 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel PEG-based solid support enables the synthesis of >50 amino-acid peptide thioesters and the total synthesis of a functional SUMO-1 peptide conjugate†
Emmanuelle
Boll
,
Hervé
Drobecq
,
Nathalie
Ollivier
,
Laurent
Raibaut
,
Rémi
Desmet
,
Jérome
Vicogne
and
Oleg
Melnyk
*
UMR CNRS 8161, Université Lille Nord de France, Institut Pasteur de Lille, 1 rue du Pr Calmette, 59021 Lille, France. E-mail: oleg.melnyk@ibl.fr; Web: http://csb.ibl.fr Tel: +33 320871214
First published on 5th February 2014
Abstract
A bis(2-sulfanylethyl)amino PEG-based resin enabled the synthesis of large (∼50 Aa) SEA or thioester peptides using Fmoc-SPPS. These peptide segments permitted the first total synthesis of a 97 amino-acid long SUMO-1-SEA peptide thioester surrogate and of a functional and reversible SUMO-1 peptide conjugate.
Introduction
The synthesis of proteins gives access to large peptides or small proteins with an atom-by-atom control of their structure. Today, proteins are usually assembled by the sequential ligation of unprotected peptide segments.1 Native chemical ligation (NCL)2,3 or expressed protein ligation4,5 of a C-terminal peptide thioester with an N-terminal cysteinyl peptide is the most popular amide bond forming reaction used in the field, although other complementary methods are emerging.6–10The large diversity of chemical methods available now for producing modified peptides in high purity is increasingly used for the total or semisynthesis of modified proteins which cannot be easily produced using biological methods.11 Proteins featuring well-defined post-translational modifications at specific sites such as phosphorylation12 or glycosylation13 belongs to this family. Another post-translational modification which has attracted much attention recently is ubiquitylation, which has been addressed using total14–20 or semisynthetic approaches.21–23 The preparation of ubiquitin conjugates has also been achieved using non-native chemoselective ligation methods such as oxime,24 disulfide,25 1,2,3-triazole26,27 or thioether28,29 bond forming reactions.
Recent work described also the total synthesis of the 85 amino-acid long ubiquitin-fold modifier 1 (UFM1) using sequential α-ketoacid–hydroxylamine ligations.9 Besides this, the total synthesis and introduction of other protein modifiers is faced with significant challenges. In particular, the total synthesis of small ubiquitin-like modifier (SUMO30) proteins and SUMO conjugates remains to be addressed. The C-terminal glycine residue of SUMO modifier is linked to the side chain amino group of a lysine within the target protein through an isopeptidic bond. This reversible modification is implicated in many important physiological functions such as transcription and DNA repair.31 SUMO modifier is thought to act by modulating the interactions of the target protein with its partners. However, the molecular mechanisms implicated in this regulation are still poorly understood mostly because SUMO conjugates are highly labile within cells, making their isolation highly challenging.32
Up to now, SUMO-133 or SUMO-234 conjugates were prepared by semisynthesis using recombinant SUMO proteins and non-cleavable 1,2,3-triazole linkers. We show for the first time that protein total synthesis can give access to homogeneous, functional and reversible SUMO-1 conjugates.
The work reported here relies on the chemistry of bis(2-sulfanylethyl)amido (SEA) thioester surrogate (Scheme 1).35–38 First, the assembly of SUMO-1 proteins described in this work required to push the limits of current technology by addressing the Fmoc-SPPS synthesis of long (∼50 Aa) SEA peptides and peptide thioesters. This was achieved by developing novel SEA PEG-based solid supports which are compatible with standard Fmoc-SPPS. We report also the assembly of SUMO-1 isoform modified at the C-terminus by a latent SEA thioester surrogate (Scheme 1). This protein could be isolated or alternately ligated in situ with a SUMO-1 peptide consensus motif to produce a 12.5 kDa SUMO-1-peptide conjugate in a one-pot process. Importantly, the tertiary structure and functionality of the SUMO-1 modifier within the conjugate as well as its reversible nature was demonstrated by the specific cleavage of the glycyl-cysteinyl peptide bond with a recombinant SUMO-1 deconjugating enzyme.
