 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric synthesis of chiral cycloalkenone derivatives via palladium catalysis†
Barry M.
Trost
*,
James T.
Masters
,
Jean-Philip
Lumb
and
Dahlia
Fateen
Department of Chemistry, Stanford University, Stanford, CA 94305-4401, USA. E-mail: bmtrost@stanford.edu
First published on 13th January 2014
Abstract
The palladium-catalyzed oxidative desymmetrization of meso-dibenzoates yields γ-benzoyloxy cycloalkenones in good yields and with excellent levels of enantioselectivity. These compounds serve as precursors to a broad range of substituted cycloalkenones, including well-established synthetic building blocks and elaborated cycloalkanone derivatives. The ability to prepare both enantiomers of the oxidative desymmetrization products enables a unified strategy toward stereochemically diverse epoxyquinoid natural products.
Introduction
Chiral cycloalkenones are prominent structural motifs in organic synthesis.1 Chiral γ-substituted cycloalkenones in particular are important building blocks in both natural product and pharmaceutical synthesis. Heteroatom-substituted cycloalkenones such as 1–5 (Fig. 1) have been used extensively in these endeavors.2 Chemoselective elaboration of these building blocks may also provide densely substituted products in which every carbon is differentially functionalized. This is exemplified by cyclohexenone derivative 6, a general structure that encompasses a number of natural products. | ||
| Fig. 1 Diverse chiral cycloalkenones. | ||
A number of syntheses of such chiral cycloalkenones have been reported, many of which involve enzymatic processes or multi-step derivations of chiral pool materials.3 These well-established approaches can provide products with very high levels of enantioenrichment, but they may not permit access to both product enantiomers without requiring lengthier or otherwise less practical synthetic sequences. This consideration is of particular importance given that both enantiomers of certain building blocks (e.g., 1 and ent-1) have been employed in synthesis.4
Asymmetric catalysis may present an opportunity to prepare both enantiomers of a target with equal facility through catalyst control. These preparations may also benefit from high atom and step economy.5,6 Several innovative, catalytic asymmetric syntheses of γ-substituted cycloalkenones have been described.7 These methods often target or perform best for a specific cycloalkenone ring size or type of γ-substituent. A general method for the synthesis of chiral cycloalkenones of various ring sizes and with different γ-substituents would complement these approaches.
Herein we report such a strategy using asymmetric palladium catalysis. This process affords enantioenriched cyclopentenones, cyclohexenones, and cycloheptenones bearing various heteroatom substituents at the γ-position. This protocol also enables the efficient, asymmetric synthesis of more densely functionalized cyclohexenone-derived natural products.
Our group has demonstrated that palladium-catalyzed allylic alkylation presents a unique method for the oxidation of allylic esters (Fig. 2).8 In this process, ionization of allylic ester 7 generates π-allylpalladium intermediate 8, which undergoes selective O-alkylation with nitronate 9 to yield 10. Fragmentation then provides α,β-unsaturated product 11 and oxime 12, which can be recycled to 9.9
 | ||
| Fig. 2 Pd-catalyzed allylic oxidation. | ||
Meso-1,4-allylic dibenzoates are excellent substrates for this process. When compounds 13–15 (Fig. 3) are subjected to the reaction conditions in the presence of our chiral ligand L1, an oxidative desymmetrization proceeds. This delivers γ-benzoyloxy cycloalkenones 16–18 in good yields and with excellent enantioselectivity. Importantly, either enantiomer of the desired product can be obtained from the same meso precursor simply by selecting either (R,R)- or (S,S)-L1.
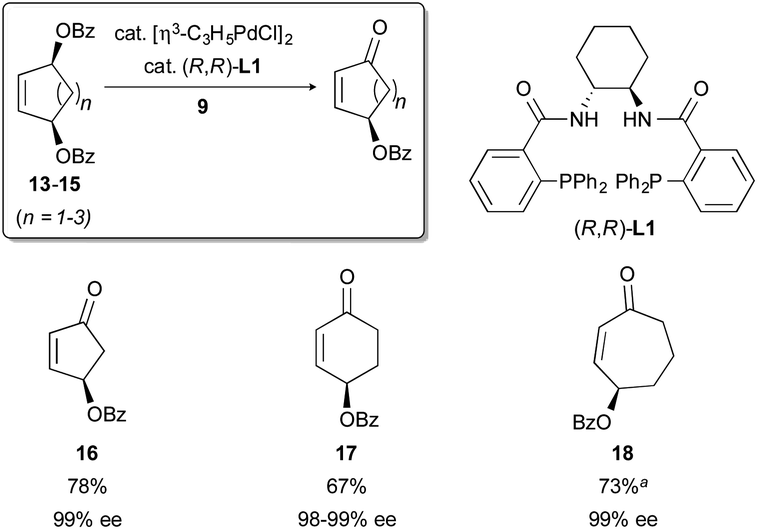 | ||
| Fig. 3 Oxidative desymmetrization of meso electrophiles. (a) Yield at 68% conversion (b.r.s.m.). | ||
With the aforementioned building blocks and natural product motifs serving as motivation, we sought to expand upon this entry into chiral γ-substituted cycloalkenones. We envisioned pairing this oxidative desymmetrization with the palladium-catalyzed allylic alkylation of heteroatom nucleophiles. This would enable access to a diverse set of γ-substituted cycloalkenones (Fig. 4). Complementing this, ester hydrolysis would provide γ-hydroxy cycloalkenones.
