 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rationalising the role of solid-acid sites in the design of versatile single-site heterogeneous catalysts for targeted acid-catalysed transformations†
Enrica
Gianotti
a,
Maela
Manzoli
b,
Matthew E.
Potter
c,
Vasudev N.
Shetti
d,
Danni
Sun
c,
James
Paterson
c,
Thomas M.
Mezza
e,
Alan
Levy
f and
Robert
Raja
*c
aDipartimento di Scienze e Innovazione Tecnologica, Centro Interdisciplinare Nano-SiSTeMI, Università del Piemonte Orientale, via T. Michel 11, I-15100, Alessandria, Italy
bDipartimento di Chimica e NIS-Centro di Eccellenza, Universitá di Torino, V. P. Giuria, 7-10125 Torino, Italy
cSchool of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: R.Raja@soton.ac.uk
dRefinery R&D, Reliance Industries Ltd. Jamnagar, 361142, Gujarat, India
eUOP, a Honeywell company, Des Plaines, IL 60017, USA
fHoneywell Int., 101 Columbia Road, Morristown, NJ 07962, USA
First published on 31st January 2014
Abstract
A versatile design strategy for rationalising the role of well-defined and isolated multifunctional solid-acid active centres, employing Mg(II)Si(IV)AlPO-5 nanoporous architectures has been demonstrated, with a view to affording structure–property correlations compared to its corresponding mono-substituted analogues (Mg(II)AlPO-5 and Si(IV)AlPO-5). The simultaneous incorporation of Mg(II) and Si(IV) ions, as isomorphous replacements for Al(III) and P(V) ions in the microporous architecture, plays an important role in modulating the nature and strength of the solid-acid active sites in the industrially-important, vapour-phase Beckmann rearrangement of cyclohexanone oxime to produce ε-caprolactam (the precursor for renewable nylon-6) and in the isopropylation of benzene to cumene. The structural integrity, coordination geometry and local environment of the active (Brønsted-acid) sites could be rationalised at the molecular level, using in situ spectroscopic techniques, for tailoring the catalytic synergy by adroit design of the framework architecture.
Introduction
Engineering multifunctional active sites in nanoporous solids is an intriguing area of research that exploits the fundamental principles of solid-state chemistry. Aluminophosphates (AlPOs) are a class of nanomaterials, where single-site catalytic entities can be judiciously designed, to create novel nano-architectures.1 In fact, isolated active centres (discrete single-sites) in AlPOs can be meticulously engineered via the isomorphous substitution of metal-ions into the alternating AlO4 and PO4 framework tetrahedra during synthesis.2 By adopting this method it is possible to generate a diverse range of active sites (solid-acid and/or redox centres) within the microporous AlPO framework. In particular, tailored Brønsted acid centres can be designed in AlPOs, wherein a charge-compensating proton can instigate the accommodation of small quantities of divalent transition-metal-ions, such as Mg(II), Zn(II), etc., as isomorphous replacements for the trivalent framework Al(III) ions; or by analogously introducing tetravalent Si(IV) ions to isomorphously substitute the P(V) ions in the neighbouring PO4 tetrahedra. Through this design strategy, it is also possible to expediently bestow multi-functionality within the same catalyst, through the concomitant introduction of redox-active and Brønsted acid centres, adroitly creating a bifunctional catalyst, for the ‘one-pot’ synthesis of ε-caprolactam directly from cyclohexanone.3–8 More recently, we have demonstrated the ability to engineer synergistic interactions between specific metal-sites by designing redox-active centres in close proximity to oxophilic entities (e.g. VVTiIVAlPO-5 and CoIIITiIVAlPO-5),9,10 thereby creating functional bimetallic solid catalysts for enhancing the activity and selectivity in acid-catalysed and redox processes.9–11Owing to the availability of a large-range of metal dopants that could be effectively incorporated (using the above strategies)3,4,9–11 into a diverse-range of nanoporous architectures containing wide-range of pore apertures, it is possible to adroitly amalgamate a specific active site, with an appropriate framework, to yield a targeted catalyst capable of efficiently catalysing the industrially-important, acid-catalysed Beckmann rearrangement of cyclohexanone oxime to ε-caprolactam. ε-caprolactam is a vital commodity for the bulk chemical industry, with production exceeding 4 million metric tons per annum,12 the vast majority of which is used as a precursor for the production of Nylon-6. In the polymer and fibre industry, Nylon-6 is generated from the reversible ring-opening polymerisation of the monomer, ε-caprolactam; whereas Nylon-6,6 is formed by the irreversible dehydration polymerisation of adipic acid and hexamethylene diamine.12,13 Notwithstanding the minor physical attributes in terms of the durability and stability of these polymers, the use of the monomer, ε-caprolactam, in the synthesis of Nylon-6, facilities the effective recovery and recycle15 of carpet and other fibres that are derived from Nylon-6, in stark contrast to Nylon-6,6, which predominantly ends up in landfill sites.
Despite the environmentally conscious age fuelling a demand for cleaner technologies, the industrial production of the ε-caprolactam poses a number of challenges to the environment5,14,15 and there is adequate scope for research tailored at mitigating some of these effects. Classically, ε-caprolactam is exclusively produced through the modified Raschig process (Scheme 1); a method which utilises hydroxylamine sulphate and stoichiometric quantities of oleum, to yield ε-caprolactam from cyclohexanone via a cyclohexanone oxime intermediate. Whilst the chemical yields and selectivities are noteworthy,15 the overall atom efficiencies (and E-factor) are inferior, as 4.4 tons of ammonium sulphate waste is generated for every ton of ε-caprolactam produced. Industrial methods that mitigate the production of ammonium sulphate to 2.6 and 1.6 tons have been reported,16,17 but a more fundamental understanding of the precise nature of the active sites at the molecular level and its associated mechanistic implications will be required for modulating the intrinsic activity and selectivity, using benign reagents under energy-efficient conditions.6–8,18
 | ||
| Scheme 1 Classical Raschig process. | ||
The use of heterogeneous solid-acid catalysts for the vapour-phase Beckmann rearrangement of cyclohexanone oxime is a highly desirable alternative, as it generates minimal waste and utilises benign reagents.19,20 Furthermore, the heterogeneous nature of the solid-acid catalysts is an added advantage from the view point of costly separations associated with the Raschig method, and circumvents the need for the neutralisation step in the work-up process. In light of this, a wide-range of materials such as TiO2,21 B2O3,22 Nb oxides,19 tungsten oxides23 and rare earth pyrophosphates24 have been reported as effective catalysts for this important transformation. In particular, the use of porous zeolitic materials has been prolific, as the combination of the crystalline structure and defect silanol groups render these materials as highly active catalysts for this process.25–28 The attractive features of zeotypes29 and related mesoporous solids30 stimulated a great deal of research into understanding the nature of the active site,31 the role of organic solvents,32 the importance of particular framework types,33–36 influence of metal dopants37 and diffusion properties linked to deactivation and lifetime of the catalyst.38 These fundamental studies have led to the development and establishment of an industrial process that utilises a high silica MFI catalyst34 for the commercial production of ε-caprolactam.