 | ||
| Scheme 1 Synthesis of long SEA peptide thioester surrogates enabled the assembly of a functional SUMO-1 conjugate. | ||
Results and discussion
The primary sequence of SUMO-1 protein features only one internal cysteine residue in position 51 (Scheme 1). A Cys residue was appended to the target lysine side-chain within the SUMO-1 peptide consensus motif to enable the assembly of the SUMO-1 peptide conjugate in three pieces using the NCL or related reactions. Lys(Cys) peptides have also been used successfully by other groups for accessing to ubiquitinated proteins.39,40 An alternative would be to use a target peptide featuring a γ-41,42 or δ-mercaptolysine14,15,18 residue. However, the access to a native SUMO-1 conjugate using this latter strategy would require protecting the Cys51 thiol group during the desulfurization of the mercaptolysine residue.Given the position of the cysteinyl residues in the target SUMO-1 conjugate, the assembly can start with the SUMO-1 peptide consensus motif or alternately with the SUMO-1 (2–50) peptide segment. The latter approach was selected in this work to enable the isolation of SUMO-1 protein featuring a latent thioester surrogate functionality at the C-terminus. We chose the bis(2-sulfanylethyl)amido35–38 (SEA) N,S-acyl shift system43 as latent thioester surrogate, which in its cyclic form called SEAoff is compatible with the NCL reaction used for assembling the SUMO-1 protein.37,38 This N-to-C assembly strategy resembles that developed by Brik and co-workers for the synthesis of a ubiquitin latent thioester.17 These authors used a C-terminal S-protected N-methylcysteine as latent thioester,44 which was deprotected and exchanged by 3-mercaptopropionic acid (MPA) in a subsequent step.
The strategy depicted in Scheme 1 is faced with the challenge of synthesizing 50 amino-acid long peptide segments featuring a C-terminal SEA or alkylthioester group. The synthesis of large peptide thioesters using standard Fmoc-SPPS is one of the major bottlenecks for addressing the synthesis of large proteins or complex scaffolds such as SUMO-1 conjugates.45 C-terminal bis(2-sulfanylethyl)amido (SEA) peptides can be synthesized by Fmoc-SPPS35 and used as precursors for the synthesis of peptide alkylthioesters.46–48 Up to now, we used a bis(2-sulfanylethyl)amino polystyrene (SEA PS) resin for the Fmoc-SPPS of SEA peptides.35,49 Although SEA PS resin permitted the synthesis of various SEA peptides composed of up to 35 amino acids, we anticipated that the synthesis of the large SUMO-1 peptide segments would require a modern PEG-based solid support instead, which is more adapted for the synthesis of long peptides.
SEA PEG-based solid supports were prepared by reacting the trifluoroacetate salt of bis(2-sulfanylethyl)amine 2 with commercially available trityl alcohol PEG-based resins as shown in Scheme 2. We targeted solid supports of type 5 for which both thiol groups are linked to the solid support and protected by a trityl group as for SEA PS resin. However, SEA PS resin was prepared by reacting the bis(2-sulfanylethyl)amine 2 with trityl chloride polystyrene resin in N,N-dimethylformamide (DMF).49 In this case, an efficient protection of both thiol groups and the formation of cross-links within the PS beads was facilitated by the large excess and high loading of the starting trityl chloride polystyrene resin (∼1.4 mmol g−1, 10 equiv.). The loading of commercially available trityl alcohol PEG-based resins (0.25–0.6 mmol g−1) is significantly less than the loading of trityl chloride polystyrene resins. Therefore, partial protection and incomplete cross-linking of the bis(2-sulfanylethyl)amine 2 leading to the formation of solid supports of type 3 was a potential issue. Indeed, the reaction of dithiol 2 with PEG-based solid supports 1a,b in the presence of trifluoroacetic acid yielded solid supports of type 3 as shown by the development of a positive Ellman assay. The presence of free thiols during the Fmoc-SPPS can be troublesome. Therefore, solid supports 3a,b were treated with N,N,N′,N′-tetramethyl-azodicarboxamide (TMAD) at pH 7 to mask the thiol groups as internal disulfides. Indeed, the resulting solid supports 4a,b furnished a negative Ellman assay, while the chloranil assay was strongly positive. The coupling of Fmoc-Ala-OH using HATU/DIEA activation followed by the quantification of the Fmoc group indicated a loading of 0.15–0.21 mmol g−1. The usefulness of solid supports 4a,b for the Fmoc-SPPS of SEA peptides was verified by the automated synthesis of model peptide ILKEPVHGA-SEA (49–54% crude, 18–21% isolated by HPLC).