 | ||
| Fig. 4 Strategy for the synthesis of diverse cycloalkenone derivatives. | ||
We also anticipated that the γ-substituents of the cycloalkenones so produced would direct the diastereoselectivity of subsequent intra- and intermolecular reactions involving the enone moiety. This would enable the rapid assembly of molecular complexity with excellent stereocontrol. Further manipulations would install additional functionality at the remaining positions, leading to fully elaborated structures of the type 6.
Results and discussion
In pursuit of these goals, it was necessary to perform the synthesis of cyclohexenone 17 from meso-diester 14 on larger (multi-gram) scale. It was soon discovered that the isolated yields on such a scale were highly variable and generally lower than yields on smaller scale. This was the case even when the consumption of 14 was high (TLC). Contemporaneously, other operations involving 17 revealed its sensitivity toward aqueous or alcoholic base. Suspecting that such a base (e.g., KOH, from the KH used to generate nitronate 9) may have been the cause of the lowered yields on larger scale, we instituted several operational modifications to improve the robustness of the oxidative desymmetrization reaction. Specifically, in the preparation of 9, the commercial KH dispersion in mineral oil was washed with THF, rather than the more typical hexanes, to remove not only the oil but also any KOH present in the material. In addition, the reactions were limited in duration while monitoring for the consumption of the meso-dibenzoate (TLC). Once the dibenzoate was fully or largely consumed, the reaction mixture was thoroughly quenched with pH 7 buffer prior to extractive workup.Validation of these modifications came in the form of increased and more consistent reaction yields. For example, the oxidation of 14 → 17 proceeded in a reproducible ca. 67% yield on multi-gram scale (9 mmol, a 35-fold increase in scale from our original report) while maintaining excellent enantioselectivity (Fig. 5). Applying this modified protocol to 13 and 15 was similarly successful.10
 | ||
| Fig. 5 Multi-gram scale synthesis of 17. | ||
The palladium-catalyzed allylic alkylation (Pd-AA) of cycloalkenones 16–18 with heteroatom nucleophiles proved highly effective.11 Chiral γ-substituted cyclopentenone, cyclohexenone, and cycloheptenone products were obtained in good yields and with very good to excellent levels of enantioselectivity (Table 1). Alkylation with carboxylic, phenolic, and alcoholic nucleophiles successfully delivered oxygenated building blocks ent-2 and 4 as well as products 19–21. This synthesis of ent-2 is particularly significant, as alkyl alcohols are typically poor nucleophiles in palladium-catalyzed allylic alkylation.11a The preparation of acetoxyenone 19 is also noteworthy, as our attempts to engage the diacetate analogue of 14 in the oxidative desymmetrization were unsuccessful. Thus, this strategy of Pd-catalyzed oxidative desymmetrization and Pd-catalyzed transesterification represents a new and useful solution to this limitation. Alkylation with nitrogen nucleophiles was also successful, with potassium phthalimide as well as alkyl amines reacting smoothly to provide products 22–25.
