adv. Mat., 2010, 22, 1196 CrossRef CAS PubMed.
- (a) J. D. Holbrey and K. R. Seddon, Clean Technol. Environ. Policy, 1999, 1, 223 CrossRef; (b) N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- H. Niedermeyer, J. P. Hallett, I. J. Villar-Garcia, P. A. Hunt and T. Welton, Chem. Soc. Rev., 2012, 41, 7780 RSC.
- (a) M. Egashira, M. Tanaka-Nakagawa, I. Watanabe, S. Okada and J. I. Yamaki, J. Power Sources, 2006, 160, 1387 CrossRef CAS PubMed; (b) P. M. Bayley, A. S. Best, D. R. MacFarlane and M. Forsyth, Phys. Chem. Chem. Phys., 2011, 13, 4632 RSC; (c) Q. Zhou, W. A. Henderson, G. B. Appetecchi and S. Passerini, J. Phys. Chem. C, 2010, 114, 6201 CrossRef CAS.
- (a) T. Kakibe, N. Yoshimoto, M. Egashira and M. Morita, Electrochem. Commun., 2010, 12, 1630l CrossRef PubMed; (b) T. Kakibe, J. Y. Hishii, N. Yoshimoto, M. Egashira and M. Morita, J. Power Sources, 2012, 203, 195 CrossRef CAS PubMed.
- P. Wang, B. Wenger, R. Humphry-Baker, J. E. Moser, J. Teuscher, W. Kantlehner, J. Mezger, E. V. Stoyanov, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 6850 CrossRef CAS PubMed.
- P. Wang, Y. N. NuLi, J. Yang and Z. Z. Feng, Surf. Coat. Technol., 2006, 201, 3783 CrossRef CAS PubMed.
- Q. Q. Baltazar, S. K. Leininger and J. L. Anderson, J. Chromatogr. A, 2008, 1182, 119 CrossRef CAS PubMed.
- T. Inoue and T. Misono, J. Colloid Interface Sci., 2008, 326, 483 CrossRef CAS PubMed.
- S. H. Lee, S. H. Ha, N. M. Hiep, W. J. Chang and Y. M. Koo, J. Biotechnol., 2008, 133, 486 CrossRef CAS PubMed.
- (a) J. N. Canongia Lopes, T. C. Cordeiro, J. Esperança, H. J. R. Guedes, S. Huq, L. P. N. Rebelo and K. R. Seddon, J. Phys. Chem. B, 2005, 109, 3519 CrossRef PubMed; (b) A. Jarosik, S. R. Krajewski, A. Lewandowski and P. Radzimski, J. Mol. Liq., 2006, 123, 43 CrossRef CAS PubMed; (c) F. Castiglione, G. Raos, G. B. Appetecchi, M. Montanino, S. Passerini, M. Moreno, A. Famulari and A. Mele, Phys. Chem. Chem. Phys., 2010, 12, 1784 RSC; (d) P. Navia, J. Troncoso and L. Romani, J. Solution Chem., 2008, 37, 677 CrossRef CAS; (e) P. Navia, J. Troncoso and L. Romani, J. Chem. Eng. Data, 2007, 52, 1369 CrossRef CAS.
- (a) H. Every, A. G. Bishop, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2000, 45, 1279 CrossRef CAS; (b) A. Stoppa, R. Buchner and G. Hefter, J. Mol. Liq., 2010, 153, 46 CrossRef CAS PubMed; (c) J. Kagimoto, K. Noguchi, K. Murata, K. Fukumoto, N. Nakamura and H. Ohno, Chem. Lett., 2008, 37, 1026 CrossRef CAS; (d) K. A. Fletcher, S. N. Baker, G. A. Baker and S. Pandey, New J. Chem., 2003, 27, 1706 RSC; (e) A. Finotello, J. E. Bara, S. Narayan, D. Camper and R. D. Noble, J. Phys. Chem. B, 2008, 112, 2335 CrossRef CAS PubMed.
- (a) M. Y. Lui, L. Crowhurst, J. P. Hallett, P. A. Hunt, H. Niedermeyer and T. Welton, Chem. Sci., 2011, 2, 1491 RSC; (b) J. M. Andanson, M. J. Beier and A. Baiker, J. Phys. Chem. Lett., 2011, 2, 2959 CrossRef CAS; (c) K. Shimizu, M. Tariq, L. P. N. Rebelo and J. N. Canongia Lopes, J. Mol. Liq., 2010, 153, 52 CrossRef CAS PubMed; (d) M. Brüssel, M. Brehm, T. Voigt and B. Kirchner, Phys. Chem. Chem. Phys., 2011, 13, 13617 RSC.
- A. B. Pereiro, J. M. M. Araujo, F. S. Oliveira, C. E. S. Bernardes, J. Esperança, J. N. Canongia Lopes, I. M. Marrucho and L. P. N. Rebelo, Chem. Commun., 2012, 48, 3656 RSC.
- (a) O. Borodin, J. Phys. Chem. B, 2009, 113, 11463 CrossRef CAS PubMed; (b) E. J. Maginn, J. Phys.: Condens. Matter, 2009, 21 CrossRef CAS PubMed; (c) F. Dommert, K. Wendler, R. Berger, L. Delle Site and C. Holm, ChemPhysChem, 2012, 13, 1625 CrossRef CAS PubMed.