 | ||
| Scheme 2 Synthesis of SEA PEG-based solid supports. | ||
Interestingly, solid supports 1c,d furnished directly SEA resins of type 5 as shown by the development of a negative Ellman assay and of a strongly positive chloranil assay, with a loading of ∼0.11–0.2 mmol g−1. Solid supports 5c,d proved to be useful for the Fmoc-SPPS of long SEA peptides as shown later. Consequently, the study of solid supports 4a,b was discontinued.
Four different peptides were synthesized to illustrate the usefulness of SEA PEG resins 5c,d for accessing to large SEA peptides (Scheme 3, Table 1). Peptides 7a and 7b correspond to SUMO-1 (2–51) and SUMO-1 (52–97) respectively and were used in this work for the assembly of SUMO-1 proteins. Peptides 6d and 7c are derived from hepatocyte growth factor (HGF). Peptide 7c illustrates the compatibility of SEA PEG resins 5d for the automated batch peptide synthesis, whereas the other peptides including the 52 amino-acid peptide 6d were assembled using an automated column peptide synthesizer. Usually, the oxidation of SEA peptides of type 6 (SEAon form) into cyclic disulfides 7 (SEAoff) is performed prior to the HPLC purification to avoid the potential N,S-acyl shift of SEAon group during the elution step.49 The oxidation of peptides 6a–c by iodine yielded successfully peptides 7a–c. In contrast, this procedure induced the aggregation of peptide 6d, which was therefore purified directly by HPLC.
 | ||
| Scheme 3 Fmoc-SPPS of SEA peptides. | ||
SEA peptide 6d was further ligated with a 72 amino-acid divalent cysteinyl peptide 8 in the presence of 4-mercaptophenylacetic acid (MPAA,50 200 mM) and tris(2-carboxyethyl)phosphine (TCEP, 200 mM) as shown in Scheme 4 to give the ∼20 kDa branched peptide 9 (Fig. 1). Although the reaction was complicated by the partial aggregation of 6d and of the ligation product 9 (see Fig. S29†), thereby explaining the modest yield for the ligation reaction (5%, 22% per step), this example illustrates the utility of SEA ligation for accessing large peptide scaffolds.
 | ||
| Scheme 4 SEA ligation of peptide 6d with divalent Cys peptide 8 yielded the ∼20 kDa branched peptide 9. | ||
 | ||
| Fig. 1 HPLC and ESI characterization of peptide 9. | ||
The successful SPPS of SUMO-1 peptides 7a,b set the stage for the assembly of SUMO-1-SEAoff protein 11 (Scheme 5). First, the SEAoff group of peptide 7a was exchanged by MPA at pH 4 in the presence of TCEP to produce SUMO-1 (2–50)-MPA thioester segment 10. The exchange proceeded very cleanly (see Fig. S7A†). The conversion reached 87% after 24 h, after which the reaction mixture was purified by HPLC to yield 28% (3.0 mg) of peptide thioester 10. The modest yield is mainly due to the small scale of synthesis. Gratifyingly, ligation of MPA thioester 10 with SEAoff segment 7b in the presence of MPAA (200 mM) at pH 7 furnished SUMO-1-SEAoff protein 11 and the mixed disulfide SUMO-1-SEAoff-MPAA 12 as a minor component after HPLC purification (22%, Fig. 2). The presence of the mixed disulfide 12 is not problematic since the reduction of the Cys51-MPAA disulfide bond is expected to occur during the subsequent activation of the C-terminal SEAoff group in the presence of TCEP.