|
|
||||
|---|---|---|---|---|
| Nucleophile | Electrophile | Product | Yield [%] | eeb [%] |
| a Reactions performed with 0.1 or 0.2 mmol 16–18 (99% ee), 2.5 mol% (η3-C3H5)2Pd2Cl2, and 15 mol% PPh3 at 0.2 M and 23 °C for 0.5–20 h. For complete details, see the ESI.1 b Determined by chiral HPLC. c Performed at 0 °C with 2.5 mol% Pd2dba3·CHCl3 and 7.5 mol% (S,S)-L1. 10% of 16 (79% ee) was recovered. d Cs2CO3 added. e 7.5 mol% dppf used in place of PPh3. PMB = para-methoxybenzyl, TBS = tert-butyldimethylsilyl. | ||||
| NaOAc | 16 |

|
64c | 93 |
| NaOAc | 17 |

|
75 | 96 |

|
17 |

|
62 | 91 |

|
17 |

|
80d | 98 |

|
17 |
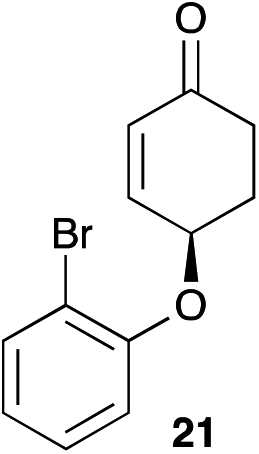
|
66d | 92 |

|
17 |

|
69 | 99 |
| HN(nPr2) | 17 |
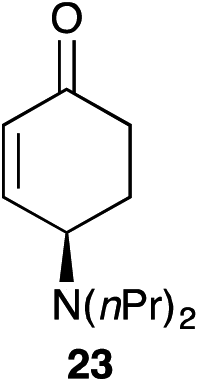
|
69 | 99 |

|
17 |

|
85 | 87 |
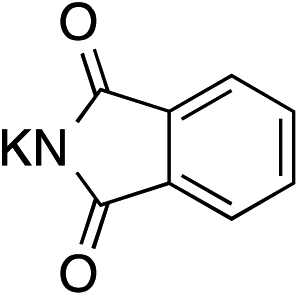
|
18 |

|
63e | 94 |

Notably, the products of conjugate addition were not observed in any of these cases. Moreover, despite the sensitivity of 17 toward hydroxide or alkoxide base, the basic nature of the reactions did not hinder us from obtaining good yields of the desired products.
The transformations proceeded with high levels of chirality transfer. This occurred despite the potential for racemization via π–σ–π interconversion involving an O-bound palladium enolate, a means by which the enantiotopic faces of the π-allylpalladium species might be equilibrated as in the case of butenolide electrophiles.12 The substitution appears to proceed primarily through overall double inversion, with the absolute stereochemistry of the starting material being conserved in the product.13
Further elaboration of these Pd-AA products to more complex cycloalkanone derivatives could be achieved through reactions that engaged the enone moiety with the γ-substituent. For example, exposure of ethanolamine derivative 24 to buffered TBAF effected desilylation and oxa-Michael addition, providing bicyclic morpholine derivative 26 (Fig. 6). The rapid, stereocontrolled synthesis of this compound further highlights the utility of this tandem oxidative desymmetrization/allylic substitution process: the former reaction sets high stereochemical purity at the γ-position that is relayed in the diastereoselective conjugate addition, and the latter enables the introduction of a useful bis-nucleophile unit for a convergent synthesis of 26.
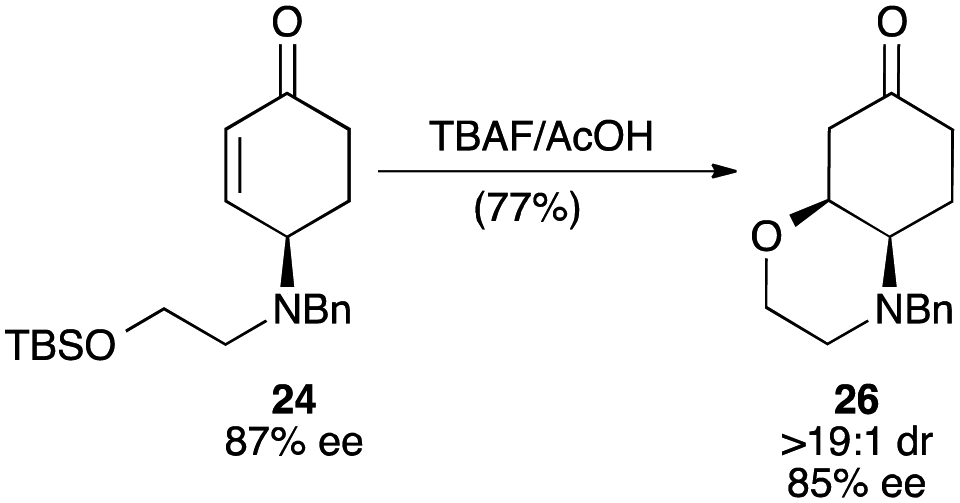 | ||
| Fig. 6 Elaboration of 24 to bicyclic ketone 26. Reagents and conditions: TBAF·3H2O, AcOH, THF, 0 → 23 °C, 2 h, 77%. | ||