Despite being structurally similar, transition-metal-substituted AlPOs have not been thoroughly investigated for this reaction (part B, Scheme 1); with a majority of reports in the literature, to the best of our knowledge, confined to the use of silicon substituted AlPOs (SAPOs).29,39 Unlike zeolites, our recent studies9,10,20,40 have indicated that the location, proximity and nature of catalytically active centers in AlPOs can be rationally designed and judiciously controlled, in stark contrast to those of related zeolites, where post-synthetic modification methods lead to the creation of surface defects, that are the loci of the intrinsic active sites.31 Earlier studies have indicated that strong Brønsted acid sites in zeolites play a crucial role in catalyzing the Beckmann rearrangement, by protonating the oxime.15,41–43 However, parallel studies44 have indicated that the presence of these strong-acid sites favor the formation of by-products, thereby decreasing the overall yield of ε-caprolactam.45 In more recent studies, it has been suggested that weaker Brønsted acid sites and silanol groups or silanol nests play a pivotal role in modulating the selectivity in the Beckmann rearrangement of cyclohexanone oxime.33,46 The degree of synthetic control afforded by AlPOs for introducing a diverse range of strong and weak Brønsted acid centers is therefore highly beneficial for gaining a deeper mechanistic insight into solid-acid catalysed transformations. The complementary catalytic tests that have been outlined in this study (e.g. the Beckmann rearrangement of cyclohexanone oxime and isopropylation of benzene) along with the detailed spectroscopic measurements with appropriate molecular probes have helped to discriminate the nature and strength of solid-acid sites required for a particular catalytic transformation.
In the present study, mono-substituted and bi-substituted Mg- and Si-containing AlPO-5 molecular sieves (MgAlPO-5, SiAlPO-5 and MgSiAlPO-5) have been devised and used as solid-acid catalysts in the vapour-phase Beckmann rearrangement of cyclohexanone oxime to produce ε-caprolactam and in the isopropylation of benzene to form cumene. Moreover, we detail how the creation of isolated active (acid) sites, through the dual (simultaneous) incorporation of Mg2+ and Si4+ metal ions, can trigger synergistic interactions within the AlPO-5 framework architecture (see Fig. 1), to enhance the catalytic activity and selectivity for the transformations shown in Fig. 1. The above metals were specifically chosen due to the relative ease with which they could be isomorphously incorporated into AlPO framework, to generate a diverse range of Brønsted acid sites. However, it is believed that the subtle differences in the degree of acidity typically achieved on incorporation, during synthesis, and the contrasting substitution mechanisms (Mg(II) undergoes type I substitution replacing Al(III), whereas Si(IV) undergoes type II and III substitutions to replace P(V), see Fig. S1†) will modulate the nature and strength of the ensuing solid-acid sites, provoking differences in catalytic behaviour. This combined with concomitant use of FTIR spectroscopy, employing both strong and weak molecular probes (2,6-dimethylpyridine and CO), further amplifies the intrinsic synergistic interactions at the molecular level, facilitating structure–property correlations to be established.
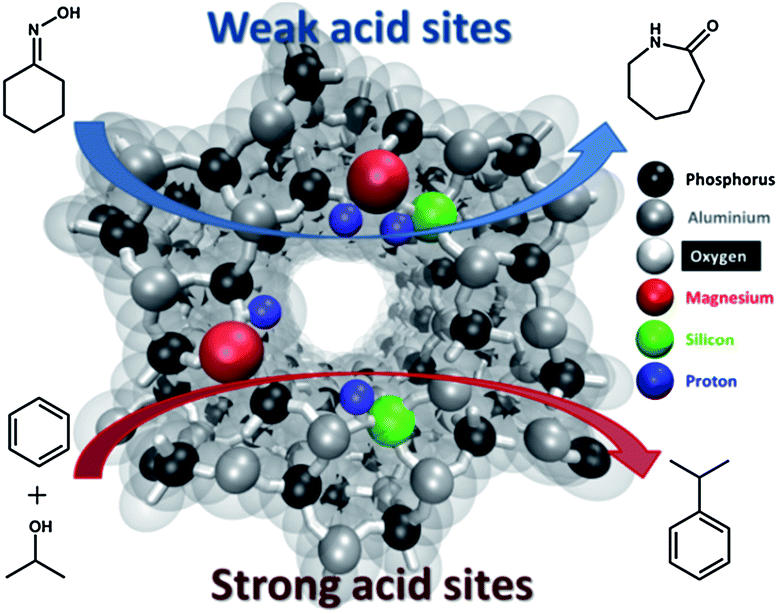 | ||
| Fig. 1 Graphical representation of the Mg(II)Si(IV)AlPO-5 catalyst highlighting the role of strong and weak Brønsted acid sites. | ||
Results and discussions
Structural and textural properties of the AlPO-5 catalysts
In order to confirm the phase-purity of the AlPO-5 architecture, a meticulous X-ray diffraction (XRD) analysis was performed on the calcined catalysts (Fig. 2). It was noted that in all three catalysts, all the observable peaks in the XRD pattern could be attributed to the AFI framework type, suggesting no significant secondary phases or metal aggregates were present either in the fresh or post-catalysis samples (Fig. S2†). It was also noteworthy that the XRD results for the isomorphously-substituted analogues were in good agreement with that of the undoped AlPO-5 catalyst. Rietveld analysis (Table 1) further supports the structurally consistency between the samples; confirming that the unit cell parameters of all four catalysts are within permitted levels of experimental deviation.47 These observations are fairly consistent, given the relatively low level of metal incorporation in the metal-substituted systems, as revealed by ICP analysis (see Table 1 and S2†). In addition, the particle size of the samples has been evaluated (Table 1) by applying the Scherrer's equation to the (100) XRD peak, which reveals that the particle size for all four catalysts falls within a narrow 4 nm range. Volumetric analyses confirmed the microporous nature of the materials (Table 1; see also Fig. S3–S8 and Table S3†): the specific surface areas of the calcined metal-substituted catalysts are in good agreement with values reported in the literature for analogous systems.48 Scanning electron microscopy (SEM) reveals spherical particles (see Fig. S9–S11†), with typical particle diameters ranging between 10 and 35 μm. | ||