 | ||
| Scheme 5 Assembly of SUMO-1-SEAoff protein 11. | ||
 | ||
| Fig. 2 Characterization of SUMO-1-SEAoff protein 11. | ||
The successful synthesis of SUMO-1-SEAoff protein 11 set the stage for the one-pot assembly of SUMO-1 conjugate 15, for which SUMO-1 modifier is attached to the side-chain of the target lysine residue through a cysteine linker (Scheme 6). Model peptide 14 features a ΨKX(E/D) sumoylation consensus motif where Ψ is a hydrophobic amino acid residue and X any amino acid. These positions are occupied in peptide 14 by Ile and Ala residues, respectively. In the one-pot assembly process, the formation of SUMO-1-SEAoff protein 11 was followed by the addition of TCEP (200 mM) and of peptide 14 in slight excess. The HPLC trace of the crude reaction mixture (Fig. 3a) shows that the target conjugate 15 is by far the major product, which was purified by HPLC with a 13% overall yield (Fig. 3b). The structure of conjugate 15 was confirmed by ESI (Fig. 3c) and chiefly MALDI-TOF mass spectrometry analysis (Fig. 3d, see also Fig. S21–23† for the in source fragmentation data).
 | ||
| Scheme 6 One-pot assembly of SUMO-1 conjugate 15. | ||
 | ||
| Fig. 3 Characterization of SUMO-1 conjugate 15. | ||
The solution NMR structure of SUMO-1 is available and shows an α-helical content of 10%.51 Consequently, the α-helical content for conjugate 15 was expected to be ∼9% if the target peptide is considered to be random coil. The solubilization of conjugate 15 in water furnished the circular dichroism (CD) spectrum shown in Fig. 4a. The ellipticity at 222 nm indicated an α-helix content of 8.6%, that is a value very close to the expected one. To unambiguously demonstrate the presence of a folded SUMO-1 domain within conjugate 15, we used ubiquitin-like specific protease 1 (Ulp1) as a sensor probe. Ulp1 is a yeast cysteine protease which cleaves specifically the isopeptidic bond linking SUMO-1 to its target proteins.32,52 The crystal structure of Ulp1 in complex with a yeast SUMO-1 protein analog has been solved and shows an extensive contact surface area of about 2400 Å2 between the two proteins.53 Thus, Ulp1 requires the SUMO-1 domain to be folded to exert its protease activity on SUMO-1 conjugates. Interestingly, incubation of SUMO-1 conjugate 15 with recombinant Ulp1 enzyme resulted in the specific cleavage of the Gly97-Cys bond within the conjugate (Fig. 4b and c). HPLC analysis of the cleavage mixture and MALDI-TOF analysis of the eluted peaks showed the exclusive formation of SUMO-1 protein and of peptide 14 (Fig. 4b).