Compounds 16–18 were also converted to the corresponding enantioenriched γ-hydroxycycloalkenones 1, 5, and 27 (Fig. 7). In evaluating methods to hydrolyze these base-sensitive compounds, Me3SnOH—which is most typically used for the hydrolysis of methyl esters to their acids14—emerged as the optimal reagent for this transformation. To the best of our knowledge, this represents its first use for the saponification of an acyloxy group at a secondary stereocenter, to yield a chiral alcohol. In these events, the γ-hydroxy products were obtained with excellent levels of enantioenrichment, and the sense of absolute stereochemistry was retained in the products.15
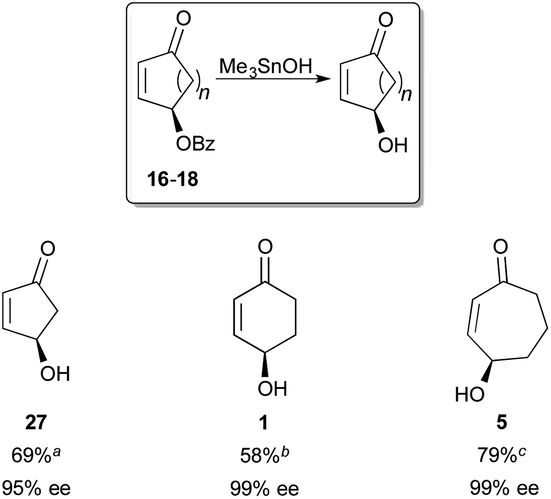 | ||
| Fig. 7 Synthesis of γ-hydroxycycloalkenones. Reagents and conditions: 0.2 mmol 16–18 (99% ee), Me3SnOH, 1,2-dichloroethane, 80 °C, 14 h. Yields at (a) 65%, (b) 73%, and (c) 66%, conversion (b.r.s.m.). | ||
Emboldened by the chemo-, enantio-, and diastereoselective preparation of the products illustrated in Table 1, Fig. 6 and 7, we next examined the synthesis of more densely functionalized cyclohexenone-derived natural products. We targeted several members of the epoxyquinoid class of natural products, compounds 28–35 (Fig. 8).16 These biologically active natural products bear an intriguing stereochemical feature: although structurally similar to each other, certain members exhibit the opposite sense of absolute stereochemistry (e.g., 28vs.29–33), while others display different relative stereochemistry between the epoxide and the alcohol (e.g., 31 and 32vs.34 and 35). Moreover, for at least one member of this family (harveynone, 29), both enantiomers are naturally occurring yet are derived from dissimilar sources and exhibit different biological activities.16a
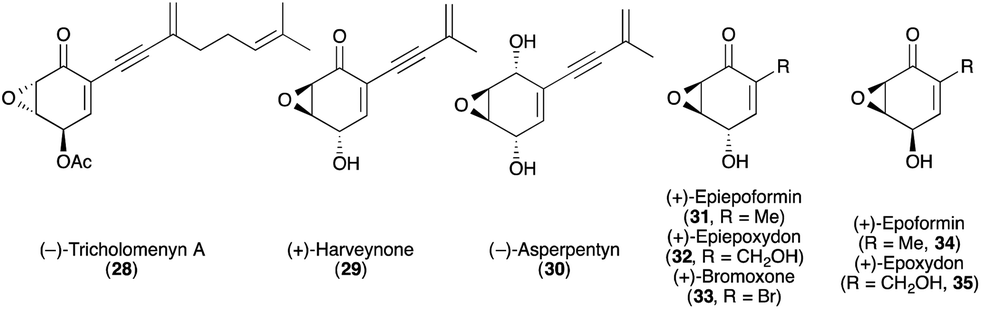 | ||
| Fig. 8 Stereochemically diverse epoxyquinoid natural products. | ||
We proposed a unified strategy toward the structural cores of 28–35 from chiral cyclohexenones 17 and ent-17 (Fig. 9). This approach would exploit the ability of an appropriately γ-substituted cyclohexenone to direct the diastereoselectivity of an intermolecular nucleophilic epoxidation. For instance, γ-hydroxycyclohexenone 1 is known to undergo hydroxyl-directed syn epoxidation.17 Thus, the preparation of 1 from 17 establishes access to cis product 36, the core structure of 34 and 35. In contrast, 17 could be expected to undergo anti-selective epoxidation. Hydrolysis would then deliver trans product 37, featuring the absolute and relative stereochemistry of 28. Performing the latter procedure with ent-17 would provide ent-37, a precursor to 29–33. Through this strategy, the core structures of all of the stereochemically diverse products 28–35 could be accessed from a common precursor, meso-diester 14.