- (a) J. N. Canongia Lopes, J. Deschamps and A. A. H. Pádua, J. Phys. Chem. B, 2004, 108, 2038 CrossRef; (b) J. N. Canongia Lopes, M. F. Costa Gomes and A. A. H. Pádua, J. Phys. Chem. B, 2006, 110, 16816 CrossRef PubMed; (c) T. Köddermann, D. Paschek and R. Ludwig, ChemPhysChem, 2007, 8, 2464 CrossRef PubMed; (d) I. J. Villar-Garcia, E. F. Smith, A. W. Taylor, F. Qiu, K. R. J. Lovelock, R. G. Jones and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 2797 RSC.
- (a) B. L. Bhargava and S. Balasubramanian, J. Chem. Phys., 2007, 127, 6 CrossRef PubMed; (b) T. I. Morrow and E. J. Maginn, J. Phys. Chem. B, 2002, 106, 12807 CrossRef CAS.
- J. Rigby and E. I. Izgorodina, Phys. Chem. Chem. Phys., 2013, 15, 1632 RSC.
- (a) P. Wasserscheid and P. Schulz, in Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, WILEY-VCH, 2nd edn, 2008, vol. 2 Search PubMed; (b) F. McLachlan, C. J. Mathews, P. J. Smith and T. Welton, Organometallics, 2003, 22, 5350 CrossRef CAS; (c) C. J. Mathews, P. J. Smith and T. Welton, Chem. Commun., 2000, 1249 RSC; (d) P. Wasserscheid, C. M. Gordon, C. Hilgers, M. J. Muldoon and I. R. Dunkin, Chem. Commun., 2001, 1186 RSC.
- L. H. J. Xiong, A. M. Fletcher, S. G. Davies, S. E. Norman, C. Hardacre and R. G. Compton, Chem. Commun., 2012, 48, 5784 RSC.
- (a) K. Siegbahn, C. Nordling, G. Johansson, J. Hedman, P. F. Heden, K. Hamrin, U. Gelius, T. Bergmark, L. O. Werme and R. Manne, ESCA Applied to Free Molecules, North Holland, Amsterdam, 1969 Search PubMed; (b) A. P. Pijpers and R. J. Meier, Chem. Soc. Rev., 1999, 28, 233 RSC; (c) R. Nordberg, R. G. Albridge, T. Bergmark, U. Ericson, J. Hedman, C. Nordling, K. Siegbahn and B. J. Lindberg, Ark. Kemi, 1968, 28, 257 Search PubMed; (d) U. Gelius, P. F. Heden, J. Hedman, B. J. Lindberg, R. Marine, R. Nordberg, C. Nordling and K. Siegbahn, Phys. Scr., 1970, 2, 70 CrossRef CAS.
- (a) B. Folkesson and R. Larsson, J. Electron Spectrosc. Relat. Phenom., 1990, 50, 251 CrossRef CAS; (b) P. Sundberg, C. Andersson, B. Folkesson and R. Larsson, J. Electron Spectrosc. Relat. Phenom., 1988, 46, 85 CrossRef CAS; (c) P. Sundberg, R. Larsson and B. Folkesson, J. Electron Spectrosc. Relat. Phenom., 1988, 46, 19 CrossRef CAS; (d) C. Sleigh, A. P. Pijpers, A. Jaspers, B. Coussens and R. J. Meier, J. Electron Spectrosc. Relat. Phenom., 1996, 77, 41 CrossRef CAS.
- K. R. J. Lovelock, I. J. Villar-Garcia, F. Maier, H. P. Steinrück and P. Licence, Chem. Rev., 2010, 110, 5158 CrossRef CAS PubMed.
- (a) T. Cremer, C. Kolbeck, K. R. J. Lovelock, N. Paape, R. Wölfel, P. S. Schulz, P. Wasserscheid, H. Weber, J. Thar, B. Kirchner, F. Maier and H. P. Steinrück, Chem.–Eur. J., 2010, 16, 9018 CrossRef CAS PubMed; (b) B. B. Hurisso, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 17737 RSC; (c) S. Men, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 15244 RSC; (d) I. J. Villar-Garcia, E. F. Smith, A. W. Taylor, F. Qiu, K. R. J. Lovelock, R. G. Jones and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 2797 RSC.
- (a) V. T. Wyatt, D. Bush, J. Lu, J. P. Hallett, C. L. Liotta and C. A. Eckert, J. Supercrit. Fluids, 2005, 36, 16 CrossRef CAS PubMed; (b) E. B. Tada, L. P. Novaki and O. A. El Seoud, J. Phys. Org. Chem., 2000, 13, 679 CrossRef CAS.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790 RSC.
- (a) L. J. Xu, W. P. Chen and J. L. Xiao, Organometallics, 2000, 19, 1123 CrossRef CAS; (b) C. Chiappe, D. Pieraccini and C. S. Pomelli, in Ionic Liquids in Organic Synthesis, ed S. V. Malhotra, Amer Chemical Soc, Washington, 2007, vol. 950 Search PubMed.
- (a) E. F. Smith, I. J. Villar Garcia, D. Briggs and P. Licence, Chem. Commun., 2005, 5633 RSC; (b) A. W. Taylor, F. L. Qiu, I. J. Villar-Garcia and P. Licence, Chem. Commun., 2009, 5817 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: complete experimental information including XP spectra fitting models and supplementary figures. See DOI: 10.1039/c4sc00106k |
| This journal is © The Royal Society of Chemistry 2014 |