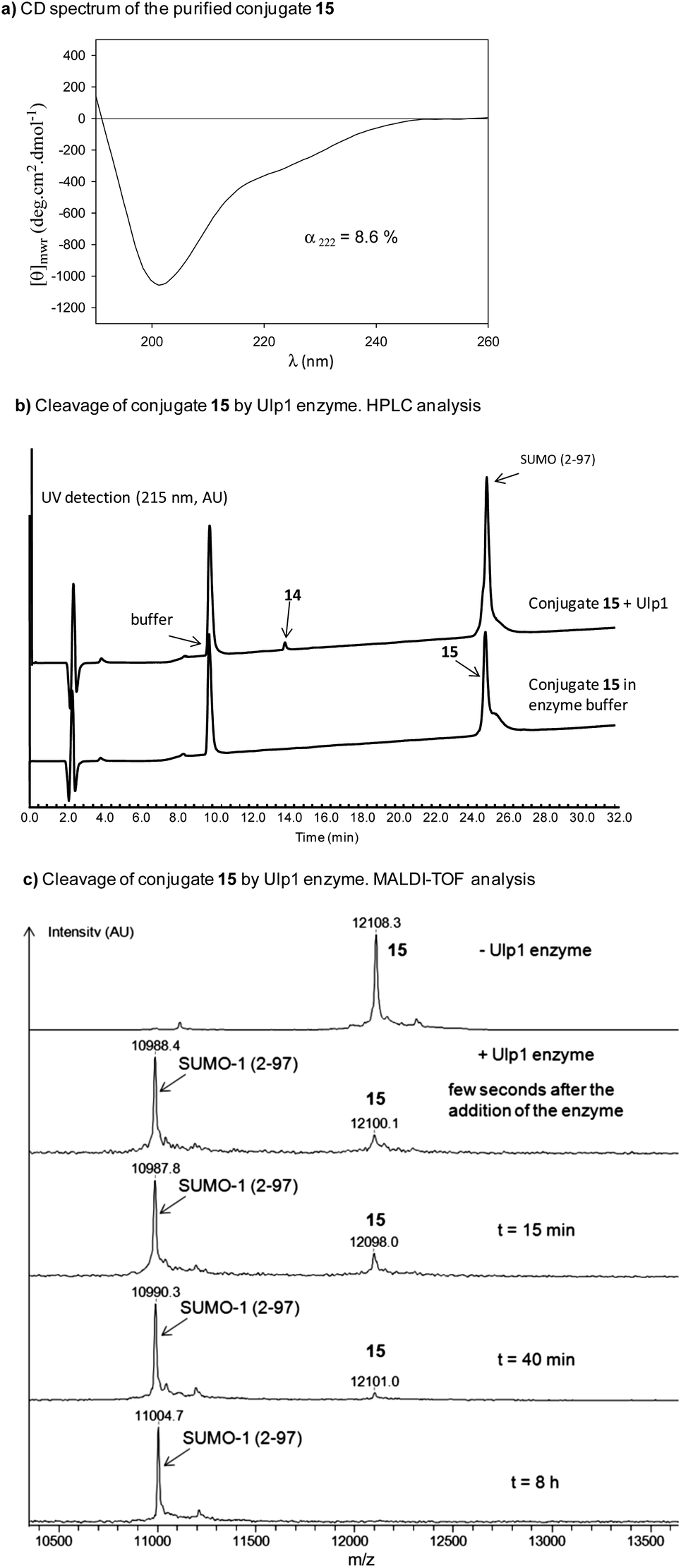 | ||
| Fig. 4 CD and biochemical characterization of SUMO-1 conjugate 15: (a) CD spectrum in water, (b) microLC analysis of the cleavage of 15 by Ulp1 enzyme, (c) MALDI-TOF monitoring of the cleavage of 15 by Ulp1 enzyme. | ||
Taken together, these data show the functionality of the SUMO-1 domain within conjugate 15 and the efficiency of the synthetic strategy depicted in Scheme 1.
Conclusions
In conclusion, we report a novel bis(2-sulfanylethyl)amino (SEA) PEG resin which enables the Fmoc-SPPS of >50 amino-acid long SEA peptide thioester surrogates. The access to such long peptide thioester surrogates enables the synthesis of complex protein targets and extends the limits of protein total synthesis. In particular, we describe for the first time the total synthesis of a folded, functional and reversible SUMO-1 conjugate. Access to SUMO conjugates proteins should facilitate the biochemical and structural implications of this post-translational modification.Acknowledgements
This work was supported financially by Cancéropôle Nord Ouest, SIRIC OncoLille, Région Nord pas de Calais and by the European Community.Notes and references
- L. Raibaut, N. Ollivier and O. Melnyk, Chem. Soc. Rev., 2012, 41, 7001–7015 RSC.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CAS.
- S. B. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS.
- R. R. Flavell and T. W. Muir, Acc. Chem. Res., 2009, 42, 107–116 CrossRef CAS PubMed.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141–2143 CrossRef CAS PubMed.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939–1941 CrossRef CAS PubMed.
- J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS PubMed.
- A. O. Ogunkoya, V. R. Pattabiraman and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9693–9697 CrossRef CAS PubMed.
- Y. Zhang, C. Xu, H. Y. Lam, C. L. Lee and X. Li, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6657–6662 CrossRef CAS PubMed.
- J. P. Pellois and T. W. Muir, Curr. Opin. Chem. Biol., 2006, 10, 487–491 CrossRef CAS PubMed.