 | ||
| Fig. 9 Stereodivergent access to epoxyquinoid precursors. | ||
Focusing on the synthesis of (−)-tricholomenyn A (28), we investigated the epoxidation of 17 (Fig. 10). Pleasingly, upon exposure to H2O2 and catalytic benzyltrimethylammonium hydroxide (Triton B),1817 cleanly underwent epoxidation to give 38 as a single diastereomer (1H NMR) and in 99% ee (chiral HPLC). Thus, the oxygenation and the absolute and relative stereochemistry of the trans epoxyquinoids were set, with excellent enantio- and diastereoselectivity, in three steps from commercial 1,3-cyclohexadiene.19 The fact that the oxidative desymmetrization delivered 17 with a γ-benzoyloxy substituent proved to be a significant synthetic advantage, as the epoxidation of the γ-acetoxy analogue led to poorer diastereoselectivity and to the formation of byproducts. These results are consistent with the observations of Bayón, Figueredo, and co-workers.20
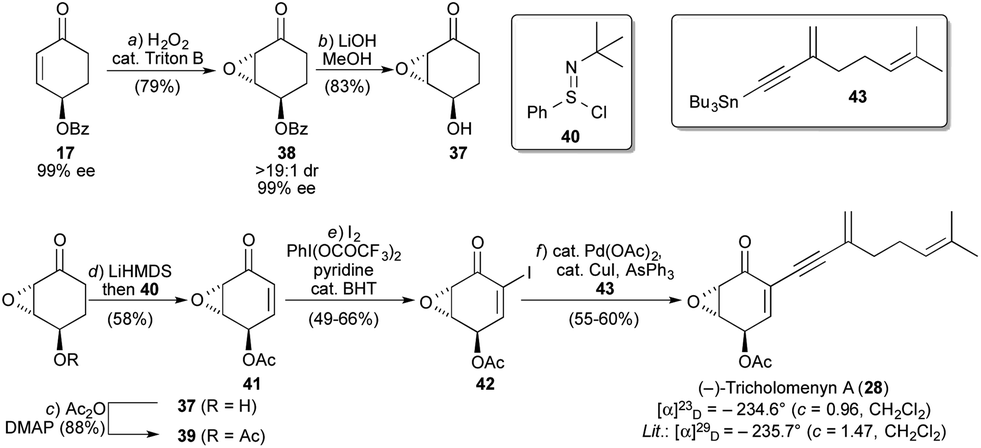 | ||
| Fig. 10 Total synthesis of (−)-tricholomenyn A (28). Reagents and conditions: (a) H2O2 (30% aq.), 10 mol% Triton B (40% aq.), THF, 0 °C, 45 min, 79%; (b) LiOH, MeOH, 0 °C, 1 h, 83%; (c) Ac2O, DMAP, MeCN, 0 → 23 °C, 10 min, 88%; (d) LiHMDS, THF, −78 °C, 30 min, then 40, −78 °C, 30 min, 58%; (e) I2, PhI(OCOCF3)2, pyridine, BHT, CH2Cl2, 23 °C, 24 h, 49–66%; (f) 10 mol% Pd(OAc)2, 10 mol% CuI, 20 mol% AsPh3, 43, THF, 0 °C, 1 h 30 min, 55–60%. Triton B = benzyltrimethylammonium hydroxide, DMAP = 4-dimethylaminopyridine, HMDS = hexamethyldisilazane, BHT = butylated hydroxytoluene. | ||
Hydrolysis and acetylation proceeded smoothly, affording 39. Completion of the synthesis then required oxidation of the ketone to an enone, α-iodination, and side chain installation. A number of oxidation conditions were investigated on both 38 and 39 (e.g., selenylation/elimination, Saegusa-Ito oxidation, bromination/elimination, IBX oxidation of a silyl enol ether,21 and Pd(TFA)2-catalyzed aerobic oxidation22), all of which led to no or trace product and/or decomposition.23 Procedures involving the use of a lithium enolate at temperatures above −78 °C proved especially problematic, leading to extensive decomposition. As oxidations of this sort have been performed on γ-silyloxy, α,β-epoxyketones,18 we suspected that the incompatibility was due to the ester moiety, the electrophilic nature of which promoted side reactions. However, rather than lengthening the synthesis with a protection/deprotection sequence involving alcohol 37, we investigated procedures in which the ketone enolate of 39 could be both formed and reacted at low temperature. We turned to the method of Mukaiyama,24 which involved sulfenylation of the lithium enolate of 39 with 40 and subsequent elimination, both of which proceeded efficiently at −78 °C to deliver 41.
The conversion of 41 to iodoenone 42 initially proved challenging, as typical conditions (I2 in CH2Cl2/pyridine25) led to decomposition. This may have been the result of interactions between the acetoxy group of 41 and these co-solvent amounts of pyridine. Success was realized by applying the conditions of Benhida26a (as applied by Hayashi26b), which employ only stoichiometric pyridine and a more activated iodinating reagent.