| Fig. 2 Powder X-ray diffraction patterns for calcined Mg(II)Si(IV)AlPO-5, Si(IV)AlPO-5, Mg(II)AlPO-5 and undoped AlPO-5. | ||
| Samples | Mg wt% | Si wt% | SSA/m2 g−1 | Particle size/nm | a/Å | c/Å |
|---|---|---|---|---|---|---|
| AlPO-5 | — | — | 295 | 56.5 | 13.69 | 8.43 |
| MgAlPO-5 | 0.85 | — | 193 | 55.5 | 13.71 | 8.40 |
| SiAlPO-5 | — | 1.69 | 182 | 54.8 | 13.70 | 8.39 |
| MgSiAlPO-5 | 0.89 | 1.70 | 168 | 52.6 | 13.68 | 8.39 |
In situ vibrational FTIR spectroscopy with molecular probes for investigating the nature of active (acid) sites
FTIR spectroscopy was used to probe the nature of the acid sites by carefully examining the O–H stretching region in the microporous AlPO-5 catalysts, as shown in Fig. 3. All four catalysts display a band at 3680 cm−1, which is assigned to the O–H stretching mode of free P–OH defects.49 In addition, a pronounced band at 3745 cm−1 is observed exclusively in the Si-containing catalysts, which is attributed to the presence of available Si–OH groups. This band is particularly distinct in the MgSiAlPO-5 catalyst, indicating the availability of a higher fraction of these sites, despite the fact that the Si-loadings are analogous when compared with the SiAlPO-5 catalyst (Table 1).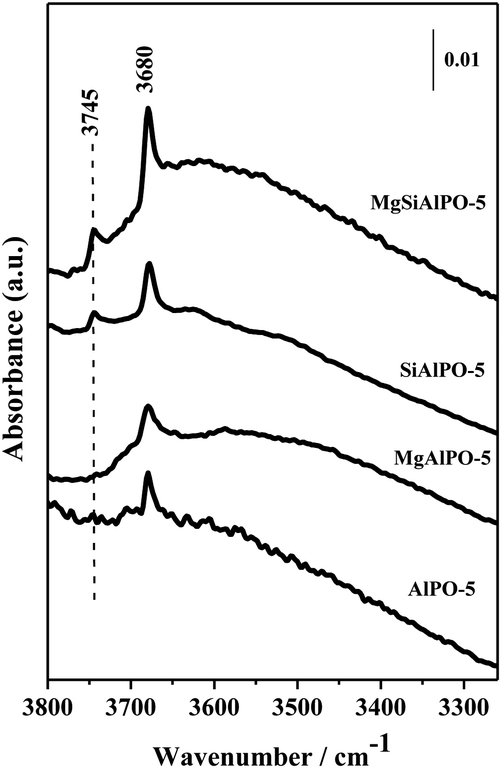 | ||
| Fig. 3 FTIR spectra highlighting the OH stretching region of the Mg-, Si-, and MgSiAlPO-5. Un-doped AlPO-5 is also shown for comparison. | ||
Due to the high scattering profile of the AlPO-5 catalysts in the characteristic Brønsted acid site region (typically 3600–3500 cm−1 in the IR spectrum), the use of probe molecules is necessary in order to gain a more detailed understanding of the Brønsted acid sites present in metal-substituted AlPO-5 catalysts. In addition, it has to be noted that, in the Si-substituted AlPO-5 catalysts, the band due to Si–OH groups (3745 cm−1) is prominently discernible; indicating that the isomorphous incorporation of Si, during synthesis, generates defects in the AFI framework. Hence in this study, a weak base (typically CO) and a strong base such as 2,6-dimethylpyridine (2,6-DMP) were employed as contrasting probe molecules, in order to elucidate the acidity of the OH groups present in the AlPO-5 catalysts.
CO was adsorbed at 80 K on all four nanoporous catalysts, in order to specifically observe and decipher its interaction with the OH groups (Fig. 4). (Note that CO is only able to interact, via H-bonding with hydroxyl groups, at low temperatures). The hydrogen-bonded C–O stretching mode emerges in the 2155–2175 cm−1 range, whereas the peak at 2138 cm−1 is attributed to liquid-like CO,50 with the latter being particularly pronounced on adsorption of 5 mbar of CO. The spectrum of undoped AlPO-5 exhibits a band at 2165 cm−1, which increases with increased CO pressure (red curve); and in conjunction with the band at 2138 cm−1 that has been previously attributed to liquid-like CO. The band at 2165 cm−1 is assigned to CO interacting exclusively with P–OH groups,51 as these are the only hydroxyl groups present in the undoped AlPO-5 catalyst. In the spectra of the Si-containing analogues, the band at 2165 cm−1 displays a shoulder (particularly visible upon adsorption of 5 mbar of CO) at ca. 2156 cm−1, which can be associated with the interaction of CO with the pendant surface silanol groups52 present on these catalysts. The metal-substituted AlPO-5 catalysts reveal a further component at 2171 cm−1, which can be assigned to the stretching mode of the CO, interacting via H-bonding, with the more acidic hydroxyl groups that can be attributed to the Si–OH–Al or the Mg–OH–P Brønsted acid centres.53–56 It is indeed noteworthy that the feature at 2171 cm−1 provides strong evidence for framework incorporation of the Mg2+ and Si4+ ions into the AlPO-5 architecture, as is evidenced from the creation of its associated Brønsted acid site (Fig. 4). As such, through the use of CO as a molecular probe, it was possible to identify three separate hydroxyl groups, which due to the high scattering profile of the AlPO-5 catalysts were not visible in the O–H stretching region (cf.Fig. 3). Nevertheless, it should be pointed out that CO is unable to discriminate between Brønsted sites of different acid strengths (see later).
 | ||
| Fig. 4 FTIR spectra of CO adsorbed at 80 K on the AlPO-5 catalysts; black curve: 1 mbar, red curve: 5 mbar of CO. | ||
In order to further elucidate the nature and the strength of the Brønsted acid sites, a stronger base such as 2,6-DMP is required. Compared to pyridine, 2,6-DMP yields the protonated species more readily, due to its stronger basicity57,58 and its weaker affinity for Lewis acid sites, owing to the steric hindrance induced by the methyl groups.59 In Scheme 2, the IR active modes of liquid and adsorbed 2,6-DMP are highlighted. In particular, the ν8a mode, which in the liquid-phase appears at 1594 cm−1, is very sensitive and allows the identification of different types of 2,6-DMP adsorption on solid catalysts. It is also striking that when the ν8a wavenumber is higher than 1625 cm−1, it characterises the presence of protonated species (2,6-DMPH+), whereas lower wavenumber correspond to coordinated or H-bonded species (DMPL).