- M. Hejjaoui, S. Butterfield, B. Fauvet, F. Vercruysse, J. Cui, I. Dikiy, M. Prudent, D. Olschewski, Y. Zhang, D. Eliezer and H. A. Lashuel, J. Am. Chem. Soc., 2012, 134, 5196–5210 CrossRef CAS PubMed.
- C. Unverzagt and Y. Kajihara, Chem. Soc. Rev., 2013, 42, 4408–4420 RSC.
- K. S. Kumar, L. Spasser, L. A. Erlich, S. N. Bavikar and A. Brik, Angew. Chem., Int. Ed., 2010, 49, 9126–9131 CrossRef CAS PubMed.
- K. S. Kumar, S. N. Bavikar, L. Spasser, T. Moyal, S. Ohayon and A. Brik, Angew. Chem., Int. Ed., 2011, 50, 6137–6141 CrossRef CAS PubMed.
- B. Fierz, C. Chatterjee, R. K. McGinty, M. Bar-Dagan, D. P. Raleigh and T. W. Muir, Nat. Chem. Biol., 2011, 7, 113–119 CrossRef CAS PubMed.
- L. A. Erlich, K. S. Kumar, M. Haj-Yahya, P. E. Dawson and A. Brik, Org. Biomol. Chem., 2010, 8, 2392–2396 CAS.
- P. Siman, S. V. Karthikeyan, M. Nikolov, W. Fischle and A. Brik, Angew. Chem., Int. Ed., 2013, 52, 8059–8063 CrossRef CAS PubMed.
- L. Spasser and A. Brik, Angew. Chem., Int. Ed., 2012, 51, 6840–6862 CrossRef CAS PubMed.
- F. El Oualid, R. Merkx, R. Ekkebus, D. S. Hameed, J. J. Smit, A. de Jong, H. Hilkmann, T. K. Sixma and H. Ovaa, Angew. Chem., Int. Ed., 2010, 49, 10149–10153 CrossRef CAS PubMed.
- C. Chatterjee, R. K. McGinty, J. P. Pellois and T. W. Muir, Angew. Chem., Int. Ed., 2007, 46, 2814–2818 CrossRef CAS PubMed.
- R. K. McGinty, J. Kim, C. Chatterjee, R. G. Roeder and T. W. Muir, Nature, 2008, 453, 812–816 CrossRef CAS PubMed.
- C. Chatterjee and T. W. Muir, J. Biol. Chem., 2010, 285, 11045–11050 CrossRef CAS PubMed.
- A. Shanmugham, A. Fish, M. P. Luna-Vargas, A. C. Faesen, F. El Oualid, T. K. Sixma and H. Ovaa, J. Am. Chem. Soc., 2010, 132, 8834–8835 CrossRef CAS PubMed.
- C. Chatterjee, R. K. McGinty, B. Fierz and T. W. Muir, Nat. Chem. Biol., 2010, 6, 267–269 CrossRef CAS PubMed.
- S. Eger, M. Scheffner, A. Marx and M. Rubini, J. Am. Chem. Soc., 2010, 132, 16337–16339 CrossRef CAS PubMed.
- N. D. Weikart, S. Sommer and H. D. Mootz, Chem. Commun., 2012, 48, 296–298 RSC.
- E. M. Valkevich, R. G. Guenette, N. A. Sanchez, Y. C. Chen, Y. Ge and E. R. Strieter, J. Am. Chem. Soc., 2012, 134, 6916–6919 CrossRef CAS PubMed.
- V. H. Trang, E. M. Valkevich, S. Minami, Y. C. Chen, Y. Ge and E. R. Strieter, Angew. Chem., Int. Ed., 2012, 51, 13085–13088 CrossRef CAS PubMed.
- R. Geiss-Friedlander and F. Melchior, Nat. Rev. Mol. Cell Biol., 2007, 8, 947–956 CrossRef CAS PubMed.
- S. Bergink and S. Jentsch, Nature, 2009, 458, 461–467 CrossRef CAS PubMed.