From 42, cross coupling with known stannane 4316e afforded (−)-tricholomenyn A (28), the analytical data for which matched literature data. The observed optical rotation ([α]23D = − 234.6° (c = 0.96, CH2Cl2)) was in line with that reported for material obtained using enzymatic resolution as the source of chirality ([α]29D = − 235.7° (c = 1.47, CH2Cl2)).27
Synthetic access to natural products 29–33 was then established through the synthesis of ent-37 from ent-17 (prepared from 14 in 98% ee using (S,S)-L1). Silylation afforded 44, from which syntheses of 31–33 are known (Fig. 11). The analytical data for 44, including optical rotation, was in line with data reported for material prepared through an enzymatic approach.18
 | ||
| Fig. 11 Syntheses of epoxyquinoids 29–33. Reagents and conditions: (a) TBSCl, imidazole, CH2Cl2, 23 °C, 16 h, 88%; (b) LiHMDS, THF, −78 °C, 30 min, then 40, −78 °C, 1 h, 66%. | ||
The asymmetric synthesis of 44 has itself been the subject of past studies, for both natural product synthesis and pharmaceutical research.18,28 Prior syntheses have employed enzymatic reduction,18 enzymatic resolution,28a chiral pool material,28b or asymmetric deprotonation,28c strategies that either required eight or more steps or, in the latter case, delivered more modest (77–85%) ee. The five-step procedure presented herein, which provides 98–99% ee, complements these approaches.
The oxidation of ketone 44 to enone 45 has previously been performed via silyl enol ether formation followed by either selenylation/elimination18 or stoichiometric Saegusa-Ito oxidation.28a Given our successful oxidation of ketone 39via the Mukaiyama protocol, we considered performing this reaction on 44 to deliver 45 directly in one step. Indeed, this protocol proved very effective, providing the enone in good yield. From 45, syntheses of 29 and 30 are known.29
Conclusions
We report a strategy for the synthesis of a diverse set of chiral cycloalkenone derivatives via asymmetric palladium catalysis. Using a newly optimized reaction procedure, the oxidative desymmetrization of meso-dibenzoates 13–15 affords cycloalkenones 16–18 in good yields and with excellent enantioselectivity. These products serve as platforms from which a variety of substituted cycloalkenones can be prepared through palladium-catalyzed allylic alkylation, including established building blocks such as ent-2 and 4. The products are obtained in good yields and with high levels of enantioenrichment. This Pd-AA approach also enables the convergent introduction of useful functionality, allowing for the synthesis of more elaborate cycloalkanone derivatives. The hydrolysis of 16–18 to enantioenriched γ-hydroxycycloalkenone building blocks is also demonstrated.Cyclohexenones 17 and ent-17 provide entry points to epoxyquinoid natural products 28–35 through stereoselective transformations and through reactions that elaborate the entire cyclohexenone structure. This approach to these stereochemically diverse products proceeds with excellent levels of enantio- and diastereoselectivity, and it establishes a concise, unified strategy starting from a single meso precursor (14).
Acknowledgements
We thank the National Institutes of Health (GM33049) and the National Science Foundation (CHE-0948222) for their generous support of our programs. J.T.M. gratefully acknowledges support from an Abbott Laboratories Stanford Graduate Fellowship. J.-P.L. is the recipient of an NIH postdoctoral fellowship (GM083428). We also thank Johnson-Matthey for their generous gift of palladium salts.Notes and references
- See: (a) T.-L. Ho, Enantioselective Synthesis: Natural Products Synthesis from Chiral Terpenes, Wiley, New York, 1992 Search PubMed; (b) G. P.-J. Hareau, M. Koiwa, S. Hikichi and F. Sato, J. Am. Chem. Soc., 1999, 121, 3640 CrossRef CAS and references therein.
- For selected examples, see: (a) A. B. Jones, M. Yamaguchi, A. Patten, S. J. Danishefsky, J. A. Ragan, D. B. Smith and S. L. Schreiber, J. Org. Chem., 1989, 54, 17 CrossRef CAS; (b) C. Spino, B. Hill, P. Dubé and S. Gingras, Can. J. Chem., 2003, 81, 81 CrossRef CAS; (c) J. D. White, Y. Li, J. Kim and M. Terienk, Org. Lett., 2013, 15, 882 CrossRef CAS PubMed; (d) T. Tachihara, H. Watanabe and T. Kitahara, Heterocycles, 2002, 57, 781 CrossRef CAS; (e) E. M. Wilson and D. Trauner, Org. Lett., 2007, 9, 1327 CrossRef CAS PubMed; (f) J.-T. Zhang, X.-L. Qi, J. Chen, B.-S. Li, Y.-B. Zhou and X.-P. Cao, J. Org. Chem., 2011, 76, 3946 CrossRef CAS PubMed; (g) M. Kawasumi, N. Kanoh and Y. Iwabuchi, Org. Lett., 2011, 13, 3620 CrossRef CAS PubMed; (h) C. C. Hughes, A. K. Miller and D. Trauner, Org. Lett., 2005, 7, 3425 CrossRef CAS PubMed . Cyclopentenones bearing various oxygen substituents at the γ-position have been used extensively in prostaglandin synthesis; for examples, see R. Noyori and M. Suzuki, Angew. Chem. Int. Ed. Engl., 1984, 23, 847 Search PubMed and references therein.