 | ||
| Scheme 2 IR active modes highlighting the different types of 2,6-DMP/surface interactions with the catalyst. | ||
In Fig. 5A, the FTIR spectra collected after adsorption of 2,6-DMP at 4 mbar at different contact times on the nanoporous AlPO-5 catalysts are presented. At the shorter contact times (2 minutes; black curves), bands at 1595 and 1582 cm−1, due to the ν8a and ν8b modes of liquid-like 2,6-DMP are visible, together with complex bands in the 1650–1600 cm−1 range, assigned to the ν8a and ν8b modes of coordinated, protonated and H-bonded 2,6-DMP species (as shown in Scheme 2). Bands in the 1500–1450 cm−1 range are also present: these are due to the ν19a and ν19b modes (as illustrated in Scheme 2) and can also be attributed to the bending modes of the –CH3 groups. It should however be noted that the peaks in this region (1500–1450 cm−1) are quite insensitive to the different coordination environments and yield very little in terms of valuable information pertaining to the nature of acid sites present in our catalysts. After prolonged contact times (90 minutes; red curves), the bands in the 1650–1600 cm−1 range increase in intensity and the bands due to the liquid-like molecule decrease. In the un-doped AlPO-5 catalyst, in which only P–OH groups are present (as evidenced earlier by CO adsorption studies; Fig. 4), the bands in the 1650–1600 cm−1 range can be assigned to 2,6-DMP, interacting via H-bonding, with the P–OH sites that are generated immediately after short exposure (2 minutes); and as expected, do not increase after prolonged contact times.
 | ||
| Fig. 5 (A) FTIR spectra of 2,6-DMP (4 mbar) adsorbed at 298 K on mono- and bi-substituted AlPO-5 catalysts; black curve: 2 min contact, red curve: 90 min contact. (B) Corresponding FTIR spectra of outgassed 2,6-DMP at 298 K on mono- and bi-substituted AlPO-5 catalysts. | ||
The ν8a mode of the protonated 2,6-DMP is present in all four catalysts (1655–1640 cm−1 range) and, from the analysis of their relative peak positions, it is possible to deduce information on the nature and strength of the Brønsted acid sites:60 the stronger the Brønsted acidity, the lower is the position of the ν8a band. With a view to clarifying further the position of this ν8a band, the FTIR spectra of the catalysts, outgassed at 298 K after 2,6-DMP adsorption, have been examined (Fig. 5B). It was noted that by outgassing the catalysts, the spectra shows only spectroscopic features of the irreversibly adsorbed 2,6-DMP species. In particular, it was noted that the position of the peak maxima of the ν8a band was markedly different (Table 2) in the four catalysts used in this study and this could be used to differentiate the nature and strength of their Brønsted acid sites. By combining the spectroscopic information outlined in Fig. 5 and Table 2, it has been possible to contrast the relative acid strengths of the undoped, mono-substituted and bi-substituted catalysts and categorise the degree of their Brønsted acidity in the following order: MgAlPO-5 > MgSiAlPO-5 > SiAlPO-5 > AlPO-5; with MgAlPO-5 possessing a greater fraction of the strong Brønsted acid sites, whilst the undoped and mono-substituted Si-analogues exhibit a greater proclivity for weak Brønsted acid sites.
| Catalysts | ν 8a(CC)/cm−1 | A/cm−1 | N/mmol g−1 |
|---|---|---|---|
| AlPO-5 | 1650 | 0.35 | 87 |
| SiAlPO-5 | 1648 | 0.37 | 92 |
| MgSiAlPO-5 | 1645 | 0.92 | 230 |
| MgAlPO-5 | 1642 | 0.74 | 185 |
NH3-TPD measurements were also performed in order to gain a better insight into the acid-site density (Table S4†). The data shows that the MgAlPO-5 catalyst possesses a significantly larger fraction of strong acid sites compared to the MgSiAlPO-5 and SiAlPO-5 catalysts; in good agreement with the FTIR findings on the position of the ν8a(CC) bands (Table 2). The NH3-TPD data, however, shows only subtle differences between the MgSiAlPO-5 and SiAlPO-5 catalysts. Given the similarity in size between the reagents used in this study (cyclohexanone oxime and benzene), it is more appropriate to use 2,6-DMP (instead as NH3) as a molecular probe for accentuating these subtle differences and for selectively probing the specific nature of the catalytically active species, in greater depth, by quantifying the FTIR signals of the protonated (2,6-DMPH+) molecules. By further resolving and fitting the peaks associated with the ν8a(CC) band (Fig. S12†), it is possible to estimate the total number of OH sites in the all samples using the Lambert–Beer law in the form A = εNρ; where A is the intensity of the band (integrated area, cm−l), ε is the molar extinction coefficient (cm μmol−1) of the ν8a(CC) band of 2,6-DMPH+ species, N is the concentration of the vibrating species (mmol g−1) and ρ is the density of the disk (mass/area ratio of the pellet, mg cm−2).55 For the calculations, a value of 4 cm μmol−1 was used for ε.60 By using this approach, we found that the amount of OH sites in the MgSiAlPO-5 catalyst is actually higher than that calculated for the SiAlPO-5 catalyst. We therefore believe that the quantification of the number and strength of acid sites using 2,6-DMP as a probe is invaluable in determining the difference in acidic density; with the TPD providing complementary evidence for these species.