- S. Muller, C. Hoege, G. Pyrowolakis and S. Jentsch, Nat. Rev. Mol. Cell Biol., 2001, 2, 202–210 CrossRef CAS PubMed.
- N. D. van Treel and H. D. Mootz, J. Pept. Sci., 2014, 20, 121–127 CrossRef PubMed.
- S. Sommer, N. D. Weikart, A. Brockmeyer, P. Janning and H. D. Mootz, Angew. Chem., Int. Ed., 2011, 50, 9888–9892 CrossRef CAS PubMed.
- N. Ollivier, J. Dheur, R. Mhidia, A. Blanpain and O. Melnyk, Org. Lett., 2010, 12, 5238–5241 CrossRef CAS PubMed.
- W. Hou, X. Zhang, F. Li and C. F. Liu, Org. Lett., 2011, 13, 386–389 CrossRef CAS PubMed.
- N. Ollivier, J. Vicogne, A. Vallin, H. Drobecq, R. Desmet, O. El-Mahdi, B. Leclercq, G. Goormachtigh, V. Fafeur and O. Melnyk, Angew. Chem., Int. Ed., 2012, 51, 209–213 CrossRef CAS PubMed.
- L. Raibaut, H. Adihou, R. Desmet, A. F. Delmas, V. Aucagne and O. Melnyk, Chem. Sci., 2013, 4, 4061–4066 RSC.
- N. Haj-Yahya, M. Haj-Yahya, C. A. Castañeda, L. Spasser, H. P. Hemantha, M. Jbara, M. Penner, A. Ciechanover, D. Fushman and A. Brik, Angew. Chem., Int. Ed., 2013, 52, 11149–11153 CrossRef CAS PubMed.
- R. K. McGinty, M. Köhn, C. Chatterjee, K. P. Chiang, M. R. Pratt and T. W. Muir, ACS Chem. Biol., 2009, 4, 958–968 CrossRef CAS PubMed.
- R. Yang, K. K. Pasunooti, F. Li, X. W. Liu and C. F. Liu, J. Am. Chem. Soc., 2009, 131, 13592–13593 CrossRef CAS PubMed.
- R. Yang, K. K. Pasunooti, F. Li, X.-W. Liu and C.-F. Liu, Chem. Commun., 2010, 46, 7199–7201 RSC.
- N. Ollivier, J. B. Behr, O. El-Mahdi, A. Blanpain and O. Melnyk, Org. Lett., 2005, 7, 2647–2650 CrossRef CAS PubMed.
- H. Hojo, Y. Onuma, Y. Akimoto, Y. Nakahara and Y. Nakahara, Tetrahedron Lett., 2007, 48, 25–28 CrossRef CAS PubMed.
- F. Mende and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 1232–1240 CrossRef CAS PubMed.
- J. Dheur, N. Ollivier, A. Vallin and O. Melnyk, J. Org. Chem., 2011, 76, 3194–3202 CrossRef CAS PubMed.
- J. Dheur, N. Ollivier and O. Melnyk, Org. Lett., 2011, 13, 1560–1563 CrossRef CAS PubMed.
- L. Raibaut, P. Seeberger and O. Melnyk, Org. Lett., 2013, 15, 5516–5519 CrossRef CAS PubMed.
- N. Ollivier, L. Raibaut, A. Blanpain, R. Desmet, J. Dheur, R. Mhidia, E. Boll, H. Drobecq, S. L. Pira and O. Melnyk, J. Pept. Sci., 2014, 20, 92–97 CrossRef CAS PubMed.
- E. C. Johnson and S. B. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS PubMed.
- P. Bayer, A. Arndt, S. Metzger, R. Mahajan, F. Melchior, R. Jaenicke and J. Becker, J. Mol. Biol., 1998, 280, 275–286 CrossRef CAS PubMed.
- S. J. Li and M. Hochstrasser, Nature, 1999, 398, 246–251 CrossRef CAS PubMed.
- E. Mossessova and C. D. Lima, Mol. Cell, 2000, 5, 865–876 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and characterization data for all compounds. See DOI: 10.1039/c3sc53509f |
| This journal is © The Royal Society of Chemistry 2014 |