- For examples, see (a) P. Bayón, G. Marjanet, G. Toribio, R. Alibés, P. de March, M. Figueredo and J. Font, J. Org. Chem., 2008, 73, 3486 CrossRef PubMed; (b) A. Demir and O. Sesenoglu, Org. Lett., 2002, 4, 2021 CrossRef CAS PubMed; (c) J. E. Audia, L. Boisvert, A. D. Patten, A. Villalobos and S. J. Danishefsky, J. Org. Chem., 1989, 54, 3738 CrossRef CAS; (d) D. R. Boyd, N. D. Sharma, M. Kaik, P. B. A. McIntyre, P. J. Stevenson and C. C. R. Allen, Org. Biomol. Chem., 2012, 10, 2774 RSC; (e) T. Kumaraguru and N. W. Fadnavis, Tetrahedron: Asymmetry, 2012, 23, 775 CrossRef CAS PubMed , and references therein; see also ref. 2d and 2h.
- As examples, see ref. 2c and 2f for the use of ent-1 and 1, respectively.
- (a) B. M. Trost, Science, 1991, 254, 1471 CAS; (b) B. M. Trost, Angew. Chem., Int. Ed., 1995, 34, 259 CrossRef CAS.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40 CrossRef CAS PubMed.
- For examples of catalytic, asymmetric approaches, see (a) G. Dickmeiss, V. De Sio, J. Udmark, T. B. Poulsen, V. Marcos and K. A. Jørgensen, Angew. Chem., Int. Ed., 2009, 48, 6650 CrossRef CAS PubMed; (b) S. Staben, X. Linghu and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 12658 CrossRef CAS PubMed; (c) C. F. Nising, U. K. Ohnemüller and S. Bräse, Synthesis, 2006, 2643 CAS; (d) H.-J. Gais, O. Bondarev and R. Hetzer, Tetrahedron Lett., 2005, 46, 6279 CrossRef CAS PubMed; (e) S. Hashiguchi, A. Fujii, K.-J. Haack, K. Matsumura, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed., 1997, 36, 288 CrossRef CAS; (f) T. Iida, N. Yamamoto, H. Sasai and M. Shibasaki, J. Am. Chem. Soc., 1997, 119, 4783 CrossRef CAS; (g) T. Fukuda and T. Katsuki, Tetrahedron Lett., 1996, 37, 4389 CrossRef CAS; (h) J. L. Leighton and E. N. Jacobsen, J. Org. Chem., 1996, 61, 389 CrossRef CAS; (i) K. Hiroya, Y. Kurihara and K. Ogasawara, Angew. Chem., Int. Ed., 1995, 34, 2287 CrossRef CAS; (j) S. Chang, R. M. Heid and E. N. Jacobsen, Tetrahedron Lett., 1994, 35, 669 CrossRef CAS; (k) M. Kitamura, K. Manabe and R. Noyori, Tetrahedron Lett., 1987, 28, 4719 CrossRef CAS ; see also M. C. Willis, J. Chem. Soc., Perkin Trans. 1, 1999, 1765 Search PubMed.
- B. M. Trost, J. Richardson and K. Yong, J. Am. Chem. Soc., 2006, 128, 2540 CrossRef CAS PubMed.
- Nitronate 9 is prepared by the oxidation of oxime 12 with NaBO3 in AcOH followed by in situ deprotonation with KHMDS, thus enabling this recycling.
- The pH 7 quench may be the most significant of these modifications: as the workup and isolation associated with a larger scale reaction necessarily takes more time compared to a smaller scale reaction, an incompletely quenched larger scale reaction mixture may translate to a longer period of exposure to reactive impurities such as base. The results listed in Fig. 3 were obtained using this modified reaction procedure (>1 mmol scale). For complete experimental details, see the ESI.†.
- For a review of allylic alkylation with heteroatom nucleophiles, see (a) B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427 RSC . For the allylic alkylation of substituted, chiral pool-derived cyclohexenone electrophiles, see: ; (b) M. Shan and G. A. O'Doherty, Org. Lett., 2010, 12, 2986 CrossRef CAS PubMed ; for the kinetic resolution of cyclopentenone electrophiles, see: ; (c) K. Ulbrin, P. Kreitmeier, T. Vilaivan and O. Reiser, J. Org. Chem., 2013, 78, 4202 CrossRef PubMed.
- (a) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1999, 121, 3543 CrossRef CAS; (b) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 3090 CrossRef CAS PubMed.