Catalysis
The varying degrees of Brønsted acid strength (as outlined from this spectroscopic study so far), which have been categorised on the basis of the interaction of 2,6-DMP with Brønsted acid centres in porous solids, prompted us to investigate the catalytic potential of these materials for solid-acid catalysed transformations. Given the intrinsic nature and strength of these solid-acid sites, we devised a series of complementary catalytic tests that would precisely implicate the role of strong, weak and intermediate Brønsted acid centres, thereby facilitating structure–property correlations to be established. With a view to achieving this goal, we investigated the Beckmann rearrangement of cyclohexanone oxime and the isopropylation of benzene to cumene, employing the mono-substituted, bi-substituted and undoped AlPO-5 catalysts.Catalytic tests for the vapour-phase Beckmann rearrangement of cyclohexanone oxime reveal that the SiAlPO-5 and MgSiAlPO-5 catalysts were significantly superior to MgAlPO-5, over a wide-range of temperatures (Fig. 6). In order to rationalise the differences in catalytic activity observed for the mono- and bi-substituted AlPO-5 catalysts, it would be prudent to consider the reaction mechanism. The reaction mechanism for the Beckmann rearrangement of cyclohexanone oxime has been extensively studied35,45,61–63 and is outlined in Scheme 3.
 | ||
| Fig. 6 Influence of temperature on the activity (A) and selectivity (B) in the Beckmann rearrangement of cyclohexanone oxime using SiAlPO-5, MgAlPO-5 and MgSiAlPO-5. Reaction conditions: Feed ≅ 300 g l−1 cyclohexanone oxime in methanol, catalyst = 0.25 g, WHSV = 3.3 h−1, helium flow rate = 20 ml min−1. | ||
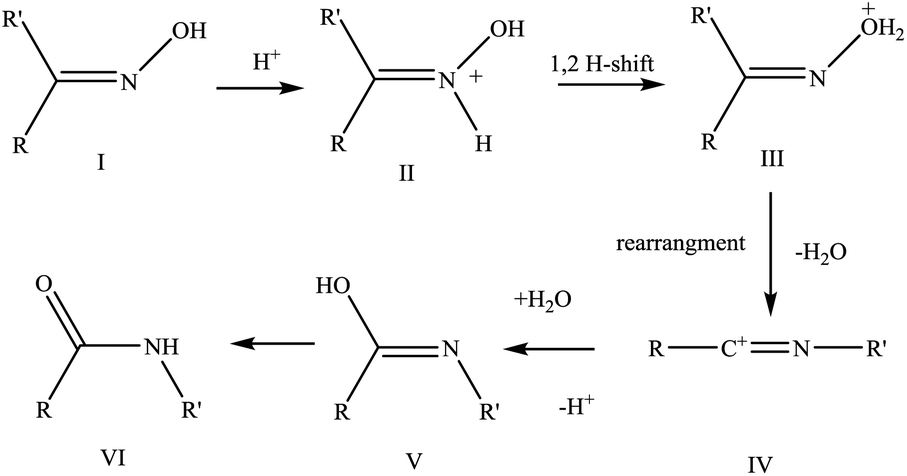 | ||
| Scheme 3 Reaction mechanism for the Beckmann rearrangement of cyclohexanone oxime. | ||
The first step is the protonation of the oxime molecule at the nitrogen atom (II); then the hydrogen is transferred from the nitrogen to the oxygen atom of the oxime (III), followed by migration of an R group (R = alkyl, aryl or hydrogen) trans to the hydroxyl group (IV), resulting in the liberation of water. Subsequently, the displaced water hydrolyses the cation (IV) to give compound (V), which finally tautomerizes to the corresponding amide (VI). Theoretical and experimental studies35,46,63 on the interaction of the oxime molecule with the acid sites (Brønsted sites or silanols), have shown that when only silanol groups are present, no hydrogen-atom transfer occurs from the catalyst surface and consequently the oxime is not protonated. On the other hand, when Brønsted acid sites are present, there is a complete transfer of the proton leading to the formation of the N-protonated oxime. These studies35,46,61–63 demonstrate that the protonation of the oxime, the first step in the Beckmann rearrangement pathway, strongly depends on the acid strength of the OH groups present on the catalyst surface; and in particular, suggests that Brønsted acid sites are the most active for this step.35,46,63 These observations strongly support the belief that the pendant silanol groups, observed during the spectroscopic FTIR characterisation, are not the active sites for this reaction; rather it is the Brønsted acid sites, resulting from the framework substitution of the heteroatoms, that initiate this reaction.
The discernible inactivity of the MgAlPO-5 catalyst further suggests that, neither the strongly acidic Mg–OH–P sites nor the weakly acidic P–OH groups observed by FTIR are the active centres for this reaction (Fig. 6A). The latter of these acid sites are too weak to facilitate a reaction, whilst the former is strongly acidic and, as such, shows a greater propensity for interacting with the solvent molecules, thereby hindering any favourable interaction between the oxime and the acid site; which hinders the production of the desired ε-caprolactam.27 This effect would be particularly pronounced within a microporous architecture, where the solvent molecules would line the pore-walls. In this scenario, the 7.3 Å cylindrical pore aperture of the AlPO-5 framework would be significantly diminished, hindering the diffusion of the bulky oxime molecule through the channels, preventing it from accessing the active sites inside the pore. It is believed that it is a combination of these factors that are responsible for the lower activity of the MgAlPO-5 catalyst.
In general it was found that the conversions increased with temperature: this behaviour can be attributed to an improved rate of diffusion of the oxime through the constrained pores and enhanced desorption of methanol with increasing temperature, resulting in a two-fold increase in the number of available active sites, which leads to the increase in catalytic efficiency. In contrast, the selectivity of the MgAlPO-5 is found to decrease with increasing temperature: this can be rationalised on the basis that the strong Brønsted acid sites facilitate the enhanced production of side-products, which can often lead to coking and deactivation of the active site.45,64 Furthermore, the stronger Brønsted acid sites present in MgAlPO-5 will anchor the ε-caprolactam product to the catalyst surface, and as such increasing the temperature will promote the formation of dimers, oligomers and ring-opening products through its prolonged exposure to the active site.26
It is observed that the MgSiAlPO-5 and SiAlPO-5 catalysts show almost an identical activity profile over a wide-range of temperatures (Fig. 6A); yet, the MgSiAlPO-5 consistently demonstrates a greater specificity for the ε-caprolactam product (Fig. 6B). These observations can be rationalised by considering the nature and type of hydroxyl groups present in the two catalysts. By employing in situ FTIR spectroscopy, it was found that both catalysts possessed a range of Brønsted acid sites, as a result of metal-incorporation, in addition to the silanol groups. As previously discussed, it is believed that it is the isolated Brønsted acid sites that are active for this reaction, and not the silanol groups. We also observed (through the adsorption of 2,6-DMP) that the Brønsted acid sites present in MgSiAlPO-5 are unique to those present in the MgAlPO-5 and SiAlPO-5 catalysts. Specifically, it was found that strength of the Brønsted acid centres present in the MgSiAlPO-5 catalyst is intermediary between that of the MgAlPO-5 and SiAlPO-5 catalysts.
It has been shown that in the MgAlPO-5 catalysts only strong Brønsted acid sites (Mg–OH–P) were present. Such sites appear to have a significant detrimental effect on the catalytic performance of the material, as evidenced by the significantly low conversions achieved. In the SiAlPO-5 catalyst, the silanols and Brønsted sites, Si–OH–Al, are both considerably weaker acid sites compared to Mg–OH–P, which vindicates its superior performance over that of MgAlPO-5 catalyst. In subtle contrast, the MgSiAlPO-5 catalyst contains both types of Brønsted acid sites (Mg–OH–P and Si–OH–Al) and silanol species. The catalytic results from above (Fig. 6) coupled with the findings from the FTIR spectroscopy using 2,6-DMP as a molecular probe, lead us to believe that the presence of both types of Brønsted acid centres prompts a synergistic cooperation between the two sites, which manifests in the MgSiAlPO-5 catalyst possessing an acid strength that is intermediary between that of MgAlPO-5 and SiAlPO-5. This results in the MgSiAlPO-5 catalyst retaining an activity profile that is comparable to that of the SiAlPO-5 catalyst, though boasting a markedly superior selectivity. Despite the MgSiAlPO-5 having a consistently higher specificity for ε-caprolactam, the selectivity of the SiAlPO-5 progressively increased with temperature. This can be attributed to thermodynamic factors, whereby the increased temperature would allow the bound ε-caprolactam to overcome the desorption energy-barrier, thereby enhancing the desorption of ε-caprolactam from the surface of the catalyst. In doing so, this would minimise the contact time between the product and the active acid sites and hence there would be a considerably lower concentration of surface-bound ε-caprolactam to partake in by-product formation; thus lowering the rates and kinetics for the formation of higher molecular weight condensation products derived from ε-caprolactam.
The influence of WHSV (weight hourly space velocity) at 673 K (the best results were obtained at this temperature – see Fig. 6) was also investigated (Fig. 7 & S13†), with a view to probing the influence of contact time on the activity and selectivity of the mono-substituted and bi-substituted catalysts. The increase in catalytic activity at lower WHSV can be attributed to the increased contact times of the substrate to the loci of the solid acid active centres at the expense of fewer solvent molecules that are in the vicinity, which play a detrimental role in blocking access to these active centres. We further note that by decreasing the WHSV (Fig. 7A & B), significant differences start to manifest, which translates to a large (>10 mol%) difference in caprolactam yield (see Fig. S13†) between the SiAlPO-5 and MgSiAlPO-5 catalysts, which is quite revealing. These results further vindicate our earlier findings that, in addition to process variables, the nature and strength of the isolated, Brønsted acid active centres play a far more critical role in influencing the activity and selectivity of solid catalysts, in the Beckmann rearrangement of cyclohexanone oxime to ε-caprolactam.
 | ||
| Fig. 7 Influence of WHSV on activity (A) and selectivity (B) in the Beckmann rearrangement of cyclohexanone oxime using Mg and Si containing catalysts. Reaction conditions: Feed ≅ 300 g l−1 cyclohexanone oxime in methanol, catalyst = 0.25 g, WHSV = 3.3 h−1; T = 673 K, helium flow rate = 20 ml min−1. | ||
Given the diverse array of strong, intermediate and weak Brønsted acid centres that are present in our mono-substituted and bi-substituted catalysts, we devised a complementary catalytic test, which has been well-established in the literature,65–67 to involve strong Brønsted acid centres. The isopropylation of benzene to cumene is catalysed by strong Brønsted acid centres65–67 and MgAlPO-5 is known to be highly active for this reaction.68 The high activity of this catalyst has been attributed to the fact that the framework incorporation of magnesium is known to produce one of the strongest Brønsted acid sites within an AlPO architecture.69 We observed that our MgAlPO-5, which was fairly inactive for the Beckmann rearrangement of cyclohexanone oxime, was reasonably active and selective (Fig. 8) for the isopropylation of benzene, justifying our rationale from the FTIR spectroscopic observations, that this catalyst contains an overwhelming majority of strong Brønsted acid active centres.20 Consistent with the above arguments, it was noted that the SiAlPO-5 catalyst, which contains a higher proportion of weak acid sites, displays an inferior catalytic performance (Fig. 8), when compared with the MgAlPO-5. It was, however, rather surprising that the MgSiAlPO-5 again demonstrates a much higher catalytic activity, particularly when compared to MgAlPO-5.
 | ||
Fig. 8 Catalytic results for the isopropylation of benzene with isopropanol using Mg and Si containing catalysts. Reaction conditions: WHSV = 3.3 h−1; T = 323 K; benzene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :isopropanol feed ratio = 6:1 mol; He flow rate ≅ 10 ml min−1. :isopropanol feed ratio = 6:1 mol; He flow rate ≅ 10 ml min−1. | ||
Given the nature of the acid sites and on the basis of the relative acid strengths (as summarised in Table 2), it was to be expected that MgAlPO-5, containing an overwhelming majority of strong Brønsted acid sites, was more active than the SiAlPO-5 analogue. Contrary to expectations, the MgSiAlPO-5, that possesses intermediate strength Brønsted acid centres, displays superior catalytic properties when compared to its mono-substituted counterparts. In line with our earlier observations, this synergistic enhancement in catalytic activity can be attributed to the presence of both the Mg(II) and Si(IV) ions, the loci of the Mg–OH–P and Si–OH–Al Brønsted acid centres, within the same AlPO framework, and this can be further substantiated by considering the reaction mechanism. The isopropylation reaction is known65–67 to proceed via a multi-step pathway, comprising of the dehydration of the 2-propanol and subsequent alkylation of benzene. An efficient catalyst should therefore comprise of the requisite active centres that are able to successfully catalyse these individual steps. It has recently emerged11,70 that Si-containing AlPOs are efficient catalysts for the dehydration of alcohols and we believe that the Si–OH–Al Brønsted acid centres in the MgSiAlPO-5 catalyst trigger the dehydration of 2-propanol. The subsequent protonation of the olefin requires strong Brønsted acid centres and the ready availability of the Mg–OH–P sites, in close proximity to the Si–OH–Al centres, we believe, is critical for the enhanced performance observed with the MgSiAlPO-5 catalyst. The overall effectiveness in catalysing these tandem reactions is fundamental to the rate-determining step that involves the electrophilic attack of the protonated olefin and benzene.