- The absolute stereochemistry of compounds 4, 19, and ent-2 was determined by comparison of their optical rotations to literature values, and the stereochemistry of the remaining Pd-AA products is assigned by analogy.
- (a) K. C. Nicolaou, A. A. Estrada, M. Zak, S. H. Lee and B. S. Safina, Angew. Chem., Int. Ed., 2005, 44, 1378 CrossRef CAS PubMed; (b) R. L. E. Furlán and E. G. Mata, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd. 2010 Search PubMed.
- The absolute stereochemistry of the alcohol products was determined by comparison of their optical rotations to literature values, and the % ee was determined by chiral HPLC after their conversion to benzoates 16–18 (BzCl, pyridine; for details, see the ESI†).
- For reviews covering epoxyquinoid natural products, including past syntheses, natural origins, and biological activities, see: (a) J. Marco-Contelles, M. T. Molina and S. Anjum, Chem. Rev., 2004, 104, 2857 CrossRef CAS PubMed; (b) K. Ogasawara, J. Synth. Org. Chem., Jpn., 1999, 57(11), 957 CrossRef CAS; (c) M. Shoji and Y. Hayashi, Eur. J. Org. Chem., 2007, 3783 CrossRef CAS . For more recent syntheses, see: ; (d) J. Li, S. Park, R. L. Miller and D. Lee, Org. Lett., 2009, 11, 571 CrossRef CAS PubMed; (e) D. M. Pinkerton, M. G. Banwell and A. C. Willis, Aust. J. Chem., 2009, 62, 1639 CrossRef CAS; (f) D. M. Pinkerton, M. G. Banwell and A. C. Willis, Org. Lett., 2009, 11, 4290 CrossRef CAS PubMed , and references therein.
- J. B. Evarts, Jr and P. L. Fuchs, Tetrahedron Lett., 1999, 40, 2703 CrossRef.
- T. Tachihara and T. Kitahara, Tetrahedron, 2003, 59, 1773 CrossRef CAS.
- Dibenzoate 14 is readily prepared in one step from 1,3-cyclohexadiene (J.-E. Bäckvall, K. L. Granberg, R. B. Hopkins, Acta Chem. Scand., 1990, 44, 492); for a multi-gram synthesis, see the ESI.†.
- G. Toribio, G. Marjanet, R. Alibés, P. De March, J. Font, P. Bayón and M. Figueredo, Eur. J. Org. Chem., 2011, 1534 CrossRef CAS.
- K. C. Nicolaou, D. L. F. Gray, T. Montagnon and S. T. Harrison, Angew. Chem., Int. Ed., 2002, 41, 996 CrossRef CAS.
- T. Diao and S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566 CrossRef CAS PubMed.
- The reaction of 39 with excess IBX and cat. p-toluenesulfonic acid (DMSO, 65 °C, 24 h) afforded 41, but in highly variable yields. For further discussion, see the ESI;† see also K. C. Nicolaou, T. Montagnon, P. S. Baran and Y.-L. Zhong, J. Am. Chem. Soc., 2002, 124, 2245 CrossRef CAS PubMed.
- T. Mukaiyama, J.-i. Matsuo and H. Kitagawa, Chem. Lett., 2000, 1250 CrossRef CAS.
- C. R. Johnson, J. P. Adams, M. P. Braun, C. B. W. Senanayake, P. M. Wovkulich and M. R. Uskoković, Tetrahedron Lett., 1992, 33, 917 CrossRef CAS.
- (a) R. Benhida, P. Blanchard and J.-L. Fourrey, Tetrahedron Lett., 1998, 39, 6849 CrossRef CAS; (b) M. Shoji, H. Imai, M. Mukaida, K. Sakai, H. Kakeya, H. Osada and Y. Hayashi, J. Org. Chem., 2005, 70, 79 CrossRef CAS PubMed.
- T. Kamikubo and K. Ogasawara, Chem. Commun., 1996, 1679 RSC.
- (a) A. O'Byrne, S. O'Reilly, C. Tighe, P. Evans, L. Ciuffini and M. G. Santoro, Tetrahedron Lett., 2012, 53, 5936 CrossRef CAS PubMed; (b) J. B. Evarts, Jr and P. L. Fuchs, Tetrahedron Lett., 2001, 42, 3673 CrossRef; (c) V. Rodeschini, P. Van de Weghe, E. Salomon, C. Tarnus and J. Eustache, J. Org. Chem., 2005, 70, 2409 CrossRef CAS PubMed.
- M. T. Barros, C. D. Maycock and M. R. Ventura, Chem. – Eur. J., 2000, 6, 3991 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and copies of NMR spectra and HPLC chromatograms. See DOI: 10.1039/c3sc53250j |
| This journal is © The Royal Society of Chemistry 2014 |