Conclusions
A resourceful strategy for the design and creation of a novel bi-substituted heterogeneous catalyst has been developed, wherein a unique array of multifunctional solid-Brønsted acid centres (comprising of Mg–OH–P and Si–OH–Al) and silanol species have been introduced through simultaneous isomorphous substitution of the AlO4 and PO4 framework tetrahedra. The precise location, geometry, nature and strength of these active centres has been probed and characterised using a combination of structural, physico-chemical, and in situ spectroscopic techniques using molecular probes, with a view to affording structure–property correlations, in two contrasting acid-catalysed transformations that require distinct Brønsted acid centres. In particular, synergistic enhancements in catalytic behaviour have been observed with the MgSiAlPO-5 catalyst in the industrially significant Beckmann rearrangement of cyclohexanone oxime to ε-caprolactam and in the isopropylation of benzene to cumene. These findings have been rationalised in terms of the loci of the active sites and their specific role in tailoring the Brønsted acid strength for influencing the desired reaction pathway, from a mechanistic perspective, at the molecular level. By gaining a thorough understanding of the precise nature and strength of the solid-acid active centres, it has been possible to predict the type of substitution mechanism that leads to the creation of isolated single-sites for targeted catalysis. We believe that this represents the genesis of predictively designing multifunctional sites for enhancing the synergy in catalytic transformations and emphasises the importance of using a multidisciplinary approach that is allied with operando spectroscopy.Acknowledgements
We wish to thank the British-Italian partnership program for funding this research. MEP is grateful to Honeywell Int. for a PhD scholarship, and we are grateful to Compagnia di San Paolo for sponsorship to NIS – Centre of Excellence (Torino). We are also grateful to Cambridge Reactor Design (CRD) for their invaluable help in reactor design and configuration. We would also like to acknowledge Marvin Shachter and Nancy Darko of UOP for their assistance in performing NH3-TPD and BET analysis respectively.Notes and references
- M. Hartmann and L. Kevan, Chem. Rev., 1999, 99, 635–664 CrossRef CAS PubMed.
- L. Gómez-Hortigüela, F. Corà, G. Sankar, C. M. Zicovich-Wilson and C. R. A. Catlow, Chem. – Eur. J., 2010, 16, 13638–13645 CrossRef PubMed.
- R. Raja, G. Sankar and J. M. Thomas, J. Am. Chem. Soc., 2001, 123, 8153–8154 CrossRef CAS.
- R. Raja and J. M. Thomas, Proc. Natl. Acad. Sci. U. S. A., 2005, 39, 13732–13736 Search PubMed.
- R. Moyaka and M. Poliakoff, Nature, 2005, 437, 1243–1244 CrossRef PubMed.
- R. Raja, US Pat., 2010/0179317 A1, 2010.
- R. Raja and J. M. Thomas, US Pat., 2013/0245323, 2013.
- R. Raja and A. J. Paterson, US Pat., 2013/0245322, 2013.
- J. Paterson, M. E. Potter, E. Gianotti and R. Raja, Chem. Commun., 2011, 47, 517–519 RSC.
- R. M. Letihall, V. N. Shetti, S. Maurelli, M. Chiesa, E. Gianotti and R. Raja, J. Am. Chem. Soc., 2013, 135, 2915–2918 CrossRef PubMed.
- M. Lefenfeld, R. Raja, A. J. Paterson and M. E. Potter, WO Pat. 2010/085708, US Pat. 2010/0249476, 2010.
- I. M. Kohan, in Nylon Plastics Handbook, Hanser/Gardner, New York, 1995, ch. 13, p. 519 Search PubMed.
- E. Dorey, Green Nylon-6, Chemistry and Industry, 2005, vol. 18, p. 4 Search PubMed.
- G. Bellussi and C. Perego, CATTECH, 2000, 4, 4–16 CrossRef CAS.
- G. Dahlhoff, J. P. M. Niederer and W. F. Hoelderich, Catal. Rev.: Sci. Eng., 2001, 43, 381–441 CAS.
- G. Petrini, G. Leofanti, M. A. Mantegazza and F. Piagnataro, Caprolactam via Ammoximation, American Chemical Society Symp. Ser., American Chemical Society, Washington, 1996, 626, p. 33 Search PubMed.
- J. Ritz, H. Fuchs, H. Kieczka and W. C. Moran, Ullmann's Encyclopaedia of Industrial Chemistry, Vol. A5, VCH Verlagsgsellschaft mbH, Germany, 1986, p. 31 Search PubMed.
- A. Levy, R. Raja and M. E. Potter, WO Pat., 2013063244 A1, US Pat., 20130109851, 2013.
- M. Anilkumar and W. F. Hoelderich, J. Catal., 2008, 260, 17–29 CrossRef CAS PubMed.
- M. E. Potter, D. Sun, E. Gianotti, M. Manzoli and R. Raja, Phys. Chem. Chem. Phys., 2013, 15, 13288–13295 RSC.
- H. Sharghi and M. H. Sarvari, J. Chem. Res., 2003, 3, 176–178 CrossRef.
- T. Curtin, J. B. McMonagle and B. K. Hodnett, Appl. Catal., A, 1992, 93, 91–101 CrossRef CAS.
- N. Kob and R. S. Drago, Catal. Lett., 1997, 49, 229–234 CrossRef CAS.
- Z. Zhang, J. Li and X. Yang, Catal. Lett., 2007, 118, 300–305 CrossRef CAS.
- W. F. Hoelderich, J. Roseler, G. Heitmann and A. T. Liebens, Catal. Today, 1997, 37, 353–366 CrossRef.
- C. Flego and L. Dalloro, Microporous Mesoporous Mater., 2003, 60, 263–271 CrossRef CAS.
- L. X. Dai, K. Koyama, M. Miyamoto and T. Tatsumi, Appl. Catal., A, 1999, 189, 237 CrossRef CAS.
- H. Kath, R. Glaser and J. Weitkamp, Chem. Eng. Technol., 2001, 24, 150–153 CrossRef CAS.
- P. S. Singh, R. Bandyopadhyay, S. G. Hegde and B. S. Rao, Appl. Catal., A, 1996, 136, 249–263 CrossRef CAS.
- K. Chaudhari, R. Bal, A. J. Chandwadkar and S. Sivasanker, J. Mol. Catal. A: Chem., 2002, 177, 247–253 CrossRef CAS.
- G. P. Heitmann, G. Dahlhoff and W. F. Hoelderich, Appl. Catal., A, 1999, 185, 99–108 CrossRef CAS.
- T. Komatsu, T. Maeda and T. Yashima, Microporous Mesoporous Mater., 2000, 35, 173–180 CrossRef.
- J. Sirjiaraensre, T. N. Truong and J. Limtrakul, J. Phys. Chem. B, 2005, 109, 12099–12106 CrossRef PubMed.
- Y. Izumi, H. Ichihashi, Y. Shimazu, M. Kitamura and H. Sato, Bull. Chem. Soc. Jpn., 2007, 80, 1280–1287 CrossRef CAS.
- A. B. Fernandez, M. Boronat, T. Blasco and A. Corma, Angew. Chem., Int. Ed., 2005, 44, 2370–2373 CrossRef CAS PubMed.
- J. Sirijaraensre and J. Limtrakul, ChemPhysChem, 2006, 7, 2424–2432 CrossRef CAS PubMed.
- R. Anand, R. B. Khomane, B. S. Rao and B. D. Kulkarni, Catal. Lett., 2002, 78, 189–194 CrossRef CAS.
- A. Cesana, S. Palmery, R. Buzzoni, G. Spano, F. Rivetti and L. Carnelli, Catal. Today, 2010, 154, 264–270 CrossRef CAS PubMed.
- K. D. Olson, US Pat., 251168, 1988.
- M. E. Potter, A. J. Paterson and R. Raja, ACS Catal., 2012, 12, 2446–2451 CrossRef.
- H. Sato, Catal. Rev.: Sci. Eng., 1997, 39, 395–424 CAS.
- A. Aucejo, M. C. Burguet, A. Corma and V. Fornes, J. Catal., 1986, 22, 187–200 CAS.
- A. Camblor, A. Corma, H. Garcia, V. Semmer-Herledan and S. Valencia, J. Catal., 1998, 177, 267–272 CrossRef.
- H. Ichihashi and M. Kitamura, Catal. Today, 2002, 73, 23–28 CrossRef CAS.
- V. R. Reddy Marthala, Y. Jiang, J. Huang, W. Wang, R. Glaser and M. Hunger, J. Am. Chem. Soc., 2006, 128, 14812–14813 CrossRef PubMed.
- J. Sirijaraensre and J. Limtrakul, Phys. Chem. Chem. Phys., 2009, 11, 578–585 RSC.
- M. M. J. Treacey and J. B. Higgins, Collection of simulated XRD powder patterns for Zeolites, Elsevier Science and Technology, 5th edn, 2007 Search PubMed.
- L. Zhou, J. Xu, C. Chen, F. Wang and X. Li, J. Porous Mater., 2008, 15, 7–12 CrossRef CAS.
- L. Marchese, J. Chen, J. M. Thomas, S. Coluccia and A. Zecchina, J. Phys. Chem., 1994, 98, 13350–13356 CrossRef CAS.
- G. E. Ewing, J. Chem. Phys., 1962, 37, 2250–2256 CrossRef CAS PubMed.
- E. Gianotti, V. Dellarocca, E. C. Oliveira, S. Coluccia, H. O. Pastore and L. Marchese, Stud. Surf. Sci. Catal., 2002, 142, 1419–1426 CrossRef.
- E. Gianotti, C. Bisio, L. Marchese, M. Guidotti, N. Ravasio, R. Psaro and S. Coluccia, J. Phys. Chem. C, 2007, 111, 5083–5089 CAS.
- S. Bordiga, C. Lamberti, F. Geobaldo, A. Zecchina, G. Turnes Palomino and A. Zecchina, Langmuir, 1995, 11, 527–533 CrossRef CAS.
- O. Cairon, T. Chevreau and J.-C. Lavalley, J. Chem. Soc. Faraday Trans., 1998, 94, 3039–3047 RSC.
- G. V. A. Martins, G. Berlier, C. Bisio, S. Coluccia, H. O. Pastore and L. Marchese, J. Phys. Chem. C, 2008, 112, 7193–7200 CAS.
- K. Chakarova and K. Hadjiivanov, J. Phys. Chem. C, 2011, 115, 4806–4817 CAS.
- C. Lahousse, A. Aboulayt, F. Maugé, J. Bachelier and J. C. Lavalley, J. Mol. Catal., 1993, 84, 283–297 CrossRef CAS.
- E. P. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS PubMed.
- P. A. Jacobs and C. F. Heylen, J. Catal., 1974, 34, 267–274 CrossRef CAS.
- L. Oliviero, A. Vimont, J.-C. Lavalley, F. Romero Sarria, M. Gaillard and F. Maugé, Phys. Chem. Chem. Phys., 2005, 7, 1861–1809 RSC.
- P. S. Landis and P. B. Venuto, J. Catal., 1966, 6, 245–252 CrossRef CAS.
- A. B. Fernandez, I. Lezcano-Gonzales, M. Boronat, T. Blasco and A. Corma, J. Catal., 2007, 249, 116–119 CrossRef CAS PubMed.
- I. Lezcano-Gonzales, A. Vidal-Moya, M. Boronat, T. Blasco and A. Corma, Phys. Chem. Chem. Phys., 2010, 12, 6396–6403 RSC.
- H. Ichihiashi and M. Kiamura, Catal. Today, 2002, 73, 23–28 CrossRef.
- S. K. Saha, S. B. Waghmode, H. Maekawa, K. Koruma, Y. Kubota, Y. Sugi, Y. Oumi and T. Sano, Microporous Mesoporous Mater., 2005, 81, 277–287 CrossRef CAS PubMed.
- S. Barman, J. Appl. Sci., 2010, 10, 2602–2607 CrossRef CAS.
- J. Joni, M. Haumann and P. Wasserscheid, Appl. Catal., A, 2010, 372, 8–15 CrossRef CAS PubMed.
- S. K. Saha, S. B. Waghmode, H. Maekawa, K. Komura, Y. Kubota, Y. Sugi, Y. Oumi and T. Sano, Microporous Mesoporous Mater., 2005, 81, 289–303 CrossRef CAS PubMed.
- F. Cora, M. Alfredsson, C. M. Barker, R. G. Bell, M. D. Foster, I. Saadoune, A. Simplerer and C. R. A. Catlow, J. Solid State Chem., 2003, 176, 496–529 CrossRef CAS.
- J. N. Kondo, K. Ito, E. Yoda, F. Wakabayashi and K. Domen, J. Phys. Chem. B, 2005, 109, 10969–10972 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: This includes full details on the synthesis of the various catalysts, characterisation and catalytic protocols employed, figures detailing various substitution mechanisms in AlPO frameworks, Powder XRD analysis, Full ICP results, BET isotherms, SEM images, further FTIR spectra, TPD data, carbon balance and further graphical representation of catalytic data. See DOI: 10.1039/c3sc53088d |
| This journal is © The Royal Society of Chemistry 2014 |